
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Prédiction computationnelle des préférences en acides aminés de domaines de liaison à des peptides potentiellement multispécifiques impliqués dans des interactions protéine-protéine
Dans cet article
Résumé
Nous décrivons une méthodologie basée sur la diversification des séquences pour estimer les préférences en acides aminés des sites de liaison multispécifiques dans les interactions protéine-protéine (IPP). Dans cette stratégie, des milliers de ligands peptidiques potentiels sont générés et criblés in silico, surmontant ainsi certaines limites des méthodes expérimentales disponibles.
Résumé
De nombreuses interactions protéine-protéine impliquent la liaison de courts segments de protéines à des domaines de liaison aux peptides. Habituellement, de telles interactions nécessitent la reconnaissance de motifs linéaires à conservation variable. La combinaison de régions hautement conservées et plus variables dans les mêmes ligands contribue souvent à la multispécificité de la liaison, une propriété commune des enzymes et des protéines de signalisation cellulaire. La caractérisation des préférences en acides aminés des domaines de liaison aux peptides est importante pour la conception de médiateurs des interactions protéine-protéine (IPP). Les méthodes de calcul sont une alternative efficace aux techniques expérimentales souvent coûteuses et lourdes, permettant de concevoir des médiateurs potentiels qui peuvent ensuite être validés dans des expériences en aval. Ici, nous avons décrit une méthodologie utilisant l’application Pepspec du package de modélisation moléculaire Rosetta pour prédire les préférences en acides aminés des domaines de liaison aux peptides. Cette méthodologie est utile lorsque la structure de la protéine réceptrice et la nature du ligand peptidique sont toutes deux connues ou peuvent être déduites. La méthodologie commence par une ancre bien caractérisée du ligand, qui est prolongée par l’ajout aléatoire de résidus d’acides aminés. L’affinité de liaison des peptides générés de cette manière est ensuite évaluée par l’amarrage des peptides de squelette flexible afin de sélectionner les peptides avec les meilleurs scores de liaison prédits. Ces peptides sont ensuite utilisés pour calculer les préférences en acides aminés et pour calculer éventuellement une matrice position-poids (PWM) qui peut être utilisée dans d’autres études. Pour illustrer l’application de cette méthodologie, nous avons utilisé l’interaction entre les sous-unités du facteur de régulation 5 de l’interféron humain (IRF5), précédemment connu pour être multispécifique mais globalement guidé par un motif court conservé appelé pLxIS. Les préférences estimées en acides aminés étaient cohérentes avec les connaissances antérieures sur la surface de liaison d’IRF5. Les positions occupées par les résidus de sérine phosphorylables présentaient une fréquence élevée d’aspartate et de glutamate, probablement parce que leurs chaînes latérales chargées négativement sont similaires à celles de la phosphosérine.
Introduction
L’interaction entre deux protéines implique souvent la liaison de courts segments d’acides aminés à des domaines de liaison peptidiques, ressemblant à des interfaces protéine-peptide. Les protéines réceptrices impliquées dans de telles interactions protéine-protéine (IPP) ont souvent la capacité de reconnaître un certain ensemble de séquences de ligands qui se chevauchent mais divergent, une propriété connue sous le nom de multispécificité 1,2. La reconnaissance multispécifique est une caractéristique de nombreuses protéines cellulaires, mais elle est particulièrement remarquable dans les enzymes et les protéines de signalisation cellulaire3. Les protéines qui interagissent avec des sites de liaison multispécifiques ont souvent une combinaison de régions plus ou moins conservées dans leur séquence 4,5,6. Dans ce scénario, les motifs de séquence les plus conservés sont impliqués dans des interactions moléculaires strictes. À l’inverse, les séquences les plus variables interagissent avec des surfaces permissives dans le site de liaison du récepteur. Habituellement, ces segments moins conservés mais toujours pertinents sur le plan fonctionnel sont des boucles dépourvues de modèles de structure secondaire définis ou ont des conformations encore plus dynamiques, telles que celles typiques des protéines intrinsèquement désordonnées7.
L’identification de ligands peptidiques potentiels de sites de liaison est généralement la première étape de la conception de médiateurs capables d’interférer avec les IPP correspondants8. Cependant, il est souvent peu probable de trouver un seul résidu d’acide aminé le plus fréquent à la plupart des positions de séquence dans les ligands des sites de liaison multispécifiques. Au lieu de cela, ces sites peuvent avoir des préférences particulières pour une classe spécifique d’acides aminés en fonction de leurs propriétés chimiques, par exemple, les acides aminés acides et chargés négativement tels que l’aspartate ou le glutamate, les acides aminés aromatiques volumineux tels que la phénylalanine ou les résidus plus hydrophobes tels que les acides aminés aliphatiques alanine, valine, leucine ou isoleucine3. Plusieurs méthodes expérimentales peuvent fournir des informations sur les préférences en acides aminés des sites de liaison aux protéines, notamment l’évolution dirigée9, la mutagenèse à balayage multi-codons10 et le balayage mutationnel profond11. Toutes ces méthodes suivent l’approche de la diversification des séquences, qui est basée sur l’introduction de mutations dans les ligands originaux et l’analyse plus approfondie de leur effet sur la fonction de la protéine réceptrice (voir Bratulic et Badran12 pour une revue complète). Cependant, ces méthodes nécessitent souvent l’étude de grandes bibliothèques de séquences, ce qui les rend plus lourdes, plus coûteuses et plus longues.
Les méthodes informatiques permettant de déduire les préférences en acides aminés des sites de liaison multispécifiques ont le potentiel de contourner les limites des méthodes de laboratoire humide. Parmi celles-ci, l’approche de diversification de séquences in silico évalue l’impact énergétique d’une large gamme de remplacements d’acides aminés dans la séquence de ligands afin de caractériser la plasticité structurale de l’IPP13. Cette méthode commence par la structure ou le modèle du ligand peptidique lié au site de liaison du récepteur et introduit ensuite des mutations dans la séquence du ligand. Des fonctions statistiques et de scoring énergétique sont ensuite utilisées pour évaluer l’impact de ces mutations sur la stabilité et l’affinité de liaison. L’ensemble des séquences de ligands les mieux notées résultant de la phase d’évaluation peut ensuite être utilisé pour calculer les préférences en acides aminés. Cette stratégie a le potentiel de traiter un très grand nombre de séquences de ligands de manière efficace. Par conséquent, il peut fournir une inférence plus complète et cohérente des préférences en acides aminés par rapport à celles calculées à partir du nombre plus limité de séquences qui peuvent généralement être traitées dans les approches de laboratoire humide.
L’application Pepspec de la suite de modélisation moléculaireRosetta 14 est un outil qui effectue la diversification des séquences comme une étape clé de son mode de conception peptidique. Cette application nécessite une structure ou un modèle de la protéine réceptrice avec un peptide lié jusqu’à un seul résidu d’acide aminé de longueur, qui est utilisé comme point d’ancrage pour les étapes suivantes. La séquence du peptide lié est ensuite étendue (si nécessaire) et diversifiée pour générer un grand nombre de ligands peptidiques peptidiques présumés. L’affinité de liaison de ces peptides est ensuite évaluée par l’amarrage de peptides de squelette flexible afin de sélectionner ceux dont les scores de liaison sont les mieux prédits. Bien que le principal résultat de cette application soit les meilleurs candidats peptidiques sélectionnés à la fin de la phase de conception, l’ensemble beaucoup plus large de peptides acceptés au cours de cette phase peut également être utilisé pour calculer les préférences en acides aminés du site de liaison cible. Les préférences en acides aminés sont calculées comme la fréquence de chaque résidu d’acide aminé par position de la séquence de ligand, représentée soit par une matrice de poids de position (PWM), soit par un logo de séquence plus visuel.
Dans cet article, nous décrivons un protocole permettant d’estimer les préférences en acides aminés de la surface de liaison d’une protéine réceptrice impliquée dans un IPP. Le protocole se concentre sur les IPP dans lesquels un segment linéaire du ligand protéique est connu pour se lier à la protéine réceptrice, de sorte que le scénario peut être modélisé comme une interface protéine-peptide. Dans ce scénario, les motifs conservés du ligand interagissent généralement avec des poches définies dans le site de liaison du récepteur, bien que l’ensemble du segment du ligand impliqué dans l’IPP puisse contenir des régions moins conservées. La figure 1 présente un organigramme résumant les principales étapes du protocole. Le protocole commence par la structure 3D du complexe protéine-protéine et réduit davantage la protéine ligand au segment potentiel qui interagit le mieux, laissant la protéine réceptrice intacte. Le segment qui interagit le mieux est déduit en utilisant le serveur BUDE Alanine Scan15, qui effectue une mutagenèse computationnelle par balayage de l’alanine pour identifier les résidus de points chauds entre les deux protéines en interaction. Dans cette approche, les résidus du ligand sont remplacés individuellement par l’alanine, et la variation estimée de l’énergie libre ou de la stabilité du complexe (ΔΔG) est ensuite utilisée pour déduire la pertinence du résidu correspondant pour l’IPP cible. Une fois que le segment qui interagit le mieux est déduit, son complexe avec la protéine réceptrice est utilisé comme structure de base soumise à Pepspec pour effectuer la diversification de la séquence.

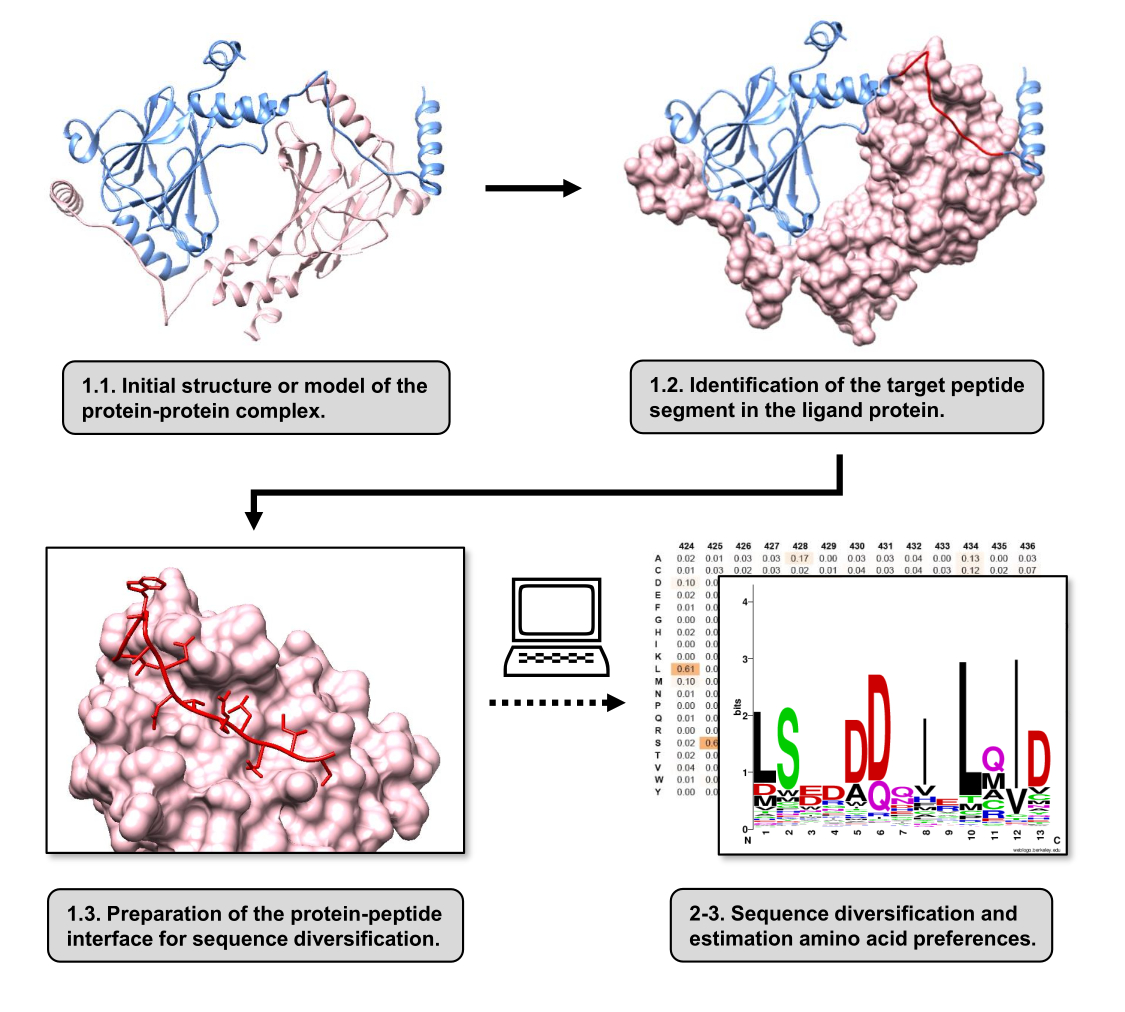
Figure 1 : Vue d’ensemble des principales étapes du protocole proposé dans ce travail. Les numéros correspondent aux numéros d’étape dans la section protocole. Les figures ont été faites avec le complexe protéine-protéine utilisé comme exemple décrit dans le texte. Dans ce complexe, la chaîne protéique considérée comme le récepteur est représentée en rose, tandis que la chaîne considérée comme le ligand est représentée en bleu clair avec son segment prédit qui interagit le mieux mis en évidence en rouge. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’une des limites du protocole suggéré est l’exigence d’une structure résolue de l’interface protéine-peptide. Le protocole peut également commencer par un modèle de l’interface protéine-peptide cible, bien que les étapes de modélisation spécifiques ne soient pas décrites dans le présent document. De plus, bien que le protocole puisse être exécuté sur un ordinateur personnel exécutant n’importe quel système d’exploitation, un environnement Linux est nécessaire pour les étapes impliquant les applications Rosetta. Un cluster d’ordinateurs est également fortement recommandé pour l’étape de diversification de séquences en raison du grand nombre d’itérations généralement effectuées par Pepspec.
L’application du protocole suggéré est illustrée par l’estimation des préférences en acides aminés de la surface d’attente d’IRF5, un membre de la famille des facteurs de régulation de l’interféron humain (IRF). Nous avons choisi cette protéine comme exemple car, lors de son activation, deux sous-unités se lient pour former un dimère dont la structure est bien caractérisée16. Dans les dimères IRF, la liaison peut être modélisée comme une interface protéine-peptide dans laquelle une sous-unité fournit la surface de liaison et l’autre interagit à travers une région contenant un court motif conservé appelé pLxIS17,18. De plus, la liaison aux sous-unités IRF est multispécifique ; Par conséquent, ils peuvent former des homodimères, des hétérodimères et des complexes avec d’autres protéines cellulaires appelées coactivateurs18.
Access restricted. Please log in or start a trial to view this content.
Protocole
1. Préparation initiale de l’interface protéine-peptide
- Téléchargement de la structure du complexe protéine-protéine
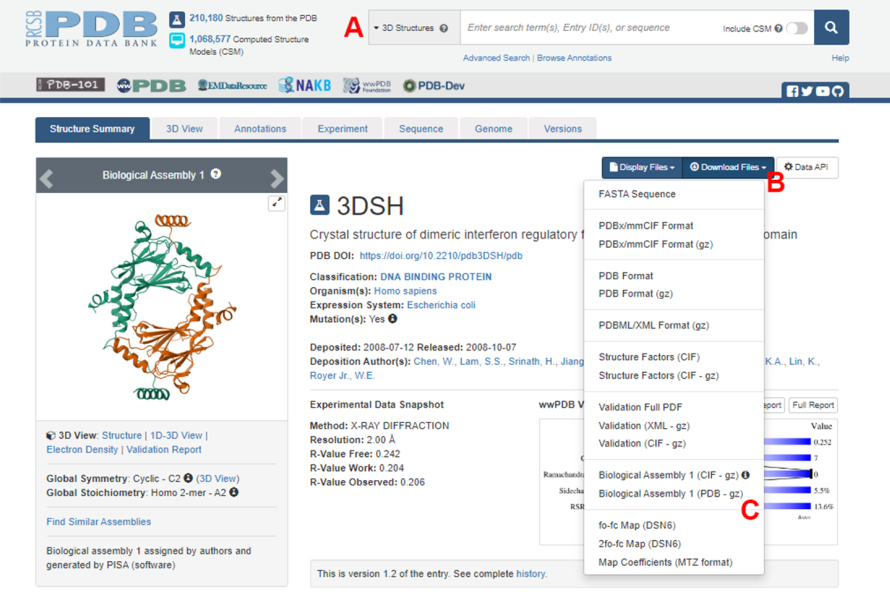
- Accédez à la page d’accueil de la Banque de données sur les protéines (BDP) (https://www.rcsb.org/) et tapez l’ID de la base de données sur la structure du complexe protéine-protéine dans la boîte de recherche principale (Figure 2A). L’ID PDB pour la structure du dimère IRF5, utilisé comme exemple dans ce travail, est 3DSH19.
- Dans la page principale de la structure souhaitée, cliquez sur Télécharger les fichiers (Figure 2B) puis sur Assemblage biologique 1 (PDB - gz) (Figure 2C).
REMARQUE : Dans la base de données PDB, les structures de nombreux complexes protéiques formés par des monomères identiques sont représentées sous forme d’assemblages biologiques, dans lesquels seule la structure d’un monomère (unité asymétrique) est stockée dans le fichier PDB. La structure du multimère, dans ce cas, le dimère IRF5, doit être téléchargée en tant qu’assemblage biologique contenant deux instances de l’unité asymétrique. Pour faciliter les prochaines étapes de ce protocole, les deux monomères sont d’abord séparés et différents ID de chaîne leur sont attribués. - Ouvrez la structure téléchargée dans UCSF Chimera20 et cliquez sur Outils > l’édition de la structure > Modifier les ID de chaîne. Dans cet exemple, les deux chaînes de l’assemblage biologique sont nommées A. Renommez la deuxième chaîne (étiquetée #0.2) en B et cliquez sur OK.
- Cliquez sur Favoris > panneau Modèle , puis sélectionnez le modèle contenant les deux chaînes. Cliquez sur le bouton Grouper/Dissocier pour séparer chaque chaîne dans un modèle différent. Ensuite, sélectionnez les deux modèles et cliquez sur le bouton Copier/Combiner . Entrez un nouveau nom pour le modèle combiné, cochez la case Fermer les modèles source, puis cliquez sur OK.
- Cliquez sur Sélectionner > chaîne et confirmez que chaque chaîne du dimère est maintenant identifiée par une lettre différente, à savoir A et B.
- Utilisez Fichier > Enregistrer PDB pour enregistrer la structure modifiée dans un autre fichier PDB, qui sera utilisé dans les prochaines étapes du protocole (ici, le nom IRF5_dimer.pdb a été utilisé).

Figure 2 : La page de la banque de données sur les protéines (PDB) pour la structure utilisée comme exemple représentatif dans ce travail. (A) Boîte de recherche pour introduire le code d’accès PDB de la structure cible. (B) Menu pour télécharger la structure en plusieurs formats. (C) Options permettant de télécharger des assemblages biologiques lorsque la structure a été enregistrée en tant qu’unité asymétrique (voir l’étape 1.1.2 pour plus de détails). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Identification du segment cible dans la protéine ligand
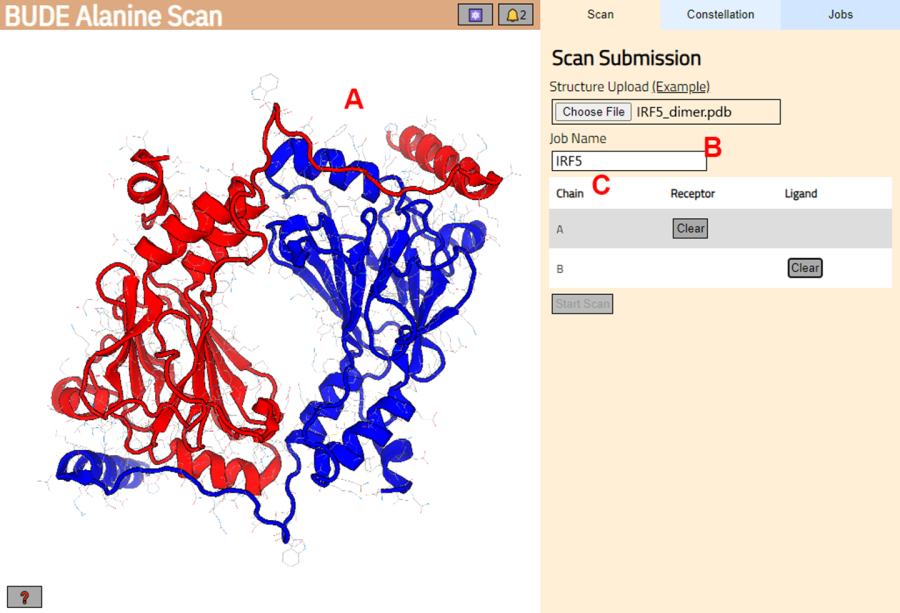
- Accédez au serveur BUDE Alanine Scan (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Cliquez sur le bouton Choisir un fichier sous Téléchargement de structure et sélectionnez le fichier PDB enregistré à l’étape 1.1.6.
- Sur la page suivante, vérifiez que la structure a été correctement chargée (Figure 3A) et entrez un nom pour le travail sur le serveur (Figure 3B).
- Définissez les chaînes de la PDB qui seront traitées comme le récepteur (A) et le ligand (B) (Figure 3C). Ensuite, cliquez sur le bouton Démarrer l’analyse pour soumettre le travail.
- Une fois le travail terminé, cliquez sur Afficher les résultats pour ouvrir la page des résultats (Figure 4).
REMARQUE : Dans la page de résultats, les résidus de la structure du ligand sont colorés en fonction de leur variation estimée de l’énergie libre (ΔΔG), et ceux avec des valeurs plus élevées sont colorés en rouge. - Dans la liste des résidus, sélectionnez le tronçon de résidus prévu pour mieux interagir avec la surface de liaison cible. Assurez-vous que ces résidus regroupent les valeurs les plus élevées pour la différence d’énergie libre (ΔΔG). Dans cet exemple, le segment entre les résidus Leu424 et Ser436 a été sélectionné (mis en évidence par un encadré rouge dans le panneau de droite de la figure 4).
- Préparation de l’interface protéine-peptide pour la diversification des séquences
- Ouvrez le fichier PDB enregistré à l’étape 1.1.6 dans Chimera et vérifiez qu’il ne manque pas d’atomes ou de liaisons dans la structure des sous-unités cibles.
- Supprimez toutes les petites molécules, ions et solvants qui ont été cocristallisés avec la structure d’origine. Pour ce faire, cliquez sur Sélectionner > résidus , puis sélectionnez toutes les molécules autres que les acides aminés standard. Ensuite, cliquez sur Actions > Atomes/Liaisons et Supprimer.
- Recadrez la chaîne de ligands jusqu’au segment qui interagit le mieux choisi à l’étape 1.2.5. Pour ce faire, cliquez sur Favoris et Séquence , puis sur la chaîne considérée comme le ligand (B). Dans le panneau Séquence , faites glisser la souris pour sélectionner tous les résidus, à l’exception de ceux situés entre les positions 424 et 436. Pour supprimer ces résidus, cliquez sur Actions > Atomes/Liaisons et Supprimer.
- Utilisez Fichier > Enregistrer PDB pour enregistrer la structure modifiée dans un autre fichier PDB, qui est utilisé dans les étapes suivantes du protocole (ici, le nom IRF5_interface.pdb a été utilisé).

Figure 3 : Sélection du récepteur et du ligand dans le serveur BUDE Alanine Scan. (A) Représentation graphique du complexe protéine-protéine. (B) Zone de texte pour entrer le nom de la tâche sur le serveur. (C) Panel pour sélectionner de manière interactive les chaînes qui seront considérées comme récepteur et ligand (voir l’étape 1.2 pour plus de détails). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Page de résultats du serveur BUDE Alanine Scan. Le segment potentiel qui interagit le mieux dans la séquence de ligand est indiqué par une case rouge. Dans le panneau de gauche, le résidu avec la contribution énergétique prévue la plus élevée (Leu433) est surligné en vert. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Diversification séquentielle
REMARQUE : dans les étapes suivantes, rosetta_main fait référence au répertoire d’installation principal de Rosetta, qui se trouve généralement dans /opt/rosetta_src__bundle/main/, où indique la version de Rosetta installée. En outre, il est supposé que les applications Rosetta sont accessibles à l’ensemble du système ; Si ce n’est pas le cas, le chemin complet vers les exécutables doit être fourni. Lorsqu’ils sont compilés à partir du source, ces exécutables se trouvent dans le répertoire /rosetta_main/source/bin/ .
- Optimisation initiale des chaînes latérales d’acides aminés
- Copiez la structure modifiée enregistrée à l’étape 1.3.4 dans un emplacement Linux accessible par les applications Rosetta.
- Utilisez l’application FixBB de Rosetta pour effectuer un reconditionnement de toutes les chaînes latérales d’acides aminés de la structure de base avant la diversification de la séquence. Dans cette opération, l’orientation de toutes les chaînes latérales d’acides aminés est optimisée pour minimiser l’énergie et améliorer la stabilité du complexe. Pour ce faire, exécutez la commande suivante :

REMARQUE : Cette commande génère un fichier PDB nommé d’après la structure d’origine avec un suffixe numérique supplémentaire (IRF5_interface_0001.pdb dans cet exemple). - Pour faciliter l’étape suivante du protocole, renommez le fichier PDB recompressé avec le suffixe _repack à l’aide de la commande suivante :
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- Diversification des séquences
- Exécutez Pepspec en mode conception pour effectuer l’étape de diversification de séquence réelle à l’aide de la commande suivante :

Les options générales sont les suivantes :- -s indique le fichier d’entrée (le fichier PDB recompressé généré à l’étape 2.1.3).
- -o indique le préfixe pour nommer les fichiers de sortie.
- - database indique le chemin d’accès à la base de données principale de Rosetta 3.
- -ex1, -ex2 et extrachi_cutoff sont des options de la bibliothèque de rotamer (voir la documentation Pepspec pour plus de détails).
- -overwrite indique à l’application d’écraser les sorties préexistantes possibles générées par les itérations précédentes.
- -pepspec :pep_chain indique les chaînes PDB considérées comme ligand ('b' dans cet exemple).
- -pepspec :native_pep_anchor indique le résidu d’acide aminé utilisé comme ancre (dans cet exemple, le résidu Leu à la position 10 du peptide ligand).
- -pepspec :n_peptides indique le nombre de structures peptidiques à afficher.
- -pepspec :no_prepack_prot indique à l’application d’ignorer le reconditionnement dans la structure de base d’entrée (puisque cela a été précédemment effectué à l’étape 2.1).
REMARQUE : La sortie principale de Pepspec est un répertoire contenant les fichiers PDB pour les peptides résultant de la phase de conception, nommés à l’aide du préfixe de sortie avec le suffixe .pdbs (IRF5.pdbs dans l’exemple). De plus, Pepspec affiche toutes les séquences peptidiques acceptées testées dans le cadre de l’étape de diversification de séquence et leurs scores d’énergie Rosetta correspondants dans un fichier texte délimité par des tabulations nommé d’après le préfixe de sortie, avec l’option . spec (IRF5.spec dans l’exemple). Étant donné que le protocole décrit dans ce travail vise à estimer les préférences en acides aminés plutôt que la conception réelle des peptides, les étapes suivantes utilisent IRF5.spec au lieu des structures PDB dans le répertoire .pdbs .
- Exécutez Pepspec en mode conception pour effectuer l’étape de diversification de séquence réelle à l’aide de la commande suivante :
3. Estimation des préférences en acides aminés
- Calcul d’un PWM
- Pour générer un PWM, utilisez le script gen_pepspec_pwm.py inclus dans la suite Rosetta. Pour exécuter ce script, utilisez la commande suivante :

où:- IRF5.spec est le fichier de sortie Pepspec généré à l’étape 2.2.
- -1 indique qu’il n’y a pas de résidus N-terminaux supplémentaires dans la séquence et, par conséquent, les positions dans le PWM sont basées sur 1.
- 0.2 indique au script de ne prendre en compte que les 20 % de peptides les mieux notés de la sortie Pepspec (la valeur par défaut est 0.1, correspondant aux 10 %)
- interface_score indique au script de classer les peptides en fonction du score de l’interface, qui est l’un des différents scores Rosetta inclus dans le fichier de sortie Pepspec.
REMARQUE : Ce script génère deux fichiers de sortie, l’un pour le PWM calculé (avec le suffixe .pwm ) et l’autre pour les séquences du sous-ensemble de peptides utilisés pour calculer le PWM (avec le suffixe .seq ). Les noms de ces fichiers incluent également le score et la fraction de peptides utilisée pour le classement. Dans cet exemple, ces fichiers sont respectivement nommés IRF5_interface_score_0.2.pwm et IRF5_interface_score_0.2.seq.
- Pour générer un PWM, utilisez le script gen_pepspec_pwm.py inclus dans la suite Rosetta. Pour exécuter ce script, utilisez la commande suivante :
- Génération d’un logo de séquence
- Accédez au serveur WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 et cliquez sur le bouton Choisir un fichier à côté de Télécharger les données de séquence. Téléchargez le fichier contenant les séquences peptidiques générées à l’étape 3.1.1 (IRF5_interface_score_0.2.seq dans cet exemple).
- Choisissez le format et la taille du logo souhaités en fonction de la longueur d’entrée. L’exemple utilise le format PDF et une taille de 15 cm x 12 cm. Cliquez sur Créer un logo.
Access restricted. Please log in or start a trial to view this content.
Résultats
Dans cet article, nous avons décrit un protocole permettant de prédire les préférences en acides aminés de la surface de liaison d’IRF5, membre d’une famille de facteurs de transcription connus sous le nom de facteurs régulateurs de l’interféron humain. Ces protéines sont des régulateurs des réponses immunitaires innées et adaptatives et participent à la différenciation et à l’activation de plusieurs cellules immunitaires. Les sous-unités IRF ont des surfaces de li...
Access restricted. Please log in or start a trial to view this content.
Discussion
Le présent article décrit un protocole permettant d’estimer les préférences en acides aminés de sites de liaison potentiellement multispécifiques basés sur la diversification de séquences in silico. Peu d’outils informatiques ont été développés pour estimer les préférences en acides aminés des interfaces protéine-peptide 14,25,26. Ces outils ont un caractère prédictif, mais...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions vivement le Sistema Nacional de Investigación (SNI) (subventions SNI-043-2023 et SNI-170-2021), le Secretaría Nacional de Ciencia, le Tecnología e Innovación (SENACYT) du Panama et l’Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU). Les auteurs tiennent à remercier le Dr Miguel Rodríguez pour l’examen minutieux du manuscrit.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Références
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627(2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), London, England. 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267(2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33(2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999(2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451(2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614(2017).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.