
Bu içeriği görüntülemek için JoVE aboneliği gereklidir. Oturum açın veya ücretsiz deneme sürümünü başlatın.
Method Article
Protein-Protein Etkileşimlerinde Görev Alan Potansiyel Olarak Çok Spesifik Peptit Bağlayıcı Alanların Amino Asit Tercihlerinin Hesaplamalı Tahmini
Bu Makalede
Özet
Protein-protein etkileşimlerinde (PPI'ler) çok spesifik bağlanma bölgelerinin amino asit tercihlerini tahmin etmek için dizi çeşitlendirmesine dayalı bir metodoloji açıklıyoruz. Bu stratejide, binlerce potansiyel peptit ligandı üretilir ve in silico olarak taranır, böylece mevcut deneysel yöntemlerin bazı sınırlamalarının üstesinden gelinir.
Özet
Birçok protein-protein etkileşimi, kısa protein segmentlerinin peptit bağlama alanlarına bağlanmasını içerir. Genellikle, bu tür etkileşimler, değişken koruma ile doğrusal motiflerin tanınmasını gerektirir. Aynı ligandlarda yüksek oranda korunmuş ve daha değişken bölgelerin kombinasyonu, genellikle enzimlerin ve hücre sinyal proteinlerinin ortak bir özelliği olan bağlanmanın çok özgüllüğüne katkıda bulunur. Peptit bağlayıcı alanların amino asit tercihlerinin karakterizasyonu, protein-protein etkileşimlerinin (PPI'ler) aracılarının tasarımı için önemlidir. Hesaplamalı yöntemler, genellikle maliyetli ve hantal deneysel tekniklere etkili bir alternatiftir ve daha sonra aşağı akış deneylerinde doğrulanabilecek potansiyel aracıların tasarımını sağlar. Burada, peptit bağlama alanlarının amino asit tercihlerini tahmin etmek için Rosetta moleküler modelleme paketinin Pepspec uygulamasını kullanan bir metodoloji tanımladık. Bu metodoloji, reseptör proteininin yapısı ve peptit ligandının doğası bilindiğinde veya çıkarılabildiğinde yararlıdır. Metodoloji, rastgele amino asit kalıntıları eklenerek uzatılan liganddan iyi karakterize edilmiş bir ankraj ile başlar. Bu şekilde üretilen peptitlerin bağlanma afinitesi daha sonra en iyi tahmin edilen bağlanma skorlarına sahip peptitleri seçmek için esnek omurga peptit yerleştirme ile değerlendirilir. Bu peptitler daha sonra amino asit tercihlerini hesaplamak ve isteğe bağlı olarak daha sonraki çalışmalarda kullanılabilecek bir konum-ağırlık matrisini (PWM) hesaplamak için kullanılır. Bu metodolojinin uygulanmasını göstermek için, daha önce çok spesifik olduğu bilinen, ancak küresel olarak pLxIS adı verilen kısa korunmuş bir motif tarafından yönlendirilen insan interferon düzenleyici faktör 5'in (IRF5) alt birimleri arasındaki etkileşimi kullandık. Tahmini amino asit tercihleri, IRF5 bağlanma yüzeyi hakkındaki önceki bilgilerle tutarlıydı. Fosforile edilebilir serin kalıntıları tarafından işgal edilen pozisyonlar, muhtemelen negatif yüklü yan zincirlerinin fosfoserine benzer olması nedeniyle yüksek bir aspartat ve glutamat frekansı sergiledi.
Giriş
İki protein arasındaki etkileşim genellikle kısa amino asit segmentlerinin, protein-peptit arayüzlerine benzeyen peptit bağlama alanlarına bağlanmasını içerir. Bu tür protein-protein etkileşimlerinde (PPI) yer alan reseptör proteinleri, genellikle multispesifisite 1,2 olarak bilinen bir özellik olan belirli bir örtüşen ancak farklı ligand dizileri setini tanıma yeteneğine sahiptir. Multispesifik tanıma, birçok hücresel proteinin bir özelliğidir, ancak enzimlerde ve hücre sinyal proteinlerinde özellikle dikkat çekicidir3. Multispesifik bağlanma bölgeleri ile etkileşime giren proteinler genellikle dizilimlerinde daha fazla ve daha az korunmuş bölgelerin bir kombinasyonuna sahiptir 4,5,6. Bu senaryoda, daha fazla korunmuş dizi motifleri, sıkı moleküler etkileşimlerde yer alır. Tersine, daha değişken diziler, reseptör bağlanma bölgesindeki bir şekilde izin verilen yüzeylerle etkileşime girer. Genellikle, bu daha az korunmuş ancak yine de işlevsel olarak ilgili segmentler, tanımlanmış ikincil yapı modellerinden yoksun döngülerdir veya içsel olarak düzensiz proteinlerin tipik olanları gibi daha dinamik konformasyonlara sahiptir7.
Bağlanma bölgelerinin potansiyel peptit ligandlarının tanımlanması, genellikle karşılık gelen PPI'lere müdahale edebilen aracıların tasarımında ilk adımdır8. Bununla birlikte, çok spesifik bağlanma bölgelerinin ligandlarında çoğu dizi pozisyonunda en sık görülen tek bir amino asit kalıntısı bulmak genellikle olası değildir. Bunun yerine, bu bölgeler kimyasal özelliklerine göre belirli bir amino asit sınıfı için özel tercihlere sahip olabilir, örneğin aspartat veya glutamat gibi asidik ve negatif yüklü amino asitler, fenilalanin gibi hacimli aromatik amino asitler veya alifatik amino asitler alanin, valin, lösin veya izolösin gibi daha hidrofobik kalıntılar3. Yönlendirilmiş evrim9, çoklu kodon tarama mutajenez10 ve derin mutasyon taraması11 dahil olmak üzere çeşitli deneysel yöntemler, protein bağlanma bölgelerinin amino asit tercihleri hakkında bilgi sağlayabilir. Bu yöntemlerin tümü, orijinal ligandlara mutasyonların eklenmesine ve bunların reseptör proteininin işlevi üzerindeki etkilerinin daha fazla analiz edilmesine dayanan dizi çeşitlendirmesi yaklaşımını takip eder (kapsamlı bir inceleme için Bratulic ve Badran12'ye bakınız). Bununla birlikte, bu yöntemler genellikle büyük dizi kitaplıklarının araştırılmasını gerektirir ve bu da onları daha hantal, maliyetli ve zaman alıcı hale getirir.
Çok spesifik bağlanma bölgelerinin amino asit tercihlerini çıkarmak için kullanılan hesaplama yöntemleri, ıslak laboratuvar yöntemlerinin sınırlamalarını aşma potansiyeline sahiptir. Bunlar arasında, in silico dizi çeşitlendirme yaklaşımı, PPI13'ün yapısal plastisitesini karakterize etmenin bir yolu olarak ligand dizisindeki çok çeşitli amino asit replasmanlarının enerjik etkisini değerlendirir. Bu yöntem, reseptör bağlanma bölgesine bağlı peptit ligandının yapısı veya modeli ile başlar ve daha sonra ligand dizisine mutasyonlar getirir. İstatistiksel ve enerji puanlama fonksiyonları daha sonra bu mutasyonların stabilite ve bağlanma afinitesi üzerindeki etkisini değerlendirmek için kullanılır. Değerlendirme aşamasından elde edilen en iyi skor ligand dizileri seti daha sonra amino asit tercihlerini hesaplamak için kullanılabilir. Bu strateji, çok yüksek sayıda ligand dizisini verimli bir şekilde işleme potansiyeline sahiptir. Bu nedenle, genellikle ıslak laboratuvar yaklaşımlarında işlenebilen daha sınırlı sayıda diziden hesaplananlara kıyasla amino asit tercihlerinin daha eksiksiz ve tutarlı bir çıkarımını sağlayabilir.
Rosetta moleküler modelleme paketi14'ün Pepspec uygulaması, peptit tasarım modunun önemli bir adımı olarak dizi çeşitlendirmesini gerçekleştiren bir araçtır. Bu uygulama, sonraki adımlar için bir çapa olarak kullanılan, uzunluk olarak tek bir amino asit kalıntısına kadar bağlı bir peptit ile reseptör proteininin bir yapısını veya modelini gerektirir. Bağlı peptitin dizisi daha sonra genişletilir (gerekirse) ve çok sayıda varsayılan peptit ligandı oluşturmak için çeşitlendirilir. Bu peptitlerin bağlanma afinitesi daha sonra en iyi tahmin edilen bağlanma skorlarına sahip olanları seçmek için esnek omurga peptit yerleştirme ile değerlendirilir. Bu uygulamanın ana çıktısı, tasarım aşamasının sonunda seçilen en iyi peptit adayları olmasına rağmen, bu aşamada kabul edilen çok daha büyük peptit seti, hedef bağlanma bölgesinin amino asit tercihlerini hesaplamak için de kullanılabilir. Amino asit tercihleri, bir konum ağırlık matrisi (PWM) veya daha görsel bir dizi logosu olarak temsil edilen ligand dizisinin konumu başına her bir amino asit kalıntısının frekansı olarak hesaplanır.
Bu makalede, bir PPI'da yer alan bir reseptör proteininin bağlanma yüzeyinin amino asit tercihlerini tahmin etmek için bir protokol açıklıyoruz. Protokol, protein-ligandın doğrusal bir segmentinin reseptör proteinine bağlandığı bilinen PPI'lara odaklanmıştır, bu nedenle senaryo bir protein-peptit arayüzü olarak modellenebilir. Bu senaryoda, liganddan korunan motifler tipik olarak reseptör bağlanma bölgesindeki tanımlanmış ceplerle etkileşime girer, ancak PPI'da yer alan tüm ligand segmenti daha az korunmuş bölgeler içerebilir. Protokolün ana adımlarını özetleyen bir akış şeması Şekil 1'de gösterilmektedir. Protokol, protein-protein kompleksinin 3D yapısı ile başlar ve ligand proteinini potansiyel olarak en iyi etkileşime giren segmente indirgeyerek reseptör proteinini sağlam bırakır. En iyi etkileşime giren segment, etkileşen iki protein arasındaki sıcak nokta kalıntılarını tanımlamak için hesaplamalı alanin tarama mutajenezi gerçekleştiren BUDE Alanin Tarama sunucusu15 kullanılarak çıkarılır. Bu yaklaşımda, liganddan gelen kalıntılar ayrı ayrı alanin ile değiştirilir ve daha sonra serbest enerjideki veya kompleksin stabilitesindeki (ΔΔG) tahmini değişiklik, hedef PPI için karşılık gelen kalıntının alaka düzeyini çıkarmak için kullanılır. En iyi etkileşime giren segment çıkarıldıktan sonra, reseptör proteini ile olan kompleksi, dizi çeşitlendirmesini gerçekleştirmek için Pepspec'e gönderilen baz yapı olarak kullanılır.

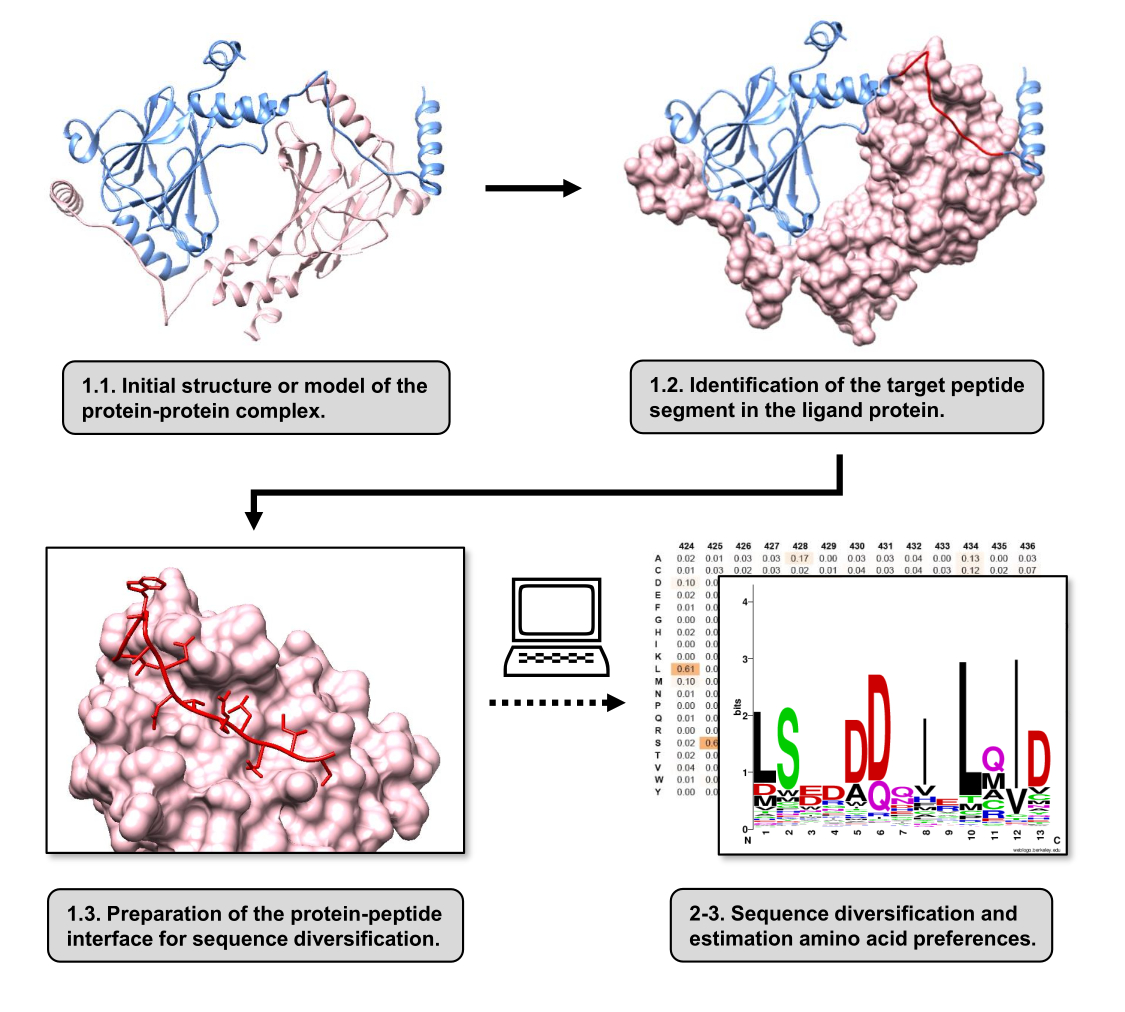
Şekil 1: Bu çalışmada önerilen protokolün ana adımlarına genel bakış. Sayılar, protokol bölümündeki adım numaralarıyla eşleşir. Metinde anlatılan örnek olarak kullanılan protein-protein kompleksi ile figürler yapılmıştır. Bu komplekste, reseptör olarak kabul edilen protein zinciri pembe renkle gösterilirken, ligand olarak kabul edilen zincir açık mavi renkle gösterilir ve tahmin edilen en iyi etkileşime giren segmenti kırmızı renkle vurgulanır. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
Önerilen protokolün sınırlamalarından biri, protein-peptit arayüzünün çözülmüş bir yapısı için gerekliliktir. Protokol alternatif olarak hedef protein-peptit arayüzünün bir modeli ile başlayabilir, ancak spesifik modelleme adımları burada açıklanmamıştır. Ayrıca, protokol herhangi bir işletim sistemini çalıştıran kişisel bir bilgisayarda yürütülebilse de, Rosetta uygulamalarını içeren adımlar için bir Linux ortamı gereklidir. Genellikle Pepspec tarafından gerçekleştirilen çok sayıda yineleme nedeniyle dizi çeşitlendirme adımı için bir bilgisayar kümesi de şiddetle tavsiye edilir.
Önerilen protokolün uygulanması, insan interferon düzenleyici faktör (IRF) ailesinin bir üyesi olan IRF5'in teklif yüzeyinin amino asit tercihlerinin tahmini ile gösterilmiştir. Bu proteini örnek olarak seçtik, çünkü aktivasyonu sırasında iki alt birim, yapısı iyi karakterize edilmiş bir dimer oluşturmak üzere bağlanır16. IRF dimerlerinde bağlanma, bir alt birimin bağlanma yüzeyini sağladığı ve diğerinin pLxIS17,18 adı verilen kısa korunmuş bir motif içeren bir bölge aracılığıyla etkileşime girdiği bir protein-peptit arayüzü olarak modellenebilir. Ek olarak, IRF alt birimlerine bağlanma çok spesifiktir; bu nedenle, koaktivatörler18 olarak bilinen diğer hücresel proteinlerle homodimerler, heterodimerler ve kompleksler oluşturabilirler.
Protokol
1. Protein-peptit arayüzünün ilk hazırlanması
- Protein-protein kompleksinin yapısının indirilmesi
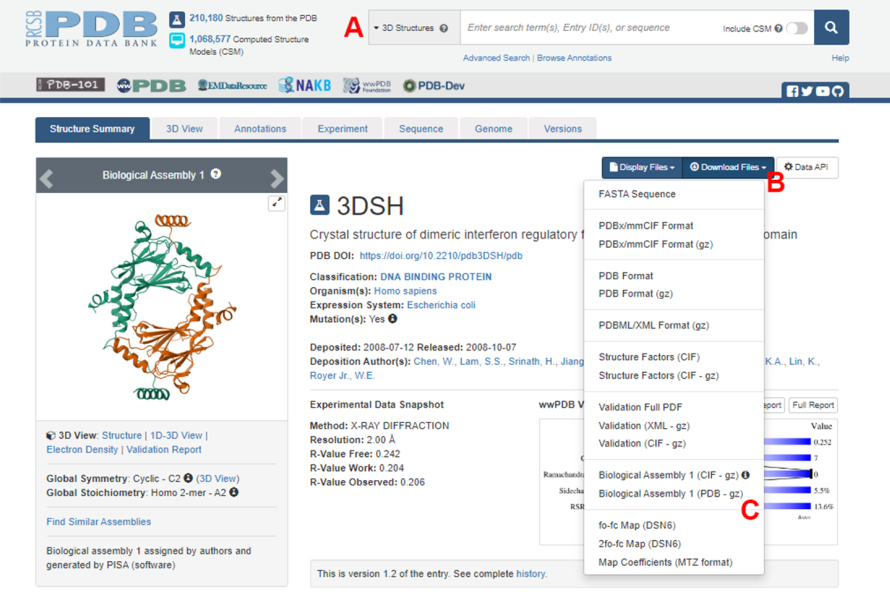
- Protein Veri Bankası (PDB) ana sayfasına (https://www.rcsb.org/) gidin ve ana arama kutusuna protein-protein kompleksinin yapısı için PDB ID'yi yazın (Şekil 2A). Bu çalışmada örnek olarak kullanılan IRF5 dimerinin yapısı için PDB ID'si 3DSH19'dur.
- İstenen yapı için ana sayfada, Dosyaları İndir'e (Şekil 2B) ve ardından Biyolojik Montaj 1'e (PDB - gz) (Şekil 2C) tıklayın.
NOT: PDB veritabanında, aynı monomerler tarafından oluşturulan birçok protein kompleksinin yapıları, PDB dosyasında yalnızca bir monomerin (asimetrik birim) yapısının saklandığı biyolojik düzenekler olarak temsil edilir. Multimerin yapısı, bu durumda, IRF5 dimer, asimetrik birimin iki örneğini içeren biyolojik düzenek olarak indirilmelidir. Bu protokolün sonraki adımlarını kolaylaştırmak için, önce iki monomer ayrılır ve bunlara farklı zincir kimlikleri atanır. - İndirilen yapıyı UCSF Chimera20'de açın ve Tools > Structure Editing > Change Chain IDS'e tıklayın. Bu örnekte, biyolojik düzenekteki her iki zincir de A olarak adlandırılmıştır. İkinci zinciri ( #0.2 etiketli) B olarak yeniden adlandırın ve Tamam'a tıklayın.
- Model Paneli> Sık Kullanılanlar'a tıklayın ve ardından iki zinciri içeren modeli seçin. Her zinciri farklı bir modele ayırmak için Grupla/Grubu Çöz düğmesine tıklayın. Ardından, iki modeli seçin ve Kopyala/Birleştir düğmesine tıklayın. Birleştirilmiş model için yeni bir ad girin, Close Source Models (Kaynak Modellerini Kapat) seçeneğini işaretleyin ve OK (Tamam) düğmesini tıklatın.
- > Zincir Seç'e tıklayın ve dimerdeki her zincirin artık A ve B olmak üzere farklı bir harfle tanımlandığını onaylayın.
- Düzenlenen yapıyı, protokolün sonraki adımlarında kullanılacak olan farklı bir PDB dosyasına kaydetmek için Dosya > PDB'yi Kaydet'i kullanın (burada IRF5_dimer.pdb adı kullanılmıştır).

Şekil 2: Bu çalışmada temsili bir örnek olarak kullanılan yapı için Protein Veri Bankası (PDB) sayfası. (A) Hedef yapının PDB katılım kodunu tanıtmak için arama kutusu. (B) Yapıyı çeşitli formatlarda indirmek için menü. (C) Yapı asimetrik bir birim olarak kaydedildiğinde biyolojik düzenekleri indirme seçenekleri (daha fazla ayrıntı için bkz. adım 1.1.2). Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
- Ligand proteinindeki hedef segmentin belirlenmesi
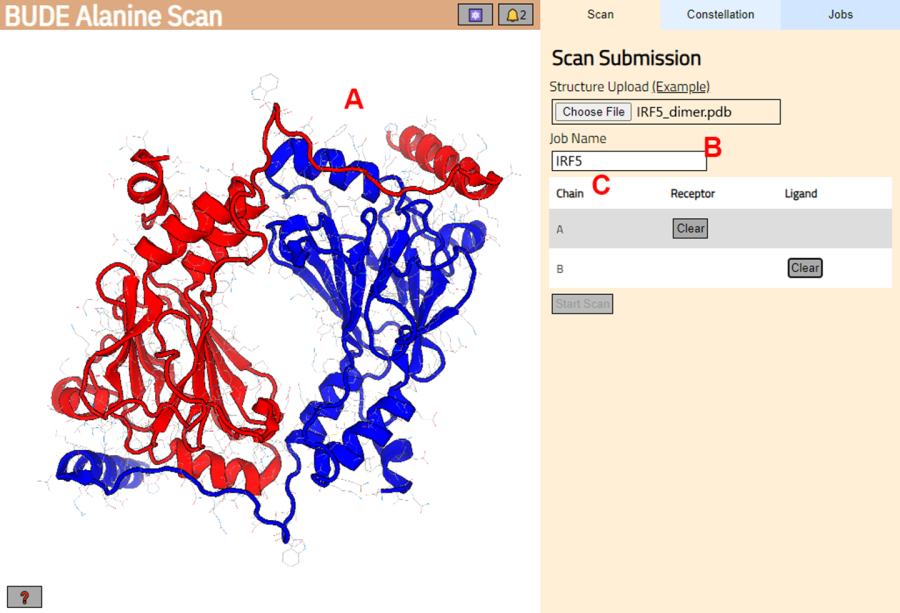
- BUDE Alanine Scan sunucusuna (https://pragmaticproteindesign.bio.ed.ac.uk/balas/) gidin. Structure Upload (Yapı Yükleme) altındaki Choose File (Dosya Seç) düğmesine tıklayın ve adım 1.1.6'da kaydedilen PDB dosyasını seçin.
- Sonraki sayfada, yapının doğru şekilde yüklenip yüklenmediğini kontrol edin (Şekil 3A) ve sunucuda iş için bir ad girin (Şekil 3B).
- Reseptör (A) ve ligand (B) olarak muamele görecek olan PDB'den zincirleri ayarlayın (Şekil 3C). Ardından, işi göndermek için Taramayı Başlat düğmesine tıklayın.
- İş bittiğinde, sonuçlar sayfasını açmak için Sonuçları Göster'e tıklayın (Şekil 4).
NOT: Sonuçlar sayfasında, ligand yapısından gelen kalıntılar, serbest enerjideki tahmini değişimlerine (ΔΔG) göre renklendirilir ve daha yüksek değerlere sahip olanlar kırmızı ile renklendirilir. - Kalıntı listesinden, hedef bağlama yüzeyi ile daha iyi etkileşime gireceği tahmin edilen kalıntı gerginliğini seçin. Bu kalıntıların serbest enerji (ΔΔG) farkı için daha yüksek değerleri kümelediğinden emin olun. Bu örnekte, Leu424 ve Ser436 kalıntıları arasındaki segment seçilmiştir ( Şekil 4'ün sağ panelinde kırmızı bir kutu ile vurgulanmıştır).
- Dizi çeşitlendirmesi için protein-peptit arayüzünün hazırlanması
- Adım 1.1.6'da kaydedilen PDB dosyasını Chimera'da açın ve hedef alt birimlerin yapısında eksik atom veya bağ olup olmadığını kontrol edin.
- Orijinal yapı ile birlikte kristalize edilmiş tüm küçük molekülleri, iyonları ve çözücüleri silin. Bunu yapmak için, Kalıntı > Seç'e tıklayın ve ardından standart amino asitler dışındaki tüm molekülleri seçin. Ardından, Atomlar/Bağlar > Eylemler'e tıklayın ve Sil'e tıklayın.
- Ligand zincirini, adım 1.2.5'te seçilen en iyi etkileşime giren segmente kırpın. Bunu yapmak için, Sık Kullanılanlar ve Sıra'ya tıklayın ve ardından ligand (B) olarak kabul edilen zincire tıklayın. Sıra panelinde, 424 ve 436 konumları arasındakiler dışındaki tüm kalıntıları seçmek için fareyi sürükleyin. Bu kalıntıları silmek için Atomlar/Bağlar > Eylemler'e ve Sil'e tıklayın.
- Düzenlenen yapıyı, protokolün sonraki adımlarında kullanılan farklı bir PDB dosyasına kaydetmek için Dosya > PDB'yi Kaydet'i kullanın (burada IRF5_interface.pdb adı kullanılmıştır).

Resim 3: BUDE Alanin Scan sunucusunda reseptör ve ligand seçimi. (A) Protein-protein kompleksinin grafik gösterimi. (B) Sunucudaki işin adını girmek için metin kutusu. (C) Reseptör ve ligand olarak kabul edilecek zincirleri etkileşimli olarak seçmek için panel (daha fazla ayrıntı için bkz. adım 1.2). Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}

Şekil 4: BUDE Alanine Scan sunucusunun sonuç sayfası. Ligand dizisindeki potansiyel en iyi etkileşime giren segment kırmızı bir kutu ile gösterilir. Sol panelde, tahmin edilen daha yüksek enerji katkısına (Leu433) sahip kalıntı yeşil renkle vurgulanmıştır. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
2. Dizi çeşitlendirmesi
NOT: Aşağıdaki adımlarda, rosetta_main , genellikle /opt/rosetta_src__bundle/main/ konumunda bulunan ana Rosetta kurulum dizinine atıfta bulunur, burada kurulu Rosetta sürümünü gösterir. Ayrıca, Rosetta uygulamalarının sistem genelinde erişilebilir olduğu varsayılmaktadır; Durum böyle değilse, yürütülebilir dosyalara giden tam yolun sağlanması gerekir. Kaynaktan derlendiğinde, bu yürütülebilir dosyalar /rosetta_main/source/bin/ dizininde bulunur.
- Amino asit yan zincirlerinin ilk optimizasyonu
- Adım 1.3.4'te kaydedilen düzenlenmiş yapıyı Rosetta uygulamaları tarafından erişilebilen bir Linux konumuna kopyalayın.
- Dizi çeşitlendirmesinden önce baz yapısının tüm amino asit yan zincirlerinin yeniden paketlenmesini gerçekleştirmek için Rosetta'nın FixBB uygulamasını kullanın. Bu işlemde, tüm amino asit yan zincirlerinin oryantasyonu, enerjiyi en aza indirmek ve kompleksin stabilitesini artırmak için optimize edilir. Bunu yapmak için aşağıdaki komutu çalıştırın:

NOT: Bu komut, ek bir sayısal sonek (bu örnekte IRF5_interface_0001.pdb ) ile orijinal yapının adını taşıyan bir PDB dosyası çıktısı verir. - Protokolün bir sonraki adımını kolaylaştırmak için, aşağıdaki komutu kullanarak yeniden paketlenmiş PDB dosyasını _repack sonekiyle yeniden adlandırın:
MV IRF5_interface_0001.PDB IRF5_repack.pdb
- Dizi çeşitlendirmesi
- Aşağıdaki komutu kullanarak gerçek dizi çeşitlendirme adımını gerçekleştirmek için Pepspec'i tasarım modunda çalıştırın:

Aşağıdakiler genel seçeneklerdir:- -s giriş dosyasını gösterir (adım 2.1.3'te oluşturulan yeniden paketlenmiş PDB dosyası).
- -o , çıktı dosyalarını adlandırmak için öneki belirtir.
- - veritabanı, ana Rosetta 3 veritabanının yolunu gösterir.
- -ex1, -ex2 ve extrachi_cutoff rotamer kitaplık seçenekleridir (daha fazla ayrıntı için Pepspec belgelerine bakın).
- -overwrite , uygulamaya önceki yinelemeler tarafından oluşturulan önceden var olan olası çıktıların üzerine yazmasını söyler.
- -pepspec:pep_chain , ligand (bu örnekte 'b') olarak kabul edilen PDB zincirlerini belirtir.
- -pepspec:native_pep_anchor , çapa olarak kullanılan amino asit kalıntısını gösterir (bu örnekte, ligand peptidinin 10. pozisyonundaki Leu kalıntısı).
- -pepspec:n_peptides çıktı alınacak peptit yapılarının sayısını gösterir.
- -pepspec:no_prepack_prot uygulamaya giriş tabanı yapısında yeniden paketlemeyi atlamasını söyler (çünkü bu daha önce adım 2.1'de gerçekleştirilmiştir).
NOT: Ana Pepspec çıktısı, .pdbs sonekiyle (örnekte IRF5.pdbs) çıkış öneki kullanılarak adlandırılan, tasarım aşamasından kaynaklanan peptitler için PDB dosyalarını içeren bir dizindir. Ek olarak, Pepspec, dizi çeşitlendirme adımının bir parçası olarak test edilen tüm kabul edilen peptit dizilerini ve bunlara karşılık gelen Rosetta enerji puanlarını, çıktı önekinden sonra adlandırılan sekmeyle ayrılmış bir metin dosyasında çıkarır. spec soneki (örnekte IRF5.spec). Bu çalışmada açıklanan protokol, gerçek peptit tasarımından ziyade amino asit tercihlerini tahmin etmeyi amaçladığından, sonraki adımlar .pdbs dizinindeki PDB yapıları yerine IRF5.spec kullanır.
- Aşağıdaki komutu kullanarak gerçek dizi çeşitlendirme adımını gerçekleştirmek için Pepspec'i tasarım modunda çalıştırın:
3. Amino asit tercihlerinin tahmini
- PWM hesaplama
- Bir PWM oluşturmak için Rosetta paketinde bulunan gen_pepspec_pwm.py betiğini kullanın. Bu komut dosyasını çalıştırmak için aşağıdaki komutu kullanın:

nerede:- IRF5.spec , adım 2.2'de oluşturulan Pepspec çıktı dosyasıdır.
- -1 , dizide ek N-terminal kalıntısı olmadığını ve bu nedenle PWM'deki konumların 1 tabanlı olduğunu gösterir.
- 0,2 , komut dosyasına yalnızca Pepspec çıktısından en iyi puan alan %20'lik peptitleri dikkate almasını söyler (varsayılan değer 0,1'dir, %10'a karşılık gelir).
- interface_score , komut dosyasına, Pepspec çıktı dosyasında bulunan çeşitli Rosetta puanlarından biri olan arayüz puanına göre peptitleri sıralamasını söyler.
NOT: Bu komut dosyası, biri hesaplanan PWM ( .pwm sonekiyle) ve diğeri PWM'yi hesaplamak için kullanılan peptitlerin alt kümesinin dizileri için ( .seq sonekiyle) olmak üzere iki çıktı dosyası oluşturur. Bu dosyaların adları, sıralama için kullanılan peptitlerin skorunu ve fraksiyonunu da içerir. Bu örnekte, bu dosyalar sırasıyla IRF5_interface_score_0.2.pwm ve IRF5_interface_score_0.2.seq olarak adlandırılmıştır.
- Bir PWM oluşturmak için Rosetta paketinde bulunan gen_pepspec_pwm.py betiğini kullanın. Bu komut dosyasını çalıştırmak için aşağıdaki komutu kullanın:
- Dizi logosu oluşturma
- WebLogo sunucusuna (https://weblogo.berkeley.edu/logo.cgi)21 gidin ve Sıra Verilerini Yükle'nin yanındaki Dosya Seç düğmesine tıklayın. Adım 3.1.1'de (bu örnekte IRF5_interface_score_0.2.seq) oluşturulan peptit dizilerini içeren dosyayı yükleyin.
- Giriş uzunluğuna göre logonun istediğiniz biçimini ve boyutunu seçin. Örnek, PDF biçimini ve 15 cm x 12 cm boyutunu kullanır. Logo Oluştur'a tıklayın.
Sonuçlar
Bu makalede, insan interferon düzenleyici faktörler olarak bilinen bir transkripsiyon faktörleri ailesinin bir üyesi olan IRF5'in bağlanma yüzeyinin amino asit tercihlerini tahmin etmek için bir protokol tanımladık. Bu proteinler, doğuştan gelen ve adaptif bağışıklık tepkilerinin düzenleyicileridir ve çeşitli bağışıklık hücrelerinin farklılaşmasına ve aktivasyonuna katılırlar. IRF alt birimleri, diğer hücresel proteinlerle homodimerler, heterodimerler ve k...
Tartışmalar
Bu makale, in silico dizi çeşitlendirmesine dayalı olarak potansiyel olarak çok spesifik bağlanma bölgelerinin amino asit tercihlerini tahmin etmek için bir protokolü açıklamaktadır. Protein-peptit arayüzlerinin amino asit tercihlerini tahmin etmek için az sayıda hesaplama aracı geliştirilmiştir 14,25,26. Bu araçlar tahmine dayalı bir yapıya sahiptir, ancak tahminlerini gerç...
Açıklamalar
Yazarların ifşa edecek hiçbir şeyi yok.
Teşekkürler
Sistema Nacional de Investigación (SNI) (hibe numaraları SNI-043-2023 ve SNI-170-2021), Panama Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) ve Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) tarafından sağlanan mali destek minnetle kabul edilmektedir. Yazarlar, makaleyi dikkatlice incelediği için Dr. Miguel Rodríguez'e teşekkür eder.
Malzemeler
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Referanslar
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiThis article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır