
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Predicción computacional de las preferencias de aminoácidos de dominios de unión a péptidos potencialmente multiespecíficos implicados en las interacciones proteína-proteína
En este artículo
Resumen
Describimos una metodología basada en la diversificación de secuencias para estimar las preferencias de aminoácidos de sitios de unión multiespecíficos en interacciones proteína-proteína (PPIs). En esta estrategia, se generan miles de ligandos peptídicos potenciales y se seleccionan in silico, superando así algunas limitaciones de los métodos experimentales disponibles.
Resumen
Muchas interacciones proteína-proteína implican la unión de segmentos cortos de proteína a dominios de unión a péptidos. Por lo general, tales interacciones requieren el reconocimiento de motivos lineales con conservación variable. La combinación de regiones altamente conservadas y más variables en los mismos ligandos a menudo contribuye a la multiespecificidad de la unión, una propiedad común de las enzimas y las proteínas de señalización celular. La caracterización de las preferencias de aminoácidos de los dominios de unión a péptidos es importante para el diseño de mediadores de las interacciones proteína-proteína (PPI). Los métodos computacionales son una alternativa eficiente a las técnicas experimentales, a menudo costosas y engorrosas, ya que permiten el diseño de mediadores potenciales que pueden validarse posteriormente en experimentos posteriores. Aquí, describimos una metodología utilizando la aplicación Pepspec del paquete de modelado molecular Rosetta para predecir las preferencias de aminoácidos de los dominios de unión a péptidos. Esta metodología es útil cuando la estructura de la proteína receptora y la naturaleza del ligando peptídico son conocidas o se pueden inferir. La metodología comienza con un anclaje bien caracterizado del ligando, que se amplía mediante la adición aleatoria de residuos de aminoácidos. La afinidad de unión de los péptidos generados de esta manera se evalúa mediante el acoplamiento de péptidos de columna vertebral flexible para seleccionar los péptidos con las mejores puntuaciones de unión predichas. Estos péptidos se utilizan para calcular las preferencias de aminoácidos y, opcionalmente, para calcular una matriz de posición-peso (PWM) que se puede utilizar en estudios posteriores. Para ilustrar la aplicación de esta metodología, utilizamos la interacción entre subunidades del factor regulador 5 del interferón humano (IRF5), previamente conocido como multiespecífico pero guiado globalmente por un motivo conservado corto llamado pLxIS. Las preferencias estimadas de aminoácidos fueron consistentes con el conocimiento previo sobre la superficie de unión de IRF5. Las posiciones ocupadas por residuos de serina fosforilatables exhibieron una alta frecuencia de aspartato y glutamato, probablemente porque sus cadenas laterales cargadas negativamente son similares a las de la fosfoserina.
Introducción
La interacción entre dos proteínas a menudo implica la unión de segmentos cortos de aminoácidos a dominios de unión a péptidos, que se asemejan a las interfaces proteína-péptido. Las proteínas receptoras involucradas en tales interacciones proteína-proteína (PPI) a menudo tienen la capacidad de reconocer un cierto conjunto de secuencias de ligandos superpuestas pero divergentes, una propiedad conocida como multiespecificidad 1,2. El reconocimiento multiespecífico es una característica de muchas proteínas celulares, pero es particularmente notable en las enzimas y las proteínas de señalización celular3. Las proteínas que interactúan con sitios de unión multiespecíficos a menudo tienen una combinación de regiones más y menos conservadas en su secuencia 4,5,6. En este escenario, los motivos de secuencia más conservados están involucrados en interacciones moleculares estrictas. Por el contrario, las secuencias más variables interactúan con superficies de alguna manera permisivas en el sitio de unión al receptor. Por lo general, estos segmentos menos conservados, pero aún funcionalmente relevantes, son bucles que carecen de patrones de estructura secundaria definidos o tienen conformaciones aún más dinámicas, como las típicas de las proteínas intrínsecamente desordenadas7.
La identificación de posibles ligandos peptídicos de los sitios de unión suele ser el primer paso en el diseño de mediadores capaces de interferir con los PPIs correspondientes8. Sin embargo, a menudo es poco probable encontrar un único residuo de aminoácidos más frecuente en la mayoría de las posiciones de secuencia en ligandos de sitios de unión multiespecíficos. En cambio, estos sitios pueden tener preferencias particulares por una clase específica de aminoácidos de acuerdo con sus propiedades químicas, por ejemplo, aminoácidos ácidos y cargados negativamente como aspartato o glutamato, aminoácidos aromáticos voluminosos como fenilalanina o residuos más hidrofóbicos como aminoácidos alifáticos alanina, valina, leucina o isoleucina3. Varios métodos experimentales pueden proporcionar información sobre las preferencias de aminoácidos de los sitios de unión a proteínas, incluida la evolución dirigida9, la mutagénesis de escaneo multicodón10 y la exploración mutacional profunda11. Todos estos métodos siguen el enfoque de diversificación de secuencias, que se basa en la introducción de mutaciones en los ligandos originales y en el análisis posterior de su efecto sobre la función de la proteína receptora (véase Bratulic y Badran12 para una revisión exhaustiva). Sin embargo, estos métodos a menudo requieren el estudio de grandes bibliotecas de secuencias, lo que los hace más engorrosos, costosos y lentos.
Los métodos computacionales para inferir las preferencias de aminoácidos de los sitios de unión multiespecíficos tienen el potencial de eludir las limitaciones de los métodos de laboratorio húmedo. Entre estos, el enfoque de diversificación de secuencia in silico evalúa el impacto energético de una amplia gama de reemplazos de aminoácidos en la secuencia del ligando como una forma de caracterizar la plasticidad estructural del PPI13. Este método comienza con la estructura o modelo del ligando peptídico unido al sitio de unión al receptor y, posteriormente, introduce mutaciones en la secuencia del ligando. A continuación, se utilizan funciones estadísticas y de puntuación de energía para evaluar el impacto de estas mutaciones en la estabilidad y la afinidad de unión. El conjunto de secuencias de ligandos con mejor puntuación resultante de la fase de evaluación se puede utilizar para calcular las preferencias de aminoácidos. Esta estrategia tiene el potencial de procesar un número muy alto de secuencias de ligandos de manera eficiente. Por lo tanto, puede proporcionar una inferencia más completa y consistente de las preferencias de aminoácidos en comparación con las calculadas a partir del número más limitado de secuencias que generalmente se pueden procesar en enfoques de laboratorio húmedo.
La aplicación Pepspec de la suite de modelado molecularRosetta 14 es una herramienta que realiza la diversificación de secuencias como un paso clave de su modo de diseño de péptidos. Esta aplicación requiere una estructura o modelo de la proteína receptora con un péptido unido a un solo residuo de aminoácido de longitud, que se utiliza como ancla para los siguientes pasos. A continuación, la secuencia del péptido unido se amplía (si es necesario) y se diversifica para generar un gran número de ligandos peptídicos putativos. A continuación, se evalúa la afinidad de unión de estos péptidos mediante el acoplamiento de péptidos de columna vertebral flexible para seleccionar aquellos con las mejores puntuaciones de unión predichas. Aunque el resultado principal de esta aplicación son los mejores candidatos a péptidos seleccionados al final de la fase de diseño, el conjunto mucho mayor de péptidos aceptados durante esta fase también se puede utilizar para calcular las preferencias de aminoácidos del sitio de unión objetivo. Las preferencias de aminoácidos se calculan como la frecuencia de cada residuo de aminoácidos por posición de la secuencia del ligando, representada como una matriz de peso de posición (PWM) o como un logotipo de secuencia más visual.
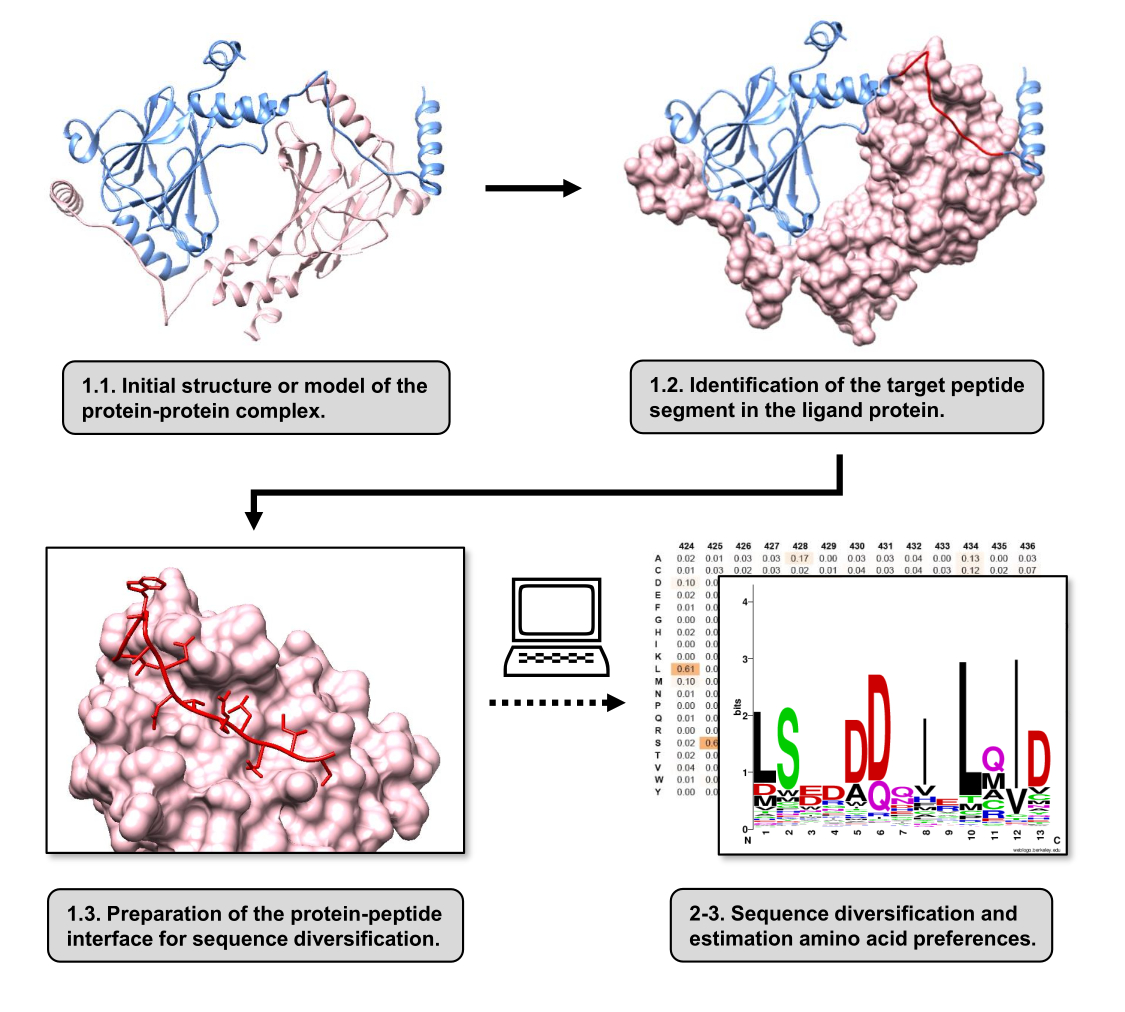
En este artículo, describimos un protocolo para estimar las preferencias de aminoácidos de la superficie de unión de una proteína receptora involucrada en un IBP. El protocolo se centra en los IBP en los que se sabe que un segmento lineal del ligando de la proteína se une a la proteína receptora, por lo que el escenario se puede modelar como una interfaz proteína-péptido. En este escenario, los motivos conservados del ligando suelen interactuar con bolsillos definidos en el sitio de unión al receptor, aunque todo el segmento del ligando implicado en el PPI puede contener regiones menos conservadas. En la Figura 1 se muestra un diagrama de flujo que resume los pasos principales del protocolo. El protocolo comienza con la estructura 3D del complejo proteína-proteína y reduce aún más la proteína ligando al segmento potencial de mejor interacción, dejando intacta la proteína receptora. El segmento que mejor interactúa se infiere mediante el uso del servidor BUDE Alanine Scan15, que realiza mutagénesis computacional de barrido con alanina para identificar residuos de puntos calientes entre las dos proteínas que interactúan. En este enfoque, los residuos del ligando se reemplazan individualmente por alanina, y el cambio estimado en la energía libre o la estabilidad del complejo (ΔΔG) se utiliza para inferir la relevancia del residuo correspondiente para el PPI objetivo. Una vez que se infiere el segmento que mejor interactúa, su complejo con la proteína receptora se utiliza como estructura base presentada a Pepspec para realizar la diversificación de secuencias.

Figura 1: Resumen de los principales pasos del protocolo propuesto en este trabajo. Los números coinciden con los números de paso de la sección de protocolo. Las figuras se realizaron con el complejo proteína-proteína utilizado como ejemplo descrito en el texto. En este complejo, la cadena de proteínas considerada como el receptor se muestra en rosa, mientras que la cadena considerada como el ligando se muestra en azul claro con su segmento de mejor interacción resaltado en rojo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Una de las limitaciones del protocolo sugerido es el requisito de una estructura resuelta de la interfaz proteína-péptido. Alternativamente, el protocolo puede comenzar con un modelo de la interfaz proteína-péptido diana, aunque los pasos específicos del modelado no se describen aquí. Además, aunque el protocolo se puede llevar a cabo en una computadora personal que ejecute cualquier sistema operativo, se requiere un entorno Linux para los pasos que involucran las aplicaciones de Rosetta. Un clúster de computadoras también es muy recomendable para el paso de diversificación de secuencias debido a la gran cantidad de iteraciones que normalmente realiza Pepspec.
La aplicación del protocolo sugerido se ilustra con la estimación de las preferencias de aminoácidos de la superficie de oferta de IRF5, un miembro de la familia del factor regulador del interferón humano (IRF). Elegimos esta proteína como ejemplo porque, durante su activación, dos subunidades se unen para formar un dímero cuya estructura está bien caracterizada16. En los dímeros IRF, la unión se puede modelar como una interfaz proteína-péptido en la que una subunidad proporciona la superficie de unión y la otra interactúa a través de una región que contiene un motivo conservado corto llamado pLxIS17,18. Además, el enlace a las subunidades IRF es multiespecífico; Por lo tanto, pueden formar homodímeros, heterodímeros y complejos con otras proteínas celulares conocidos como coactivadores18.
Protocolo
1. Preparación inicial de la interfaz proteína-péptido
- Descargando la estructura del complejo proteína-proteína
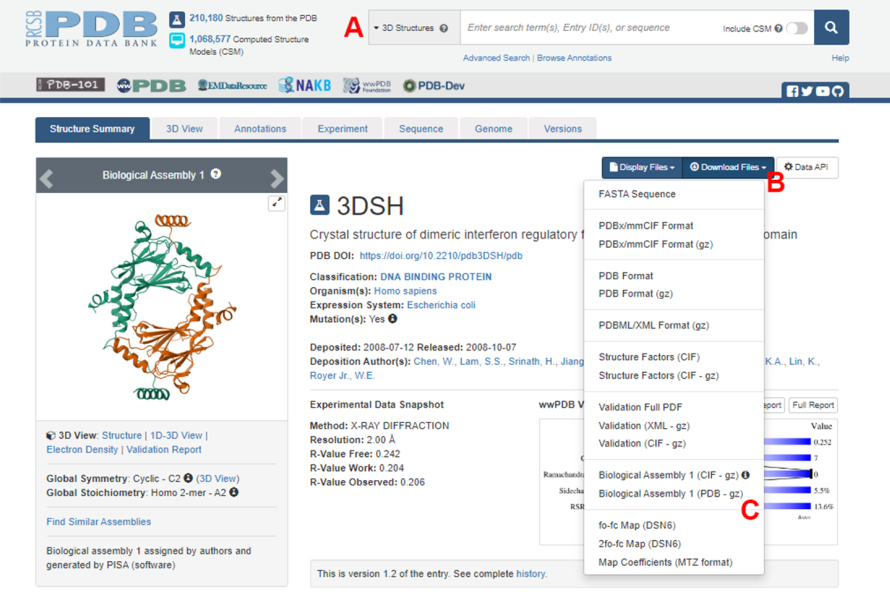
- Navegue hasta la página de inicio del Banco de Datos de Proteínas (PDB) (https://www.rcsb.org/) y escriba el ID de PDB para la estructura del complejo proteína-proteína en el cuadro de búsqueda principal (Figura 2A). El ID de PDB para la estructura del dímero IRF5, utilizado como ejemplo en este trabajo, es 3DSH19.
- En la página principal de la estructura deseada, haga clic en Descargar archivos (Figura 2B) y luego en Ensamblaje biológico 1 (PDB - gz) (Figura 2C).
NOTA: En la base de datos PDB, las estructuras de muchos complejos de proteínas formados por monómeros idénticos se representan como ensamblajes biológicos, en los que solo se almacena la estructura de un monómero (unidad asimétrica) en el archivo PDB. La estructura del multímero, en este caso, el dímero IRF5, debe descargarse como el ensamblaje biológico que contiene dos instancias de la unidad asimétrica. Para facilitar los siguientes pasos de este protocolo, primero se separan los dos monómeros y se les asignan diferentes ID de cadena. - Abra la estructura descargada en UCSF Chimera20 y haga clic en Herramientas > Edición de estructuras > cambiar ID de cadena. En este ejemplo, ambas cadenas del ensamblaje biológico se denominan A. Cambie el nombre de la segunda cadena (etiquetada como #0.2) a B y haga clic en Aceptar.
- Haga clic en Favoritos > Panel de modelos y luego seleccione el modelo que contiene las dos cadenas. Haga clic en el botón Agrupar/Desagrupar para separar cada cadena en un modelo diferente. A continuación, seleccione los dos modelos y haga clic en el botón Copiar/Combinar . Introduzca un nuevo nombre para el modelo combinado, marque Cerrar modelos de origen y haga clic en Aceptar.
- Haga clic en Seleccionar > cadena y confirme que cada cadena en el dímero ahora está identificada por una letra diferente, a saber, A y B.
- Use File > Save PDB para guardar la estructura editada en un archivo PDB diferente, que se utilizará en los siguientes pasos del protocolo (aquí, se usó el nombre IRF5_dimer.pdb ).

Figura 2: La página del Banco de Datos de Proteínas (PDB) para la estructura utilizada como ejemplo representativo en este trabajo. (A) Cuadro de búsqueda para introducir el código de acceso PDB de la estructura objetivo. (B) Menú para descargar la estructura en varios formatos. (C) Opciones para descargar ensamblajes biológicos cuando la estructura se ha guardado como una unidad asimétrica (ver paso 1.1.2 para más detalles). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Identificación del segmento diana en la proteína ligando
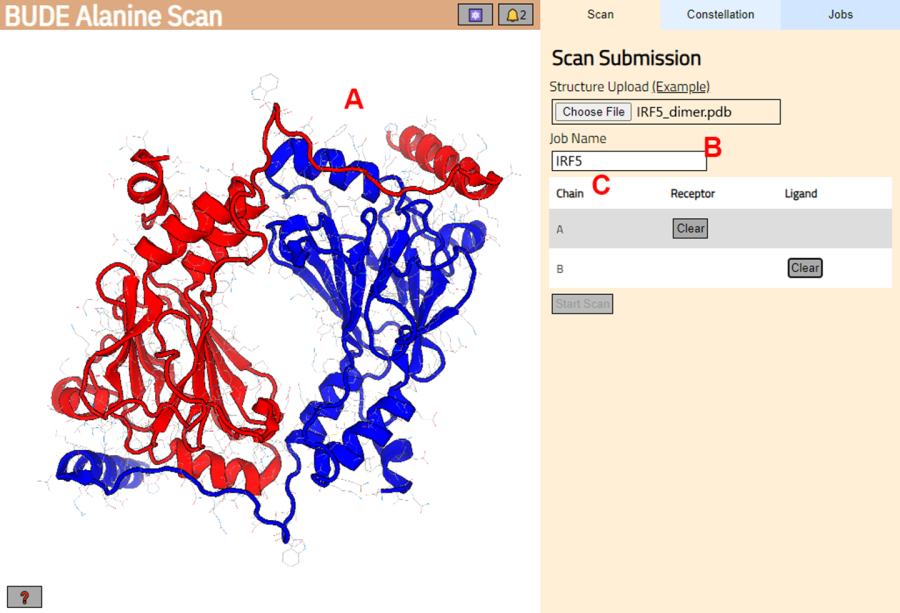
- Navegue hasta el servidor de escaneo de alanina de BUDE (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Haga clic en el botón Elegir archivo en Carga de estructura y seleccione el archivo PDB guardado en el paso 1.1.6.
- En la página siguiente, compruebe que la estructura se ha cargado correctamente (Figura 3A) y escriba un nombre para el trabajo en el servidor (Figura 3B).
- Establezca las cadenas del PDB que se tratarán como receptor (A) y ligando (B) (Figura 3C). A continuación, haga clic en el botón Iniciar análisis para enviar el trabajo.
- Una vez finalizado el trabajo, haga clic en Mostrar resultados para abrir la página de resultados (Figura 4).
NOTA: En la página de resultados, los residuos de la estructura del ligando se colorean de acuerdo con su cambio estimado en energía libre (ΔΔG), y aquellos con valores más altos se colorean en rojo. - En la lista de residuos, seleccione el tramo de residuos que se prevé que interactúe mejor con la superficie de unión objetivo. Asegúrese de que estos residuos agrupen los valores más altos de la diferencia de energía libre (ΔΔG). En este ejemplo, se seleccionó el segmento entre los residuos Leu424 y Ser436 (resaltado con un recuadro rojo en el panel derecho de la Figura 4).
- Preparación de la interfaz proteína-péptido para la diversificación de secuencias
- Abra el archivo PDB guardado en el paso 1.1.6 en Chimera y verifique que no falten átomos o enlaces en la estructura de las subunidades objetivo.
- Elimine todas las moléculas pequeñas, iones y solventes que se cocristalizaron con la estructura original. Para hacer esto, haga clic en Seleccionar > residuos y luego seleccione todas las moléculas que no sean aminoácidos estándar. A continuación, haz clic en Acciones > Átomos/Enlaces y Eliminar.
- Recorte la cadena de ligandos hasta el segmento de mejor interacción elegido en el paso 1.2.5. Para hacer esto, haga clic en Favoritos y Secuencia y luego haga clic en la cadena considerada como el ligando (B). En el panel Secuencia , arrastre el ratón para seleccionar todos los residuos excepto los que se encuentran entre las posiciones 424 y 436. Para eliminar estos residuos, haga clic en Acciones > Átomos/Enlaces y Eliminar.
- Use File > Save PDB para guardar la estructura editada en un archivo PDB diferente, que se usa en los siguientes pasos del protocolo (aquí, se usó el nombre IRF5_interface.pdb ).

Figura 3: Selección de receptor y ligando en el servidor BUDE Alanine Scan. (A) Representación gráfica del complejo proteína-proteína. (B) Cuadro de texto para introducir el nombre del trabajo en el servidor. (C) Panel para seleccionar interactivamente las cadenas que se considerarán como receptor y ligando (ver paso 1.2 para más detalles). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Página de resultados del servidor BUDE Alanine Scan. El segmento potencialmente de mejor interacción en la secuencia del ligando se indica con un recuadro rojo. En el panel izquierdo, el residuo con la mayor contribución energética prevista (Leu433) está resaltado en verde. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
2. Diversificación de secuencias
NOTA: En los siguientes pasos, rosetta_main se refiere al directorio principal de instalación de Rosetta, que normalmente se encuentra en /opt/rosetta_src__bundle/main/, donde indica la versión de Rosetta instalada. Además, se supone que las aplicaciones de Rosetta son accesibles en todo el sistema; Si este no es el caso, se debe proporcionar la ruta completa a los ejecutables. Cuando se compilan desde el origen, estos ejecutables se encuentran en el directorio /rosetta_main/source/bin/ .
- Optimización inicial de las cadenas laterales de aminoácidos
- Copie la estructura editada guardada en el paso 1.3.4 en una ubicación de Linux accesible por las aplicaciones Rosetta.
- Utilice la aplicación FixBB de Rosetta para realizar un reempaquetado de todas las cadenas laterales de aminoácidos de la estructura base antes de la diversificación de la secuencia. En esta operación, se optimiza la orientación de todas las cadenas laterales de aminoácidos para minimizar la energía y mejorar la estabilidad del complejo. Para ello, ejecute el siguiente comando:

NOTA: Este comando genera un archivo PDB con el nombre de la estructura original con un sufijo numérico adicional (IRF5_interface_0001.pdb en este ejemplo). - Para facilitar el siguiente paso del protocolo, cambie el nombre del archivo PDB reempaquetado con el sufijo _repack mediante el siguiente comando:
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- Diversificación de secuencias
- Ejecute Pepspec en modo de diseño para realizar el paso de diversificación de secuencia real utilizando el siguiente comando:

Las siguientes son opciones generales:- -s indica el archivo de entrada (el archivo PDB reempaquetado generado en el paso 2.1.3).
- -o indica el prefijo para nombrar los archivos de salida.
- - database indica la ruta a la base de datos principal de Rosetta 3.
- -ex1, -ex2 y extrachi_cutoff son opciones de biblioteca de rotadores (consulte la documentación de Pepspec para obtener más detalles).
- -overwrite indica a la aplicación que sobrescriba las posibles salidas preexistentes generadas por iteraciones anteriores.
- -pepspec:pep_chain indica las cadenas PDB consideradas como ligando ('b' en este ejemplo).
- -pepspec:native_pep_anchor indica el residuo de aminoácidos utilizado como anclaje (en este ejemplo, el residuo de Leu en la posición 10 del péptido ligando).
- -pepspec:n_peptides indica el número de estructuras peptídicas a producir.
- -pepspec:no_prepack_prot le dice a la aplicación que omita el reempaquetado en la estructura base de entrada (ya que esto se realizó anteriormente en el paso 2.1).
NOTA: La salida principal de Pepspec es un directorio que contiene los archivos PDB para los péptidos resultantes de la fase de diseño, nombrados con el prefijo de salida con el sufijo .pdbs (IRF5.pdbs en el ejemplo). Además, Pepspec genera todas las secuencias de péptidos aceptadas probadas como parte del paso de diversificación de secuencias y sus correspondientes puntuaciones de energía de Rosetta en un archivo de texto delimitado por tabulaciones que lleva el nombre del prefijo de salida, con el . spec (IRF5.spec en el ejemplo). Dado que el protocolo descrito en este trabajo tiene como objetivo estimar las preferencias de aminoácidos en lugar del diseño real del péptido, los siguientes pasos utilizan IRF5.spec en lugar de las estructuras PDB en el directorio .pdbs .
- Ejecute Pepspec en modo de diseño para realizar el paso de diversificación de secuencia real utilizando el siguiente comando:
3. Estimación de las preferencias de aminoácidos
- Cálculo de un PWM
- Para generar un PWM, utilice el script gen_pepspec_pwm.py incluido en la suite Rosetta. Para ejecutar este script, utilice el siguiente comando:

Dónde:- IRF5.spec es el archivo de salida de Pepspec generado en el paso 2.2.
- -1 indica que no hay residuos N-terminales adicionales en la secuencia y, por lo tanto, las posiciones en el PWM están basadas en 1.
- 0,2 indica al script que solo tenga en cuenta el 20% de los péptidos con mejor puntuación de la salida de Pepspec (el valor predeterminado es 0,1, correspondiente al 10%)
- interface_score le dice al script que clasifique los péptidos en función de la puntuación de la interfaz, que es una de las varias puntuaciones de Rosetta incluidas en el archivo de salida de Pepspec.
NOTA: Este script genera dos archivos de salida, uno para el PWM calculado (con el sufijo .pwm ) y el otro para las secuencias del subconjunto de péptidos utilizados para calcular el PWM (con el sufijo .seq ). Los nombres de estos archivos también incluyen la puntuación y la fracción de péptidos utilizados para la clasificación. En este ejemplo, estos archivos se denominan respectivamente IRF5_interface_score_0.2.pwm y IRF5_interface_score_0.2.seq.
- Para generar un PWM, utilice el script gen_pepspec_pwm.py incluido en la suite Rosetta. Para ejecutar este script, utilice el siguiente comando:
- Generación de un logotipo de secuencia
- Navegue hasta el servidor WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 y haga clic en el botón Elegir archivo junto a Cargar datos de secuencia. Cargue el archivo con las secuencias peptídicas generadas en el paso 3.1.1 (IRF5_interface_score_0.2.seq en este ejemplo).
- Elija el formato y el tamaño deseados del logotipo de acuerdo con la longitud de entrada. El ejemplo utiliza formato PDF y un tamaño de 15 cm x 12 cm. Haga clic en Crear logotipo.
Resultados
En este artículo, describimos un protocolo para predecir las preferencias de aminoácidos de la superficie de unión de IRF5, un miembro de una familia de factores de transcripción conocidos como factores reguladores del interferón humano. Estas proteínas son reguladoras de las respuestas inmunitarias innatas y adaptativas y participan en la diferenciación y activación de varias células inmunitarias. Las subunidades IRF tienen superficies de unión altamente plásticas y multiespe...
Discusión
El presente artículo describe un protocolo para estimar las preferencias de aminoácidos de sitios de unión potencialmente multiespecíficos basado en la diversificación de secuencias in silico. Se han desarrollado pocas herramientas computacionales para estimar las preferencias de aminoácidos de las interfaces proteína-péptido 14,25,26. Estas herramientas tienen una naturaleza predictiva,...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Se agradece el apoyo financiero del Sistema Nacional de Investigación (SNI) (subvenciones números SNI-043-2023 y SNI-170-2021), la Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) de Panamá y el Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU). Los autores desean agradecer al Dr. Miguel Rodríguez por la cuidadosa revisión del manuscrito.
Materiales
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Referencias
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados