
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
단백질-단백질 상호작용에 관여할 수 있는 다중특이적 펩타이드 결합 도메인의 아미노산 선호도에 대한 전산 예측(Computational Prediction of Amino Acid Preferences of Potentially Multispecific Peptide-binding domains involved in protein-protein interactions)
요약
단백질-단백질 상호 작용(PPI)에서 다중특이적 결합 부위의 아미노산 선호도를 추정하기 위한 염기서열 다양화에 기반한 방법론을 설명합니다. 이 전략에서는 수천 개의 잠재적인 펩타이드 리간드가 생성되고 실리코로 스크리닝되어 사용 가능한 실험 방법의 일부 한계를 극복합니다.
초록
많은 단백질-단백질 상호 작용은 짧은 단백질 세그먼트를 펩타이드 결합 도메인에 결합하는 것을 포함합니다. 일반적으로 이러한 상호 작용은 가변 보존을 가진 선형 모티프의 인식을 필요로 합니다. 동일한 리간드에서 고도로 보존된 영역과 더 가변적인 영역의 조합은 종종 효소와 세포 신호 단백질의 공통 특성인 결합의 다중 특이성에 기여합니다. 펩타이드 결합 도메인의 아미노산 선호도 특성 분석은 단백질-단백질 상호 작용(PPI)의 매개체 설계에 중요합니다. 계산 방법은 종종 비용이 많이 들고 번거로운 실험 기술에 대한 효율적인 대안으로, 나중에 다운스트림 실험에서 검증할 수 있는 잠재적 매개체를 설계할 수 있습니다. 여기에서는 Rosetta 분자 모델링 패키지의 Pepspec 애플리케이션을 사용하여 펩타이드 결합 도메인의 아미노산 선호도를 예측하는 방법론을 설명했습니다. 이 방법론은 수용체 단백질의 구조와 펩타이드 리간드의 특성이 모두 알려져 있거나 추론될 수 있는 경우에 유용합니다. 이 방법론은 리간드에서 잘 특성화된 앵커로 시작하며, 이는 아미노산 잔기를 무작위로 추가하여 확장됩니다. 이러한 방식으로 생성된 펩타이드의 결합 친화도는 그런 다음 가장 잘 예측된 결합 점수를 가진 펩타이드를 선택하기 위해 유연한 백본 펩타이드 도킹에 의해 평가됩니다. 그런 다음 이러한 펩타이드를 사용하여 아미노산 선호도를 계산하고 추가 연구에 사용할 수 있는 위치-중량 매트릭스(PWM)를 선택적으로 계산합니다. 이 방법론의 적용을 설명하기 위해 우리는 이전에 다중 특이적인 것으로 알려졌지만 pLxIS라는 짧은 보존 모티프에 의해 전 세계적으로 안내되는 인간 인터페론 조절 인자 5(IRF5)의 하위 단위 간의 상호 작용을 사용했습니다. 추정된 아미노산 선호도는 IRF5 결합 표면에 대한 이전 지식과 일치했습니다. 인산화 가능한 세린 잔기가 차지하는 위치는 아스파르테이트와 글루타메이트의 빈도가 높았는데, 이는 음전하를 띤 측쇄가 포스포세린과 유사하기 때문일 수 있습니다.
서문
두 단백질 간의 상호 작용은 종종 단백질-펩타이드 계면과 유사한 펩타이드 결합 도메인에 아미노산의 짧은 세그먼트를 결합하는 것을 포함합니다. 이러한 단백질-단백질 상호작용(PPI)에 관여하는 수용체 단백질은 종종 겹치지만 발산하는 리간드 서열의 특정 세트를 인식할 수 있는 능력을 가지고 있으며, 이러한 특성은 다중특이성(multispecificity) 1,2로 알려져 있습니다. 다중특이성 인식은 많은 세포 단백질의 특징이지만, 효소 및 세포 신호 단백질에서 특히 두드러집니다3. 다중특이적 결합 부위와 상호 작용하는 단백질은 종종 서열 4,5,6에서 더 많이 보존된 영역과 덜 보존된 영역의 조합을 가지고 있습니다. 이 시나리오에서는 더 보존된 서열 모티프가 엄격한 분자 상호 작용에 관여합니다. 반대로, 더 많은 가변 염기서열은 수용체 결합 부위의 어떻게든 허용적인 표면과 상호 작용합니다. 일반적으로, 덜 보존되었지만 여전히 기능적으로 관련성이 높은 분절은 정의된 2차 구조 패턴이 없거나 본질적으로 무질서한 단백질의 전형적인 것과 같은 훨씬 더 역동적인 구조를 가진 루프이다7.
결합 부위의 잠재적인 펩타이드 리간드를 식별하는 것은 일반적으로 해당 PPI를 방해할 수 있는 매개체 설계의 첫 번째 단계이다8. 그러나, 다중특이적 결합 부위의 리간드에서 대부분의 서열 위치에서 가장 빈번한 단일 아미노산 잔기를 발견하는 것은 종종 불가능합니다. 대신에, 이들 부위는 화학적 성질에 따라 특정 부류의 아미노산, 예를 들어, 아스파르테이트 또는 글루타메이트와 같은 산성 및 음전하를 띤 아미노산, 페닐알라닌과 같은 부피가 큰 방향족 아미노산 또는 지방족 아미노산 알라닌, 발린, 류신 또는 이소류신과 같은 더 많은 소수성 잔기에 대한 특정 선호도를 가질 수 있다3. 단백질 결합 부위의 아미노산 선호도에 대한 통찰력을 제공할 수 있는 여러 실험 방법에는 directedevolution9, multi-codon scanning mutagenesis10 및 deep mutational scanning11이 포함됩니다. 이러한 모든 방법은 원래 리간드에 돌연변이를 도입하고 수용체 단백질의 기능에 미치는 영향을 추가로 분석하는 것을 기반으로 하는 염기서열 다양화 접근 방식을 따릅니다(포괄적인 검토를 위해 Bratulic 및 Badran12 참조). 그러나 이러한 방법은 종종 대규모 시퀀스 라이브러리를 조사해야 하므로 더 번거롭고 비용이 많이 들고 시간이 많이 걸립니다.
다중특이적 결합 부위의 아미노산 선호도를 추론하는 계산 방법은 습식 실험실 방법의 한계를 우회할 수 있는 잠재력을 가지고 있습니다. 이 중 인실리코(in silico) 염기서열 다양화 접근법은 PPI13의 구조적 가소성을 특성화하는 방법으로 리간드 염기서열에서 광범위한 아미노산 치환의 에너지 영향을 평가합니다. 이 방법은 수용체 결합 부위에 결합된 펩타이드 리간드의 구조 또는 모델로 시작하여 리간드 서열에 돌연변이를 도입합니다. 그런 다음 통계 및 에너지 점수 함수를 사용하여 이러한 돌연변이가 안정성 및 결합 친화도에 미치는 영향을 평가합니다. 그런 다음 평가 단계에서 생성된 최고 점수의 리간드 서열 세트를 사용하여 아미노산 선호도를 계산할 수 있습니다. 이 전략은 효율적인 방식으로 매우 많은 수의 리간드 서열을 처리할 수 있는 잠재력을 가지고 있습니다. 따라서 일반적으로 습식 실험실 접근 방식에서 처리할 수 있는 더 제한된 수의 염기서열에서 계산된 것과 비교하여 아미노산 선호도에 대한 보다 완전하고 일관된 추론을 제공할 수 있습니다.
Rosetta 분자 모델링 제품군14 의 Pepspec 애플리케이션은 펩타이드 설계 모드의 핵심 단계로 염기서열 다양화를 수행하는 도구입니다. 이 응용 분야에는 단일 아미노산 잔기까지 결합된 펩타이드가 있는 수용체 단백질의 구조 또는 모델이 필요하며, 이는 다음 단계를 위한 앵커로 사용됩니다. 그런 다음 결합된 펩타이드의 서열을 확장하고(필요한 경우) 다양화하여 많은 수의 추정 펩타이드 리간드를 생성합니다. 그런 다음 이러한 펩타이드의 결합 친화도를 Flexible-backbone Peptide 도킹으로 평가하여 가장 잘 예측된 결합 점수를 가진 펩타이드를 선택합니다. 이 응용 프로그램의 주요 출력은 설계 단계의 끝에서 선택된 최상의 펩타이드 후보이지만, 이 단계에서 허용되는 훨씬 더 큰 펩타이드 세트를 사용하여 표적 결합 부위의 아미노산 선호도를 계산할 수도 있습니다. 아미노산 선호도는 리간드 서열의 위치당 각 아미노산 잔기의 빈도로 계산되며, 위치 가중치 매트릭스(PWM) 또는 보다 시각적인 서열 로고로 표시됩니다.
이 기사에서는 PPI에 관여하는 수용체 단백질의 결합 표면의 아미노산 선호도를 추정하는 프로토콜에 대해 설명합니다. 이 프로토콜은 단백질-리간드의 선형 세그먼트가 수용체 단백질에 결합하는 것으로 알려진 PPI에 중점을 두므로 시나리오를 단백질-펩타이드 계면으로 모델링할 수 있습니다. 이 시나리오에서, 리간드의 보존된 모티프는 전형적으로 수용체 결합 부위의 정의된 포켓과 상호 작용하지만, PPI에 관련된 전체 리간드 세그먼트는 덜 보존된 영역을 포함할 수 있습니다. 프로토콜의 주요 단계를 요약한 순서도가 그림 1에 나와 있습니다. 이 프로토콜은 단백질-단백질 복합체의 3D 구조로 시작하여 리간드 단백질을 잠재적으로 가장 상호 작용하는 세그먼트로 감소시켜 수용체 단백질을 그대로 유지합니다. 최상의 상호작용 세그먼트는 BUDE 알라닌 스캔 서버(15)를 사용하여 추론되며, 이는 두 상호작용하는 단백질 사이의 핫스팟 잔기를 식별하기 위해 계산적 알라닌 스캐닝 돌연변이 유발을 수행합니다. 이 접근법에서는 리간드의 잔류물을 개별적으로 알라닌으로 대체하고, 그런 다음 자유 에너지 또는 복합체의 안정성(ΔΔG)에서 추정된 변화를 사용하여 표적 PPI에 대한 해당 잔류물의 관련성을 추론합니다. 가장 상호 작용하는 세그먼트가 추론되면 수용체 단백질과의 복합체가 염기서열 다양화를 수행하기 위해 Pepspec에 제출된 기본 구조로 사용됩니다.

그림 1: 이 작업에서 제안된 프로토콜의 주요 단계 개요. 숫자는 프로토콜 섹션의 단계 번호와 일치합니다. 그림은 텍스트에 설명 된 예로 사용 된 단백질-단백질 복합체로 만들어졌습니다. 이 복합체에서 수용체로 간주되는 단백질 사슬은 분홍색으로 표시되는 반면, 리간드로 간주되는 사슬은 연한 파란색으로 표시되며 예측된 최상의 상호 작용 세그먼트는 빨간색으로 강조 표시됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
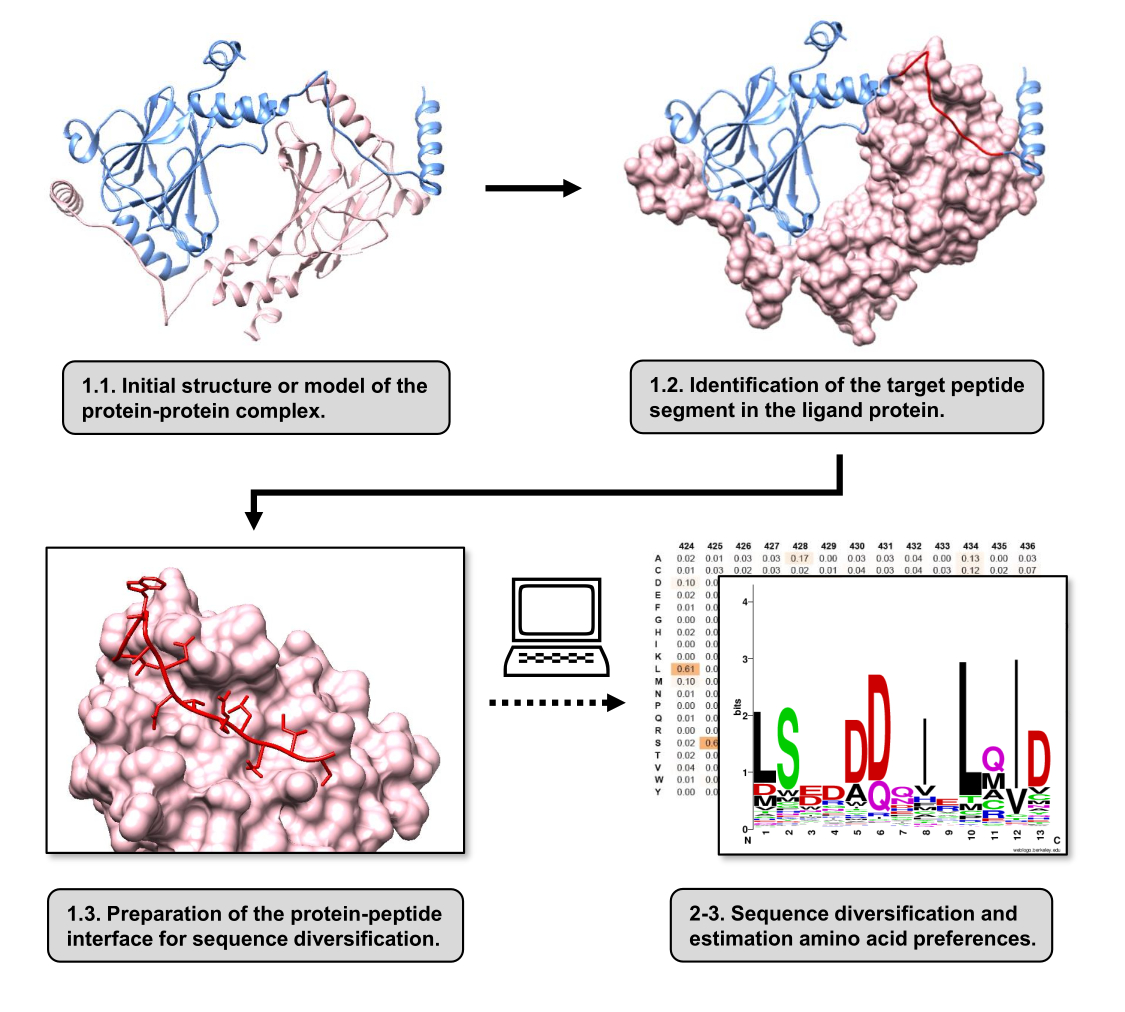{kind=link}
제안된 프로토콜의 한계 중 하나는 단백질-펩타이드 계면의 분리된 구조에 대한 요구 사항입니다. 프로토콜은 대안적으로 표적 단백질-펩티드 계면의 모델로 시작할 수 있지만, 구체적인 모델링 단계는 본원에서 설명되지 않는다. 또한 프로토콜은 모든 운영 체제를 실행하는 개인용 컴퓨터에서 수행할 수 있지만 Rosetta 응용 프로그램과 관련된 단계에는 Linux 환경이 필요합니다. 컴퓨터 클러스터는 일반적으로 Pepspec에서 수행하는 많은 수의 반복으로 인해 염기서열 다양화 단계에 적극 권장됩니다.
제안된 프로토콜의 적용은 인간 인터페론 조절 인자(IRF) 계열의 구성원인 IRF5의 입찰 표면의 아미노산 선호도 추정으로 설명됩니다. 우리가 이 단백질을 예로 든 이유는 이 단백질이 활성화되는 동안 두 개의 소단위체가 결합하여 구조가 잘 특성화된 이량체를 형성하기 때문입니다16. IRF 이량체에서 결합은 단백질-펩타이드 계면으로 모델링할 수 있으며, 여기서 하나의 소단위체는 결합 표면을 제공하고 다른 소단위체는 pLxIS17,18이라는 짧은 보존된 모티프를 포함하는 영역을 통해 상호 작용합니다. 또한 IRF 하위 단위에 대한 바인딩은 다중 특이적입니다. 따라서, 이들은 호모이량체(homodimer), 이종이량체(heterodimer) 및 코액티베이터(coactivator)로 알려진 다른 세포 단백질과 복합체를 형성할 수 있다(18).
프로토콜
1. 단백질-펩타이드 계면의 초기 준비
- 단백질-단백질 복합체의 구조 다운로드
- PDB(Protein Data Bank) 홈페이지(https://www.rcsb.org/)로 이동하여 기본 검색 상자에 단백질-단백질 복합체의 구조에 대한 PDB ID를 입력합니다(그림 2A). 본 연구에서 예시로 사용된 IRF5 이량체의 구조에 대한 PDB ID는 3DSH19이다.
- 원하는 구조의 메인 페이지에서 Download Files (그림 2B)를 클릭한 다음 Biological Assembly 1(PDB - gz) (그림 2C)을 클릭합니다.
참고: PDB 데이터베이스에서 동일한 단량체에 의해 형성된 많은 단백질 복합체의 구조는 생물학적 어셈블리로 표시되며, 여기서 하나의 단량체(비대칭 단위)의 구조만 PDB 파일에 저장됩니다. multimer의 구조(이 경우 IRF5 dimer)는 비대칭 단위의 두 인스턴스를 포함하는 생물학적 어셈블리로 다운로드해야 합니다. 이 프로토콜의 다음 단계를 용이하게 하기 위해 먼저 두 개의 단량체를 분리하고 서로 다른 체인 ID를 할당합니다. - UCSF Chimera20 에서 다운로드한 구조를 열고 Tools(도구) > Structure Editing(구조 편집) > Change Chain IDs(체인 ID 변경)를 클릭합니다. 이 예에서 생물학적 어셈블리의 두 체인 모두 A로 명명됩니다. 두 번째 체인( #0.2로 레이블이 지정됨)의 이름을 B 로 바꾸고 확인을 클릭합니다.
- 모델 패널> 즐겨찾기를 클릭한 다음 두 체인이 포함된 모델을 선택합니다. Group/Ungroup 버튼을 클릭하여 각 체인을 다른 모델로 분리합니다. 그런 다음 두 모델을 선택하고 복사/결합 버튼을 클릭합니다. 결합된 모델의 새 이름을 입력하고 Close Source Models(소스 모델 닫기)를 선택한 다음 OK(확인)를 클릭합니다.
- Select > Chain(체인 선택)을 클릭하고 이량체의 각 체인이 이제 다른 문자, 즉 A와 B로 식별되는지 확인합니다.
- 파일 > 저장 PDB를 사용하여 편집된 구조를 프로토콜의 다음 단계에서 사용할 다른 PDB 파일에 저장합니다(여기서는 IRF5_dimer.pdb라는 이름이 사용됨).

그림 2: 이 연구에서 대표적인 예로 사용된 구조에 대한 단백질 데이터 뱅크(PDB) 페이지. (A) 대상 구조체의 PDB 등록 코드를 소개하는 검색 상자. (B) 구조를 여러 형식으로 다운로드하는 메뉴. (C) 구조가 비대칭 단위로 저장된 경우 생물학적 어셈블리를 다운로드하는 옵션(자세한 내용은 1.1.2단계 참조). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
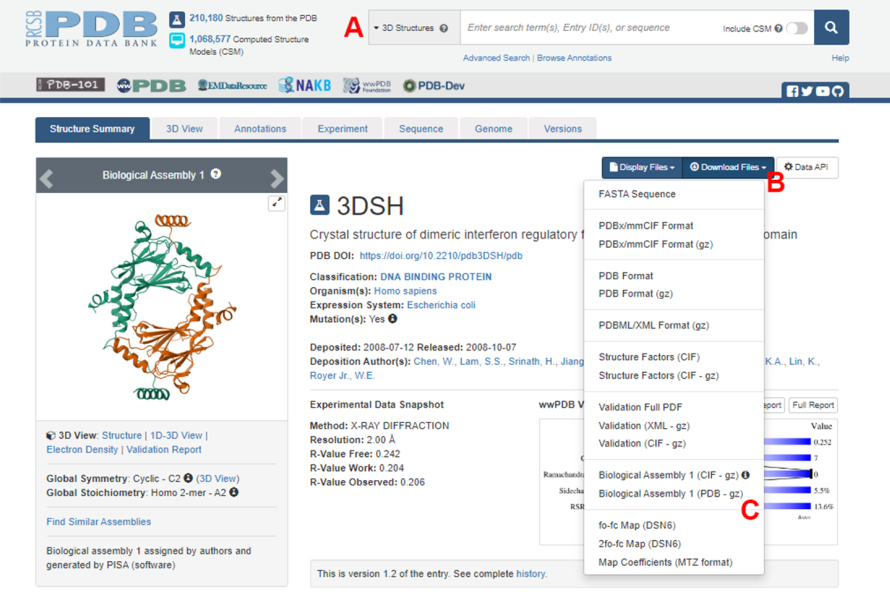{kind=link}
- 리간드 단백질의 표적 분절 식별
- BUDE 알라닌 스캔 서버(https://pragmaticproteindesign.bio.ed.ac.uk/balas/)로 이동합니다. Structure Upload(구조 업로드)에서 Choose File(파일 선택) 버튼을 클릭하고 1.1.6단계에서 저장한 PDB 파일을 선택합니다.
- 다음 페이지에서 구조가 올바르게 로드되었는지 확인하고(그림 3A) 서버에 작업 이름을 입력합니다(그림 3B).
- 수용체(A)와 리간드(B)로 처리될 PDB의 사슬을 설정합니다(그림 3C). 그런 다음 스캔 시작 버튼을 클릭하여 작업을 제출합니다.
- 작업이 완료되면 Show Results(결과 표시 )를 클릭하여 결과 페이지를 엽니다(그림 4).
참고: 결과 페이지에서 리간드 구조의 잔류물은 추정된 자유 에너지 변화(ΔΔG)에 따라 색상이 지정되고 값이 더 높은 잔차는 빨간색으로 표시됩니다. - residdue 목록에서, 타겟 binding surface와 더 잘 상호 작용할 것으로 예측된 residues의 stretch를 선택합니다. 이러한 잔차가 자유 에너지(ΔΔG)의 차이에 대해 더 높은 값을 클러스터링하는지 확인합니다. 이 예에서는 잔류물 Leu424와 Ser436 사이의 세그먼트가 선택되었습니다( 그림 4의 오른쪽 패널에서 빨간색 상자로 강조 표시됨).
- 염기서열 다양화를 위한 단백질-펩타이드 계면 준비
- 1.1.6단계에서 저장한 PDB 파일을 Chimera에서 열고 대상 subunit의 구조에 누락된 원자나 결합이 없는지 확인합니다.
- 원래 구조와 함께 결정화된 모든 작은 분자, 이온 및 용매를 삭제합니다. 이렇게 하려면 Select > Residues(잔기 선택 )를 클릭한 다음 표준 아미노산 이외의 모든 분자를 선택합니다. 그런 다음 Actions > Atoms/Bonds 및 Delete를 클릭합니다.
- 리간드 사슬을 1.2.5 단계에서 선택한 최상의 상호 작용 세그먼트로 자릅니다. 이렇게하려면 Favorites 및 Sequence 를 클릭 한 다음 ligand (B)로 간주되는 체인을 클릭하십시오. 시퀀스 패널에서 마우스를 드래그하여 위치 424와 436 사이의 잔차를 제외한 모든 잔차를 선택합니다. 이러한 잔여물을 삭제하려면 Actions > Atoms/Bonds 및 Delete를 클릭합니다.
- 파일 > 저장 PDB를 사용하여 편집된 구조를 프로토콜의 다음 단계에서 사용되는 다른 PDB 파일에 저장합니다(여기서는 IRF5_interface.pdb라는 이름이 사용됨).

그림 3: BUDE 알라닌 스캔 서버에서 수용체와 리간드의 선택. (A) 단백질-단백질 복합체의 그래픽 표현. (B) 서버의 작업 이름을 입력하는 텍스트 상자입니다. (C) 수용체와 리간드로 간주될 사슬을 대화식으로 선택하기 위한 패널(자세한 내용은 1.2단계 참조). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
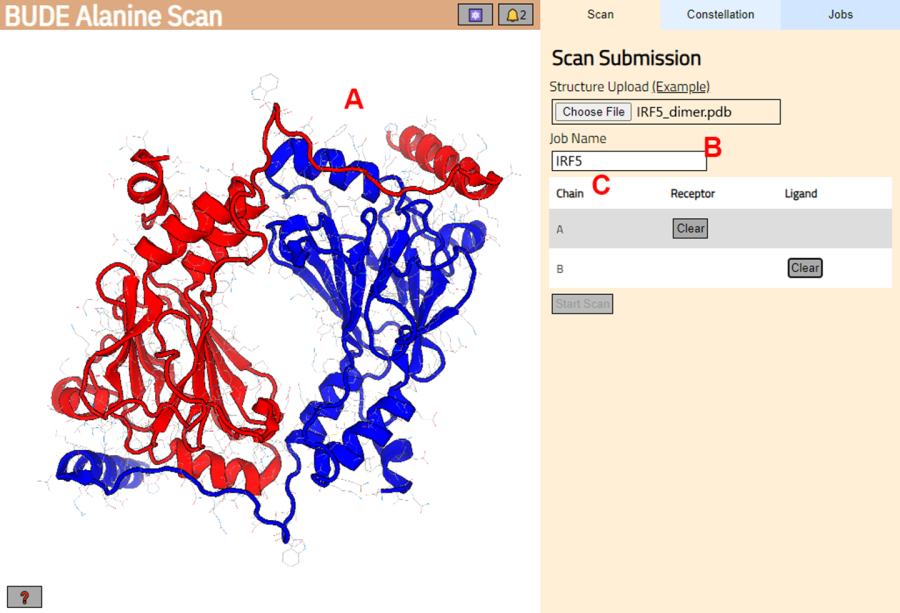{kind=link}

그림 4: BUDE Alanine Scan 서버의 결과 페이지. 리간드 서열에서 가장 잘 상호 작용할 수 있는 잠재적인 세그먼트는 빨간색 상자로 표시됩니다. 왼쪽 패널에서 예측된 에너지 기여도가 더 높은 잔류물(Leu433)이 녹색으로 강조 표시됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
2. 염기서열 다양화
참고: 다음 단계에서 rosetta_main 는 일반적으로 /opt/rosetta_src__bundle/main/에 위치한 기본 Rosetta 설치 디렉토리를 나타냅니다. 여기서 은 설치된 Rosetta 버전을 나타냅니다. 또한 Rosetta 응용 프로그램은 시스템 전체에서 액세스할 수 있다고 가정합니다. 그렇지 않은 경우 실행 파일의 전체 경로를 제공해야 합니다. 소스에서 컴파일 할 때 이러한 실행 파일은 /rosetta_main/source/bin/ 디렉토리에 있습니다.
- 아미노산 곁사슬의 초기 최적화
- 1.3.4단계에서 저장한 편집된 구조를 Rosetta 애플리케이션에서 액세스할 수 있는 Linux 위치에 복사합니다.
- Rosetta의 FixBB 애플리케이션을 사용하여 염기서열 다양화 전에 염기 구조의 모든 아미노산 곁사슬의 재포장을 수행합니다. 이 작업에서, 모든 아미노산 곁사슬의 배향은 에너지를 최소화하고 복합체의 안정성을 향상시키기 위해 최적화된다. 이렇게 하려면 다음 명령을 실행합니다.

참고: 이 명령은 원래 구조의 이름을 딴 PDB 파일에 추가 숫자 접미사(이 예에서는 IRF5_interface_0001.pdb )를 출력합니다. - 프로토콜의 다음 단계를 용이하게 하려면 다음 명령을 사용하여 _repack 접미사로 다시 압축된 PDB 파일의 이름을 바꿉니다.
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- 염기서열 다양화
- 설계 모드에서 Pepspec을 실행하여 다음 명령을 사용하여 실제 염기서열 다양화 단계를 수행합니다.

다음은 일반적인 옵션입니다.- -s 는 입력 파일(2.1.3단계에서 생성된 리패킹된 PDB 파일)을 표시합니다.
- -o 는 출력 파일의 이름을 지정하는 접두어를 나타냅니다.
- - database는 기본 Rosetta 3 데이터베이스의 경로를 나타냅니다.
- -ex1, -ex2 및 extrachi_cutoff 은 로타머 라이브러리 옵션입니다(자세한 내용은 Pepspec 설명서 참조).
- -overwrite 는 이전 반복에서 생성된 가능한 기존 출력을 덮어쓰도록 애플리케이션에 지시합니다.
- -pepspec:pep_chain 은 리간드(이 예에서는 'b')로 간주되는 PDB 체인을 나타냅니다.
- -pepspec:native_pep_anchor 은 앵커로 사용되는 아미노산 잔기를 나타냅니다(이 예에서는 리간드 펩타이드의 위치 10에 있는 Leu 잔기).
- -pepspec:n_peptides 은 출력할 펩타이드 구조의 수를 나타냅니다.
- -pepspec:no_prepack_prot 는 입력 기본 구조에서 다시 패킹을 건너뛰도록 애플리케이션에 지시합니다(이전에 2.1단계에서 수행되었으므로).
참고: 기본 Pepspec 출력은 설계 단계에서 생성된 펩타이드에 대한 PDB 파일이 포함된 디렉토리로, .pdbs 접미사(예: IRF5.pdbs)가 있는 출력 접두사를 사용하여 명명됩니다. 또한 Pepspec은 염기서열 다양화 단계의 일부로 테스트된 모든 허용 펩타이드 염기서열과 해당 Rosetta 에너지 점수를 출력 접두사의 이름을 딴 탭으로 구분된 텍스트 파일로 출력합니다. spec 접미사(예: IRF5.spec). 이 작업에서 설명하는 프로토콜은 실제 펩타이드 설계가 아닌 아미노산 선호도를 추정하는 것을 목표로 하기 때문에 다음 단계에서는 .pdbs 디렉토리의 PDB 구조 대신 IRF5.spec을 사용합니다.
- 설계 모드에서 Pepspec을 실행하여 다음 명령을 사용하여 실제 염기서열 다양화 단계를 수행합니다.
3. 아미노산 선호도 추정
- PWM 계산
- PWM을 생성하려면 Rosetta 제품군에 포함된 gen_pepspec_pwm.py 스크립트를 사용합니다. 이 스크립트를 실행하려면 다음 명령을 사용합니다.

어디:- IRF5.spec 은 2.2단계에서 생성된 Pepspec 출력 파일입니다.
- -1 은 시퀀스에 추가 N-말단 잔기가 없음을 나타내므로 PWM의 위치는 1부터 시작합니다.
- 0.2 는 Pepspec 출력에서 상위 20% 최고 점수의 펩타이드만 고려하도록 스크립트에 지시합니다(기본값은 10%에 해당하는 0.1입니다).
- interface_score 는 Pepspec 출력 파일에 포함된 다양한 Rosetta 점수 중 하나인 인터페이스 점수를 기반으로 펩타이드의 순위를 매기도록 스크립트에 지시합니다.
참고: 이 스크립트는 두 개의 출력 파일을 생성하는데, 하나는 계산된 PWM( .pwm 접미사 포함)에 대한 것이고 다른 하나는 PWM을 계산하는 데 사용되는 펩타이드 하위 집합의 시퀀스( .seq 접미사 포함)에 대한 것입니다. 이러한 파일의 이름에는 순위에 사용되는 펩타이드의 비율과 점수도 포함됩니다. 이 예에서 이러한 파일의 이름은 각각 IRF5_interface_score_0.2.pwm 및 IRF5_interface_score_0.2.seq입니다.
- PWM을 생성하려면 Rosetta 제품군에 포함된 gen_pepspec_pwm.py 스크립트를 사용합니다. 이 스크립트를 실행하려면 다음 명령을 사용합니다.
- 시퀀스 로고 생성
- WebLogo 서버 (https://weblogo.berkeley.edu/logo.cgi)21로 이동하여 시퀀스 데이터 업로드 옆에 있는 파일 선택 버튼을 클릭합니다. 3.1.1단계에서 생성된 펩타이드 서열이 포함된 파일을 업로드합니다(이 예에서는 IRF5_interface_score_0.2.seq).
- 입력 길이에 따라 원하는 로고 형식과 크기를 선택합니다. 이 예제에서는 PDF 형식과 15cm x 12cm 크기를 사용합니다. Create Logo(로고 만들기)를 클릭합니다.
결과
이 기사에서는 인간 인터페론 조절 인자(human interferon regulatory factor)로 알려진 전사 인자군(transcription factor) 계열의 구성원인 IRF5의 결합 표면의 아미노산 선호도를 예측하는 프로토콜에 대해 설명했습니다. 이 단백질은 선천성 면역 및 적응 면역 반응을 조절하며 여러 면역 세포의 분화 및 활성화에 참여합니다. IRF subunit은 매우 가소성 및 다중 특이적 결합 표면을 가지며 ...
토론
본 논문에서는 in silico 염기서열 다양화를 기반으로 잠재적으로 다중특이적 결합 부위의 아미노산 선호도를 추정하는 프로토콜에 대해 설명합니다. 단백질-펩타이드 계면 14,25,26의 아미노산 선호도를 추정하기 위해 개발된 계산 도구는 거의 없다. 이러한 도구는 예측 특성을 가지고 있지만 예측을 수?...
공개
저자는 공개할 것이 없습니다.
감사의 말
Sistema Nacional de Investigación(SNI)(보조금 번호 SNI-043-2023 및 SNI-170-2021), Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) 및 Instituto para la Formación y Aprovechamiento de Recursos Humanos(IFARHU)의 재정 지원에 감사드립니다. 저자들은 원고를 주의 깊게 검토해 준 Miguel Rodríguez 박사에게 감사의 뜻을 전합니다.
자료
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
참고문헌
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유