
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Predizione computazionale delle preferenze amminoacidiche di domini di legame peptidico potenzialmente multispecifici coinvolti nelle interazioni proteina-proteina
In questo articolo
Riepilogo
Descriviamo una metodologia basata sulla diversificazione delle sequenze per stimare le preferenze amminoacidiche dei siti di legame multispecifici nelle interazioni proteina-proteina (PPI). In questa strategia, migliaia di potenziali ligandi peptidici vengono generati e sottoposti a screening in silico, superando così alcune limitazioni dei metodi sperimentali disponibili.
Abstract
Molte interazioni proteina-proteina coinvolgono il legame di brevi segmenti proteici ai domini di legame dei peptidi. Di solito, tali interazioni richiedono il riconoscimento di motivi lineari con conservazione variabile. La combinazione di regioni altamente conservate e più variabili negli stessi ligandi spesso contribuisce alla multispecificità del legame, una proprietà comune degli enzimi e delle proteine di segnalazione cellulare. La caratterizzazione delle preferenze amminoacidiche dei domini di legame dei peptidi è importante per la progettazione di mediatori delle interazioni proteina-proteina (PPI). I metodi computazionali sono un'alternativa efficiente alle tecniche sperimentali, spesso costose e ingombranti, consentendo la progettazione di potenziali mediatori che possono essere successivamente convalidati in esperimenti a valle. Qui, abbiamo descritto una metodologia che utilizza l'applicazione Pepspec del pacchetto di modellazione molecolare Rosetta per prevedere le preferenze amminoacidiche dei domini di legame peptidico. Questa metodologia è utile quando la struttura della proteina recettore e la natura del ligando peptidico sono entrambe note o possono essere dedotte. La metodologia inizia con un'ancora ben caratterizzata dal ligando, che viene estesa aggiungendo in modo casuale residui di amminoacidi. L'affinità di legame dei peptidi generati in questo modo viene quindi valutata mediante docking del peptide della spina dorsale flessibile al fine di selezionare i peptidi con i migliori punteggi di legame previsti. Questi peptidi vengono quindi utilizzati per calcolare le preferenze degli amminoacidi e per calcolare facoltativamente una matrice posizione-peso (PWM) che può essere utilizzata in ulteriori studi. Per illustrare l'applicazione di questa metodologia, abbiamo utilizzato l'interazione tra subunità del fattore regolatore dell'interferone umano 5 (IRF5), precedentemente noto per essere multispecifico ma globalmente guidato da un breve motivo conservato chiamato pLxIS. Le preferenze stimate per gli amminoacidi erano coerenti con le conoscenze precedenti sulla superficie di legame di IRF5. Le posizioni occupate da residui di serina fosforilabili hanno mostrato un'alta frequenza di aspartato e glutammato, probabilmente perché le loro catene laterali caricate negativamente sono simili alla fosfosserina.
Introduzione
L'interazione tra due proteine spesso comporta il legame di brevi segmenti di amminoacidi a domini di legame peptidico, simili alle interfacce proteina-peptide. Le proteine recettoriali coinvolte in tali interazioni proteina-proteina (PPI) hanno spesso la capacità di riconoscere un certo insieme di sequenze di ligandi sovrapposte ma divergenti, una proprietà nota come multispecificità 1,2. Il riconoscimento multispecifico è una caratteristica di molte proteine cellulari, ma è particolarmente notevole negli enzimi e nelle proteine di segnalazione cellulare3. Le proteine che interagiscono con i siti di legame multispecifici hanno spesso una combinazione di regioni più e meno conservate nella loro sequenza 4,5,6. In questo scenario, i motivi di sequenza più conservati sono coinvolti in interazioni molecolari rigorose. Al contrario, le sequenze più variabili interagiscono con superfici in qualche modo permissive nel sito di legame del recettore. Di solito, questi segmenti meno conservati ma ancora funzionalmente rilevanti sono loop privi di modelli di struttura secondaria definiti o hanno conformazioni ancora più dinamiche, come quelle tipiche delle proteine intrinsecamente disordinate7.
L'identificazione di potenziali ligandi peptidici dei siti di legame è solitamente il primo passo nella progettazione di mediatori in grado di interferire con i corrispondenti PPI8. Tuttavia, è spesso improbabile trovare un singolo residuo di amminoacidi più frequente nella maggior parte delle posizioni di sequenza nei ligandi dei siti di legame multispecifici. Invece, questi siti possono avere particolari preferenze per una specifica classe di amminoacidi in base alle loro proprietà chimiche, ad esempio, amminoacidi acidi e caricati negativamente come l'aspartato o il glutammato, amminoacidi aromatici voluminosi come la fenilalanina o residui più idrofobici come gli amminoacidi alifatici alanina, valina, leucina o isoleucina3. Diversi metodi sperimentali possono fornire informazioni sulle preferenze aminoacidiche dei siti di legame delle proteine, tra cui l'evoluzione diretta9, la mutagenesi a scansione multi-codone10 e la scansione mutazionale profonda11. Tutti questi metodi seguono l'approccio della diversificazione delle sequenze, che si basa sull'introduzione di mutazioni nei ligandi originali e sull'ulteriore analisi del loro effetto sulla funzione della proteina recettore (vedi Bratulic e Badran12 per una revisione completa). Tuttavia, questi metodi spesso richiedono l'indagine di librerie di sequenze di grandi dimensioni, il che li rende più ingombranti, costosi e dispendiosi in termini di tempo.
I metodi computazionali per dedurre le preferenze amminoacidiche dei siti di legame multispecifici hanno il potenziale per aggirare i limiti dei metodi di laboratorio umido. Tra questi, l'approccio di diversificazione della sequenza in silico valuta l'impatto energetico di un'ampia gamma di sostituti di amminoacidi nella sequenza del ligando come un modo per caratterizzare la plasticità strutturale del PPI13. Questo metodo inizia con la struttura o il modello del ligando peptidico legato al sito di legame del recettore e successivamente introduce mutazioni nella sequenza del ligando. Le funzioni statistiche e di punteggio energetico vengono quindi utilizzate per valutare l'impatto di queste mutazioni sulla stabilità e sull'affinità di legame. L'insieme delle sequenze di ligandi con il miglior punteggio risultanti dalla fase di valutazione può quindi essere utilizzato per calcolare le preferenze degli amminoacidi. Questa strategia ha il potenziale per elaborare un numero molto elevato di sequenze di ligando in modo efficiente. Pertanto, può fornire un'inferenza più completa e coerente delle preferenze amminoacidiche rispetto a quelle calcolate dal numero più limitato di sequenze che di solito possono essere elaborate negli approcci di laboratorio umido.
L'applicazione Pepspec della suite di modellazione molecolare Rosetta14 è uno strumento che esegue la diversificazione delle sequenze come passaggio chiave della sua modalità di progettazione dei peptidi. Questa applicazione richiede una struttura o un modello della proteina del recettore con un peptide legato fino a un singolo residuo di amminoacido in lunghezza, che viene utilizzato come ancoraggio per i passaggi successivi. La sequenza del peptide legato viene quindi estesa (se necessario) e diversificata per generare un gran numero di ligandi peptidici putativi. L'affinità di legame di questi peptidi viene quindi valutata mediante docking del peptide della spina dorsale flessibile al fine di selezionare quelli con i migliori punteggi di legame previsti. Sebbene l'output principale di questa applicazione siano i migliori peptidi candidati selezionati alla fine della fase di progettazione, l'insieme molto più ampio di peptidi accettati durante questa fase può essere utilizzato anche per calcolare le preferenze amminoacidiche del sito di legame target. Le preferenze degli amminoacidi sono calcolate come la frequenza di ciascun residuo di amminoacido per posizione della sequenza del ligando, rappresentata come una matrice di peso della posizione (PWM) o come un logo di sequenza più visivo.
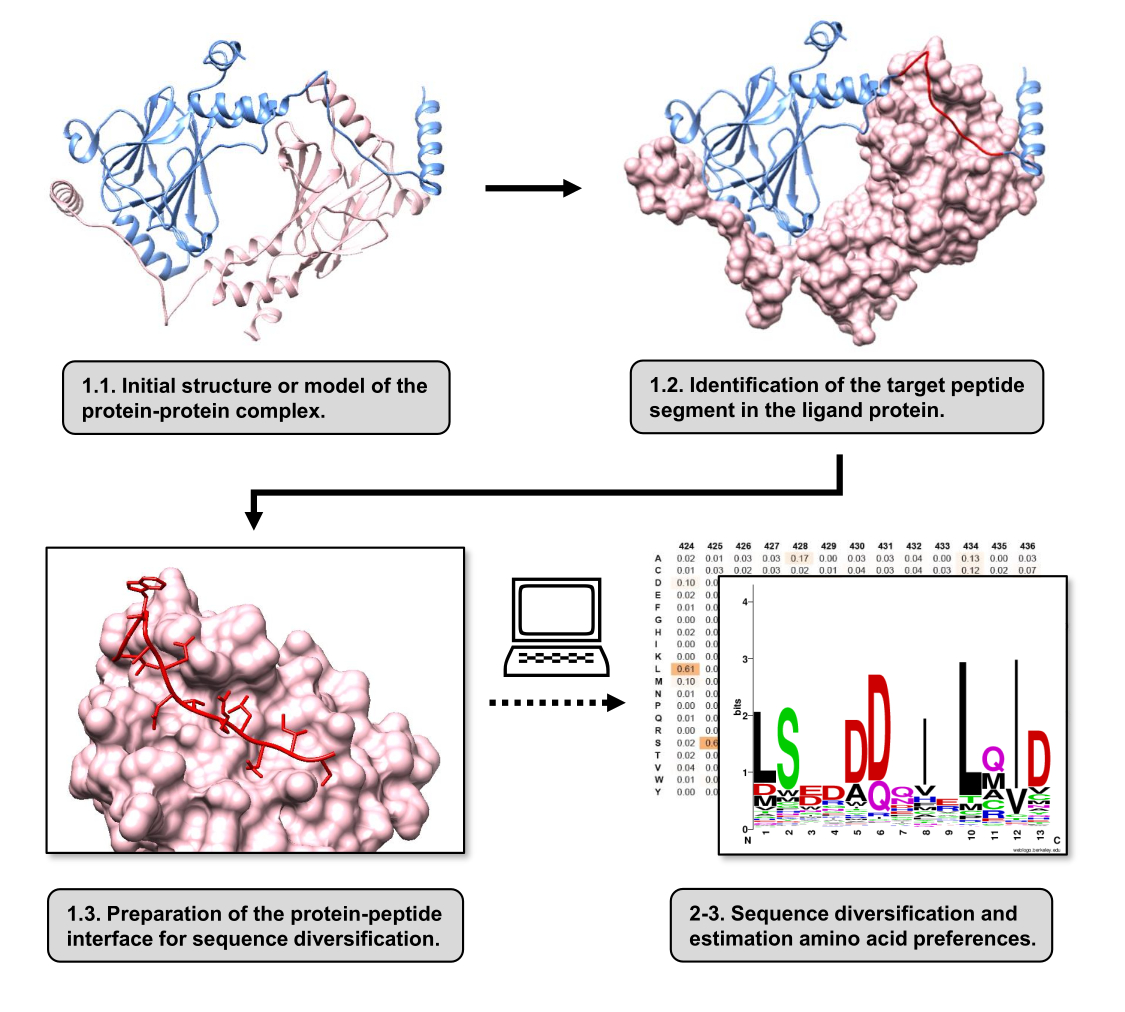
In questo articolo, descriviamo un protocollo per stimare le preferenze aminoacidiche della superficie di legame di una proteina recettore coinvolta in un PPI. Il protocollo è focalizzato su PPI in cui è noto che un segmento lineare della proteina-ligando si lega alla proteina recettore, quindi lo scenario può essere modellato come un'interfaccia proteina-peptide. In questo scenario, i motivi conservati del ligando interagiscono tipicamente con tasche definite nel sito di legame del recettore, sebbene l'intero segmento del ligando coinvolto nel PPI possa contenere regioni meno conservate. Nella Figura 1 è mostrato un diagramma di flusso che riassume le fasi principali del protocollo. Il protocollo inizia con la struttura 3D del complesso proteina-proteina e riduce ulteriormente la proteina ligando al segmento potenzialmente più interagente, lasciando intatta la proteina recettore. Il segmento con la migliore interazione viene dedotto utilizzando il server BUDE Alanine Scan15, che conduce la mutagenesi computazionale della scansione dell'alanina per identificare i residui di punti caldi tra le due proteine interagenti. In questo approccio, i residui del ligando vengono sostituiti individualmente dall'alanina e la variazione stimata dell'energia libera o della stabilità del complesso (ΔΔG) viene quindi utilizzata per dedurre la rilevanza del residuo corrispondente per il PPI target. Una volta dedotto il segmento con la migliore interazione, il suo complesso con la proteina recettore viene utilizzato come struttura di base sottoposta a Pepspec per eseguire la diversificazione della sequenza.

Figura 1: Panoramica delle fasi principali del protocollo proposto in questo lavoro. I numeri corrispondono ai numeri di passaggio nella sezione del protocollo. Le figure sono state realizzate con il complesso proteina-proteina utilizzato come esempio descritto nel testo. In questo complesso, la catena proteica considerata come recettore è mostrata in rosa, mentre la catena considerata come ligando è mostrata in azzurro con il suo segmento di migliore interazione previsto evidenziato in rosso. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Uno dei limiti del protocollo suggerito è la necessità di una struttura risolta dell'interfaccia proteina-peptide. In alternativa, il protocollo può iniziare con un modello dell'interfaccia proteina-peptide bersaglio, sebbene le fasi specifiche della modellazione non siano descritte nel presente documento. Inoltre, sebbene il protocollo possa essere eseguito su un personal computer con qualsiasi sistema operativo, è necessario un ambiente Linux per i passaggi che coinvolgono le applicazioni Rosetta. Un cluster di computer è anche altamente raccomandato per la fase di diversificazione delle sequenze a causa del gran numero di iterazioni tipicamente eseguite da Pepspec.
L'applicazione del protocollo suggerito è illustrata con la stima delle preferenze aminoacidiche della superficie di offerta di IRF5, un membro della famiglia dei fattori regolatori dell'interferone umano (IRF). Abbiamo scelto questa proteina come esempio perché, durante la sua attivazione, due subunità si legano per formare un dimero la cui struttura è ben caratterizzata16. Nei dimeri IRF, il legame può essere modellato come un'interfaccia proteina-peptide in cui una subunità fornisce la superficie di legame e l'altra interagisce attraverso una regione contenente un breve motivo conservato chiamato pLxIS17,18. Inoltre, il legame con le subunità IRF è multispecifico; Pertanto, possono formare omodimeri, eterodimeri e complessi con altre proteine cellulari note come coattivatori18.
Access restricted. Please log in or start a trial to view this content.
Protocollo
1. Preparazione iniziale dell'interfaccia proteina-peptide
- Scaricare la struttura del complesso proteina-proteina
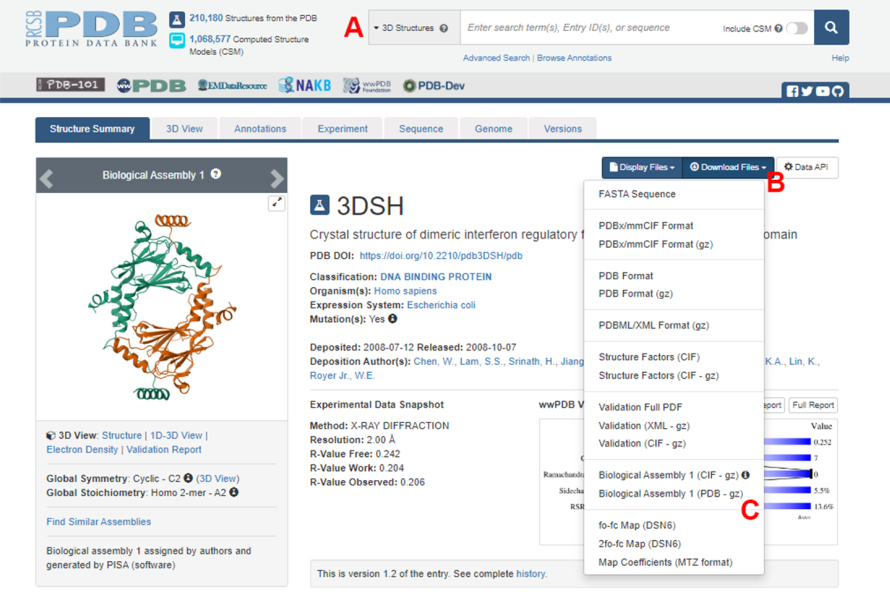
- Accedere alla home page della Protein Data Bank (PDB) (https://www.rcsb.org/) e digitare l'ID PDB per la struttura del complesso proteina-proteina nella casella di ricerca principale (Figura 2A). L'ID PDB per la struttura del dimero IRF5, utilizzato come esempio in questo lavoro, è 3DSH19.
- Nella pagina principale relativa alla struttura desiderata, cliccare su Download Files (Figura 2B) e poi su Biological Assembly 1 (PDB - gz) (Figura 2C).
NOTA: Nel database PDB, le strutture di molti complessi proteici formati da monomeri identici sono rappresentate come assemblaggi biologici, in cui solo la struttura di un monomero (unità asimmetrica) è memorizzata nel file PDB. La struttura del multimero, in questo caso il dimero IRF5, deve essere scaricata come l'assemblaggio biologico contenente due istanze dell'unità asimmetrica. Per facilitare i passaggi successivi di questo protocollo, i due monomeri vengono prima separati e vengono assegnati loro diversi ID di catena. - Aprire la struttura scaricata in UCSF Chimera20 e fare clic su Strumenti > modifica struttura > Modifica ID catena. In questo esempio, entrambe le catene nell'assemblaggio biologico sono denominate A. Rinominate la seconda catena (etichettata #0.2) in B e fate clic su OK.
- Fare clic su Preferiti > Pannello Modello e quindi selezionare il modello contenente le due catene. Fare clic sul pulsante Raggruppa/Separa per separare ogni catena in un modello diverso. Quindi, seleziona i due modelli e fai clic sul pulsante Copia/Combina. Immettere un nuovo nome per il modello combinato, selezionare Chiudi modelli di origine e fare clic su OK.
- Fare clic su Seleziona > catena e verificare che ogni catena nel dimero sia ora identificata da una lettera diversa, ovvero A e B.
- Utilizzare File > Salva PDB per salvare la struttura modificata in un file PDB diverso, che verrà utilizzato nei passaggi successivi del protocollo (in questo caso, è stato utilizzato il nome IRF5_dimer.pdb ).

Figura 2: La pagina della Protein Data Bank (PDB) per la struttura utilizzata come esempio rappresentativo in questo lavoro. (A) Casella di ricerca per introdurre il codice di accesso PDB della struttura target. (B) Menu per scaricare la struttura in diversi formati. (C) Opzioni per scaricare gli assemblaggi biologici quando la struttura è stata salvata come unità asimmetrica (vedere il passaggio 1.1.2 per maggiori dettagli). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Identificazione del segmento target nella proteina ligando
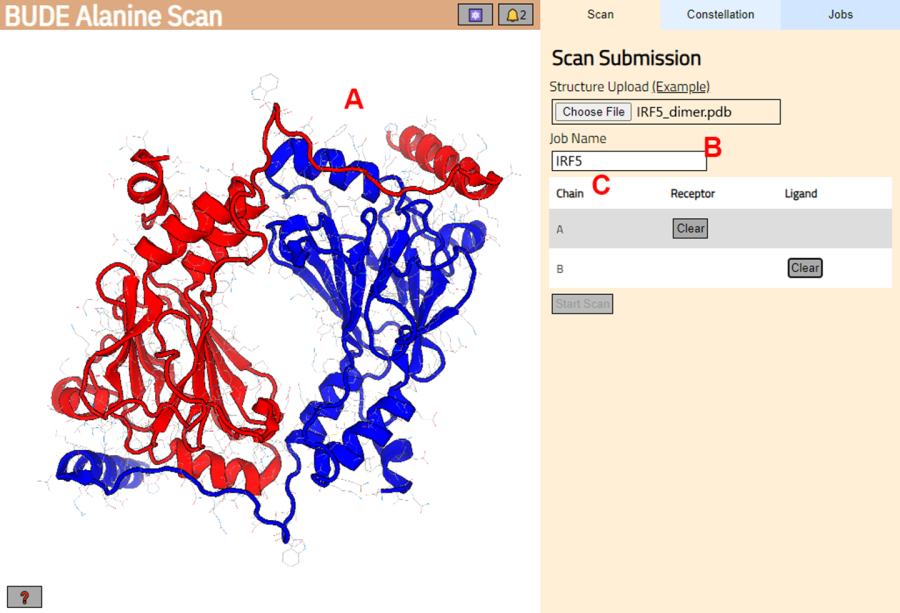
- Accedere al server di scansione alanina BUDE (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Fare clic sul pulsante Scegli file in Caricamento struttura e selezionare il file PDB salvato nel passaggio 1.1.6.
- Nella pagina successiva, verificare che la struttura sia stata caricata correttamente (Figura 3A) e inserire un nome per il lavoro nel server (Figura 3B).
- Impostare le catene dal PDB che verranno trattate come recettore (A) e ligando (B) (Figura 3C). Quindi, fare clic sul pulsante Avvia scansione per inviare il lavoro.
- Una volta terminato il lavoro, fare clic su Mostra risultati per aprire la pagina dei risultati (Figura 4).
NOTA: Nella pagina dei risultati, i residui della struttura del ligando sono colorati in base alla loro variazione stimata di energia libera (ΔΔG), e quelli con valori più alti sono colorati in rosso. - Dall'elenco dei residui, selezionare il tratto di residui che si prevede interagisca meglio con la superficie di legatura target. Assicurarsi che questi residui raggruppino i valori più alti per la differenza di energia libera (ΔΔG). In questo esempio, è stato selezionato il segmento tra i residui Leu424 e Ser436 (evidenziato con un riquadro rosso nel pannello di destra della Figura 4).
- Preparazione dell'interfaccia proteina-peptide per la diversificazione delle sequenze
- Aprire il file PDB salvato nel passaggio 1.1.6 in Chimera e verificare che non vi siano atomi o legami mancanti nella struttura delle subunità target.
- Elimina tutte le piccole molecole, gli ioni e i solventi che sono stati co-cristallizzati con la struttura originale. Per fare ciò, fai clic su Seleziona > residui e quindi seleziona tutte le molecole diverse dagli amminoacidi standard. Quindi, fai clic su Azioni > atomi/legami ed elimina.
- Ritagliare la catena di leganti al segmento con la migliore interazione scelto nel passaggio 1.2.5. Per fare ciò, fare clic su Preferiti e Sequenza e quindi fare clic sulla catena considerata come ligando (B). Nel pannello Sequenza , trascinate il mouse per selezionare tutti i residui tranne quelli compresi tra le posizioni 424 e 436. Per eliminare questi residui, fare clic su Azioni > atomi/legami ed eliminare.
- Utilizzare File > Salva PDB per salvare la struttura modificata in un file PDB diverso, che viene utilizzato nei passaggi successivi del protocollo (in questo caso, è stato utilizzato il nome IRF5_interface.pdb ).

Figura 3: Selezione del recettore e del ligando nel server di scansione BUDE Alanina. (A) Rappresentazione grafica del complesso proteina-proteina. (B) Casella di testo per immettere il nome del lavoro nel server. (C) Panel per selezionare in modo interattivo le catene che saranno considerate come recettore e ligando (vedere il passaggio 1.2 per maggiori dettagli). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Pagina dei risultati del server di scansione BUDE Alanina. Il potenziale segmento con la migliore interazione nella sequenza del ligando è indicato con una casella rossa. Nel pannello di sinistra, il residuo con il contributo energetico previsto più elevato (Leu433) è evidenziato in verde. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
2. Diversificazione delle sequenze
NOTA: Nei passaggi seguenti, rosetta_main fa riferimento alla directory di installazione principale di Rosetta, che in genere si trova in /opt/rosetta_src__bundle/main/, dove indica la versione di Rosetta installata. Inoltre, si presume che le applicazioni Rosetta siano accessibili a livello di sistema; In caso contrario, è necessario fornire il percorso completo degli eseguibili. Quando vengono compilati dal sorgente, questi eseguibili si trovano nella directory /rosetta_main/source/bin/ .
- Ottimizzazione iniziale delle catene laterali degli amminoacidi
- Copiare la struttura modificata salvata nel passaggio 1.3.4 in una posizione Linux accessibile dalle applicazioni Rosetta.
- Utilizza l'applicazione FixBB di Rosetta per eseguire un repack di tutte le catene laterali di amminoacidi della struttura di base prima della diversificazione della sequenza. In questa operazione, l'orientamento di tutte le catene laterali degli amminoacidi è ottimizzato per ridurre al minimo l'energia e migliorare la stabilità del complesso. A tale scopo, eseguire il comando seguente:

NOTA: questo comando genera un file PDB denominato in base alla struttura originale con un suffisso numerico aggiuntivo (IRF5_interface_0001.pdb in questo esempio). - Per facilitare il passaggio successivo del protocollo, rinominare il file PDB ricompresso con il suffisso _repack utilizzando il seguente comando:
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- Diversificazione delle sequenze
- Eseguire Pepspec in modalità progettazione per eseguire l'effettivo passaggio di diversificazione della sequenza utilizzando il seguente comando:

Di seguito sono riportate le opzioni generali:- -s indica il file di input (il file PDB ricompresso generato nel passaggio 2.1.3).
- -o indica il prefisso per denominare i file di output.
- - database indica il percorso del database principale di Rosetta 3.
- -ex1, -ex2 e extrachi_cutoff sono opzioni della libreria di rotameri (vedere la documentazione di Pepspec per maggiori dettagli).
- -overwrite indica all'applicazione di sovrascrivere eventuali output preesistenti generati da iterazioni precedenti.
- -pepspec:pep_chain indica le catene PDB considerate come ligando ('b' in questo esempio).
- -pepspec:native_pep_anchor indica il residuo amminoacidico utilizzato come ancoraggio (in questo esempio, il residuo di Leu in posizione 10 del peptide ligando).
- -pepspec:n_peptides indica il numero di strutture peptidiche da produrre.
- -pepspec:no_prepack_prot dice all'applicazione di saltare il reimballaggio nella struttura di base dell'input (poiché questo è stato precedentemente eseguito nel passaggio 2.1).
NOTA: L'output principale di Pepspec è una directory contenente i file PDB per i peptidi risultanti dalla fase di progettazione, denominata utilizzando il prefisso di output con il suffisso .pdbs (IRF5.pdbs nell'esempio). Inoltre, Pepspec emette tutte le sequenze peptidiche accettate testate come parte della fase di diversificazione della sequenza e i corrispondenti punteggi di energia di Rosetta in un file di testo delimitato da tabulazioni denominato in base al prefisso di output, con . spec (IRF5.spec nell'esempio). Poiché il protocollo descritto in questo lavoro mira a stimare le preferenze degli amminoacidi piuttosto che l'effettivo design del peptide, i passaggi successivi utilizzano IRF5.spec invece delle strutture PDB nella directory .pdbs .
- Eseguire Pepspec in modalità progettazione per eseguire l'effettivo passaggio di diversificazione della sequenza utilizzando il seguente comando:
3. Stima delle preferenze amminoacidiche
- Calcolo di un PWM
- Per generare un PWM, utilizzare lo script gen_pepspec_pwm.py incluso nella suite Rosetta. Per eseguire questo script, utilizzare il seguente comando:

dove:- IRF5.spec è il file di output Pepspec generato nel passaggio 2.2.
- -1 indica che non ci sono residui N-terminali aggiuntivi nella sequenza e, quindi, le posizioni nel PWM sono in base 1.
- 0,2 indica allo script di considerare solo il 20% dei peptidi con il punteggio migliore dall'output Pepspec (il valore predefinito è 0,1, corrispondente al 10%)
- interface_score indica allo script di classificare i peptidi in base al punteggio dell'interfaccia, che è uno dei vari punteggi Rosetta inclusi nel file di output Pepspec.
NOTA: Questo script genera due file di output, uno per il PWM calcolato (con il suffisso .pwm ) e l'altro per le sequenze del sottoinsieme di peptidi utilizzati per calcolare il PWM (con il suffisso .seq ). I nomi di questi file includono anche il punteggio e la frazione di peptidi utilizzati per la classificazione. In questo esempio, questi file sono denominati rispettivamente IRF5_interface_score_0.2.pwm e IRF5_interface_score_0.2.seq.
- Per generare un PWM, utilizzare lo script gen_pepspec_pwm.py incluso nella suite Rosetta. Per eseguire questo script, utilizzare il seguente comando:
- Generazione di un logo di sequenza
- Accedere al server WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 e fare clic sul pulsante Scegli file accanto a Carica dati sequenza. Caricare il file con le sequenze peptidiche generate nel passaggio 3.1.1 (IRF5_interface_score_0.2.seq in questo esempio).
- Scegli il formato e la dimensione desiderati del logo in base alla lunghezza di input. L'esempio utilizza il formato PDF e una dimensione di 15 cm x 12 cm. Fare clic su Crea logo.
Access restricted. Please log in or start a trial to view this content.
Risultati
In questo articolo, abbiamo descritto un protocollo per prevedere le preferenze aminoacidiche della superficie di legame di IRF5, un membro di una famiglia di fattori di trascrizione noti come fattori regolatori dell'interferone umano. Queste proteine sono regolatori delle risposte immunitarie innate e adattative e partecipano alla differenziazione e all'attivazione di diverse cellule immunitarie. Le subunità IRF hanno superfici di legame altamente plastiche e multispecifiche, essendo i...
Access restricted. Please log in or start a trial to view this content.
Discussione
Il presente articolo descrive un protocollo per stimare le preferenze amminoacidiche di siti di legame potenzialmente multispecifici basato sulla diversificazione di sequenze in silico. Pochi strumenti computazionali sono stati sviluppati per stimare le preferenze amminoacidiche delle interfacce proteina-peptide 14,25,26. Questi strumenti hanno una natura predittiva, ma differiscono per gli algo...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Si ringrazia il sostegno finanziario del Sistema Nacional de Investigación (SNI) (numeri di sovvenzione SNI-043-2023 e SNI-170-2021), della Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) di Panama e dell'Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU). Gli autori ringraziano il Dr. Miguel Rodríguez per l'attenta revisione del manoscritto.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Riferimenti
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627(2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), London, England. 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267(2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33(2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999(2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451(2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614(2017).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati