A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
חיזוי חישובי של העדפות חומצות אמינו של תחומים קושרי פפטיד רב-ספציפיים המעורבים באינטראקציות חלבון-חלבון
In This Article
Summary
אנו מתארים מתודולוגיה המבוססת על גיוון רצפים כדי להעריך את העדפות חומצות האמינו של אתרי קישור רב-ספציפיים באינטראקציות חלבון-חלבון (PPIs). באסטרטגיה זו, אלפי ליגנדים פפטידים פוטנציאליים נוצרים ומוקרנים בסיליקו, ובכך מתגברים על כמה מגבלות של שיטות ניסוי זמינות.
Abstract
אינטראקציות חלבון-חלבון רבות כוללות קשירה של מקטעי חלבון קצרים לתחומים קושרי פפטידים. בדרך כלל, אינטראקציות כאלה דורשות הכרה במוטיבים ליניאריים עם שימור משתנה. השילוב של אזורים שמורים מאוד ומשתנים יותר באותן ליגנדות תורם לעתים קרובות לרב-ספציפיות של הקשירה, תכונה משותפת של אנזימים וחלבוני איתות תאי. אפיון העדפות חומצות אמינו של תחומים קושרי פפטידים חשוב לתכנון מתווכים של אינטראקציות חלבון-חלבון (PPIs). שיטות חישוביות הן חלופה יעילה לטכניקות הניסוי היקרות והמסורבלות לעתים קרובות, ומאפשרות תכנון של מתווכים פוטנציאליים שניתן לאמת מאוחר יותר בניסויים במורד הזרם. במאמר זה תיארנו מתודולוגיה המשתמשת ביישום Pepspec של חבילת המידול המולקולרי של רוזטה כדי לחזות את העדפות חומצות האמינו של תחומים קושרי פפטידים. מתודולוגיה זו שימושית כאשר המבנה של חלבון הקולטן ואופי ליגנד הפפטיד ידועים או ניתנים להסקה. המתודולוגיה מתחילה עם עוגן מאופיין היטב מהליגנד, אשר מורחב על ידי הוספה אקראית של שאריות חומצות אמינו. זיקת הקשירה של פפטידים הנוצרים בדרך זו מוערכת לאחר מכן על ידי עגינה של פפטידים בעלי עמוד שדרה גמיש על מנת לבחור את הפפטידים עם ציוני הקשירה החזויים הטובים ביותר. פפטידים אלה משמשים לאחר מכן לחישוב העדפות חומצות אמינו ולחישוב אופציונלי של מטריצת מיקום-משקל (PWM) שניתן להשתמש בה במחקרים נוספים. כדי להמחיש את היישום של מתודולוגיה זו, השתמשנו באינטראקציה בין תת-יחידות של גורם רגולטורי אינטרפרון אנושי 5 (IRF5), שהיה ידוע בעבר כרב-ספציפי אך מונחה גלובלית על ידי מוטיב שימור קצר שנקרא pLxIS. ההעדפות המשוערות של חומצות אמינו עלו בקנה אחד עם ידע קודם על משטח הקישור IRF5. עמדות שנתפסו על ידי שאריות סרין זרחניות הציגו תדירות גבוהה של אספרטט וגלוטמט, ככל הנראה מכיוון ששרשראות הצד הטעונות שלילית שלהם דומות לפוספוסרין.
Introduction
אינטראקציה בין שני חלבונים כרוכה לעתים קרובות בקשירה של מקטעים קצרים של חומצות אמינו לתחומים קושרי פפטידים, הדומים לממשקים חלבון-פפטידים. חלבוני קולטן המעורבים באינטראקציות חלבון-חלבון כאלה (PPI) מסוגלים לעתים קרובות לזהות קבוצה מסוימת של רצפי ליגנדים חופפים אך מסתעפים, תכונה הידועה בשם רב-ספציפיות 1,2. זיהוי רב-ספציפי הוא תכונה של חלבונים תאיים רבים, אך הוא יוצא דופן במיוחד באנזימים ובחלבוני איתות תאי3. חלבונים המקיימים אינטראקציה עם אתרי קשירה רב-ספציפיים מכילים לעתים קרובות שילוב של אזורים שמורים יותר ופחות ברצף שלהם 4,5,6. בתרחיש זה, מוטיבים רצפים שמורים יותר מעורבים באינטראקציות מולקולריות מחמירות. לעומת זאת, הרצפים המשתנים יותר מתקשרים עם משטחים מתירניים איכשהו באתר קשירת הקולטן. בדרך כלל, מקטעים פחות שמורים אלה אך עדיין רלוונטיים מבחינה תפקודית הם לולאות חסרות תבניות מבנה משניות מוגדרות או בעלות קונפורמציות דינמיות עוד יותר, כגון אלה האופייניות לחלבונים בעלי הפרעה פנימית7.
זיהוי ליגנדות פפטידים פוטנציאליות של אתרי קשירה הוא בדרך כלל הצעד הראשון בתכנון מתווכים המסוגלים להפריע ל- PPIs8 המתאימים. עם זאת, לעתים קרובות לא סביר למצוא שארית אחת של חומצות אמינו שכיחות ביותר ברוב מיקומי הרצף בליגנדות של אתרי קישור רב-ספציפיים. במקום זאת, אתרים אלה עשויים להיות העדפות מיוחדות עבור סוג מסוים של חומצות אמינו על פי התכונות הכימיות שלהם, למשל, חומצות אמינו חומציות טעונות שלילית כגון אספרטט או גלוטמט, חומצות אמינו ארומטיות מגושמות כגון פנילאלנין או שאריות הידרופוביות יותר כגון חומצות אמינו אליפטיות אלנין, ואלין, לאוצין או איזולאוצין3. מספר שיטות ניסוי יכולות לספק תובנות לגבי העדפות חומצות אמינו של אתרי קשירת חלבונים, כולל אבולוציה מכוונת9, מוטגנזה של סריקה מרובת קודונים10 וסריקה מוטציונית עמוקה11. כל השיטות הללו נוקטות בגישה של גיוון רצפים, המבוססת על החדרת מוטציות לליגנדות המקוריות וניתוח נוסף של השפעתן על תפקוד חלבון הקולטן (ראו Bratulic and Badran12 לסקירה מקיפה). עם זאת, שיטות אלה דורשות לעתים קרובות סקר של ספריות רצפים גדולות, מה שהופך אותן למסורבלות, יקרות וגוזלות זמן רב יותר.
לשיטות חישוביות להסיק את העדפות חומצות האמינו של אתרי קישור רב-ספציפיים יש פוטנציאל לעקוף את המגבלות של שיטות מעבדה רטובות. בין אלה, גישת גיוון רצף הסיליקו מעריכה את ההשפעה האנרגטית של מגוון רחב של תחליפי חומצות אמינו ברצף הליגנד כדרך לאפיין את הפלסטיות המבנית של PPI13. שיטה זו מתחילה במבנה או במודל של ליגנד הפפטיד הקשור לאתר קשירת הקולטן ולאחר מכן מציגה מוטציות לרצף הליגנד. פונקציות סטטיסטיות וניקוד אנרגיה משמשות לאחר מכן כדי להעריך את ההשפעה של מוטציות אלה על יציבות וזיקה קשירה. לאחר מכן ניתן להשתמש בקבוצת רצפי הליגנד בעלי הניקוד הטוב ביותר הנובעים משלב ההערכה כדי לחשב את העדפות חומצות האמינו. לאסטרטגיה זו יש פוטנציאל לעבד מספר גבוה מאוד של רצפי ליגנדים בצורה יעילה. לכן, הוא יכול לספק הסקה מלאה ועקבית יותר של העדפות חומצות אמינו בהשוואה לאלה המחושבות ממספר מצומצם יותר של רצפים שבדרך כלל ניתן לעבד בגישות מעבדה רטובות.
יישום Pepspec של Rosetta molecular modeling suite14 הוא כלי המבצע גיוון רצף כשלב מפתח במצב העיצוב הפפטידי שלו. יישום זה דורש מבנה או מודל של חלבון הקולטן עם פפטיד קשור עד לשארית חומצת אמינו בודדת באורך, אשר משמש כעוגן לשלבים הבאים. לאחר מכן מרחיבים את רצף הפפטיד הכבול (במידת הצורך) ומגוונים אותו כדי ליצור מספר רב של ליגנדות פפטידיות משוערות. זיקת הקשירה של פפטידים אלה מוערכת לאחר מכן על ידי עגינה פפטידית גמישה בעמוד השדרה על מנת לבחור את אלה עם ציוני הקשירה החזויים הטובים ביותר. למרות שהפלט העיקרי של יישום זה הוא המועמדים הפפטידים הטובים ביותר שנבחרו בסוף שלב התכנון, ניתן להשתמש בקבוצה הגדולה בהרבה של פפטידים שהתקבלה בשלב זה גם כדי לחשב את העדפות חומצות האמינו של אתר קשירת המטרה. העדפות חומצות אמינו מחושבות כתדירות של כל שארית חומצת אמינו לכל מיקום של רצף הליגנד המיוצג כמטריצת משקל מיקום (PWM) או כלוגו של רצף חזותי יותר.
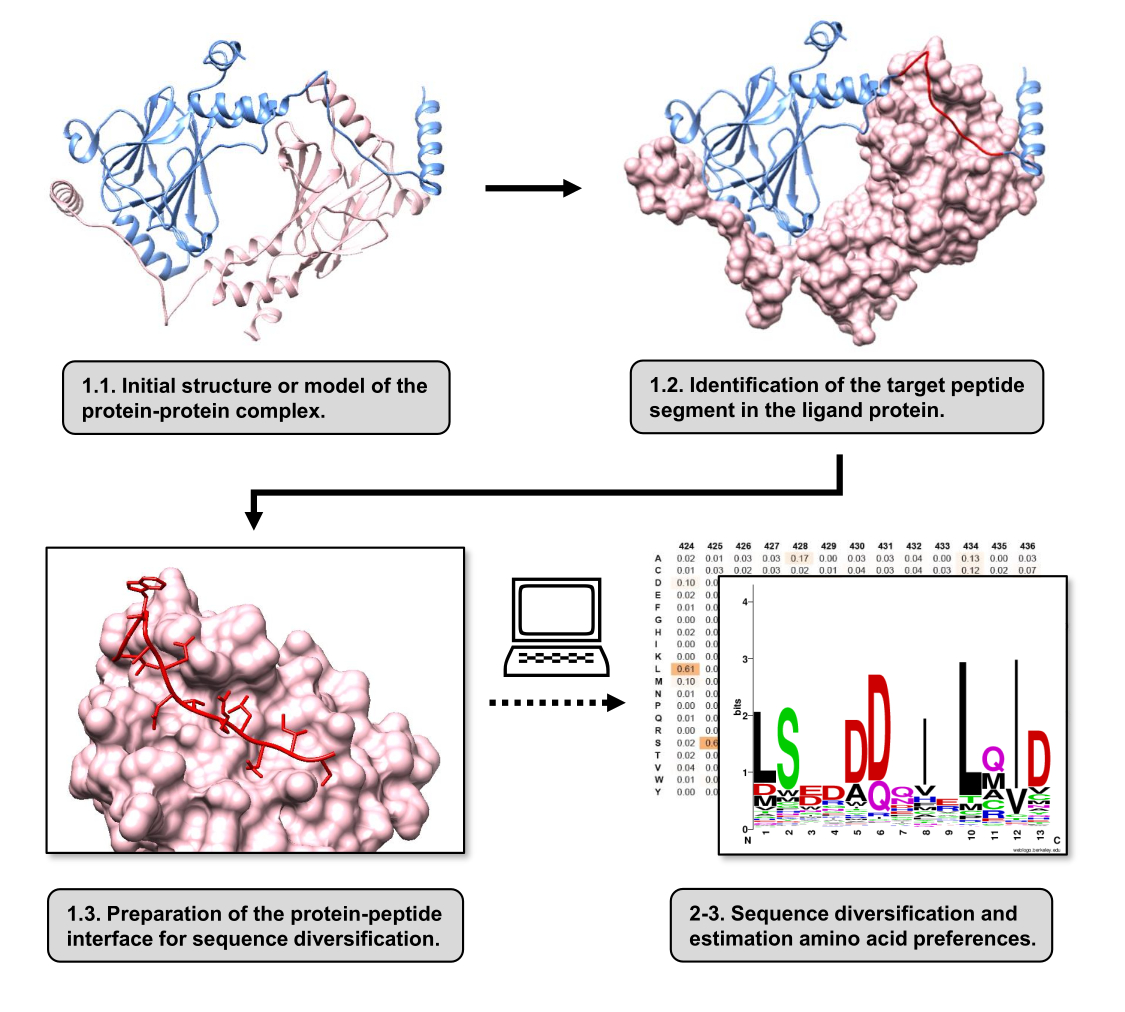
במאמר זה, אנו מתארים פרוטוקול להערכת העדפות חומצות האמינו של משטח הקישור של חלבון קולטן המעורב ב- PPI. הפרוטוקול מתמקד ב-PPIs שבהם ידוע שמקטע ליניארי של ליגנד החלבונים נקשר לחלבון הקולטן, כך שניתן למדל את התרחיש כממשק חלבון-פפטידי. בתרחיש זה, מוטיבים שמורים מהליגנד מתקשרים בדרך כלל עם כיסים מוגדרים באתר קשירת הקולטן, אם כי כל מקטע הליגנד המעורב ב- PPI עשוי להכיל אזורים פחות שמורים. תרשים זרימה המסכם את השלבים העיקריים של הפרוטוקול מוצג באיור 1. הפרוטוקול מתחיל במבנה התלת-ממדי של קומפלקס החלבון-חלבון ומפחית עוד יותר את חלבון הליגנד למקטע הפוטנציאלי בעל האינטראקציה הטובה ביותר, ומשאיר את חלבון הקולטן שלם. המקטע בעל האינטראקציה הטובה ביותר מוסק על ידי שימוש בשרת BUDE Alanine Scan15, המבצע מוטגנזה חישובית של סריקת אלנין כדי לזהות שאריות של נקודות חמות בין שני החלבונים המקיימים אינטראקציה. בגישה זו, שאריות מהליגנד מוחלפות בנפרד באלנין, והשינוי המשוער באנרגיה החופשית או ביציבות של הקומפלקס (ΔΔG) משמש לאחר מכן כדי להסיק את הרלוונטיות של השאריות המתאימות עבור PPI המטרה. לאחר הסקת המקטע בעל האינטראקציה הטובה ביותר, הקומפלקס שלו עם חלבון הקולטן משמש כמבנה הבסיס שהוגש לפפספקט לביצוע גיוון רצפים.

איור 1: סקירה כללית של השלבים העיקריים של הפרוטוקול המוצע בעבודה זו. המספרים תואמים למספרי השלבים במקטע הפרוטוקול. האיורים נעשו עם קומפלקס חלבון-חלבון המשמש כדוגמה המתוארת בטקסט. בקומפלקס זה, שרשרת החלבונים הנחשבת לקולטן מוצגת בוורוד, בעוד שהשרשרת הנחשבת לליגנד מוצגת בכחול בהיר כאשר המקטע החזוי בעל האינטראקציה הטובה ביותר מודגש באדום. אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
אחת המגבלות של הפרוטוקול המוצע היא הדרישה למבנה פתור של ממשק חלבון-פפטיד . לחלופין, הפרוטוקול עשוי להתחיל במודל של ממשק חלבון-פפטיד מטרה, אם כי שלבי המידול הספציפיים אינם מתוארים כאן. יתר על כן, למרות שניתן לנהל את הפרוטוקול במחשב אישי המריץ כל מערכת הפעלה, נדרשת סביבת לינוקס לשלבים המערבים את יישומי רוזטה. אשכול מחשבים מומלץ מאוד גם לשלב גיוון הרצף בשל המספר הגדול של חזרות המבוצעות בדרך כלל על ידי Pepspec.
יישום הפרוטוקול המוצע מומחש באמצעות הערכת העדפות חומצות אמינו של משטח המכרז של IRF5, חבר במשפחת הגורמים הרגולטוריים אינטרפרון אנושיים (IRF). בחרנו בחלבון זה כדוגמה, משום שבמהלך הפעלתו, שתי תת-יחידות נקשרות ליצירת דימר שמבנהו מאופיין היטב16. בדימרים IRF, ניתן למדל קשירה כממשק חלבון-פפטידי שבו תת-יחידה אחת מספקת את משטח הקישור והשנייה מתקשרת דרך אזור המכיל מוטיב שימור קצר הנקרא pLxIS17,18. בנוסף, כריכה ליחידות משנה IRF היא רב-ספציפית; לכן, הם יכולים ליצור הומודימרים, הטרודימרים וקומפלקסים עם חלבונים תאיים אחרים המכונים קו-אקטיבטורים18.
Protocol
1. הכנה ראשונית של ממשק חלבון-פפטיד
- הורדת המבנה של קומפלקס חלבון-חלבון
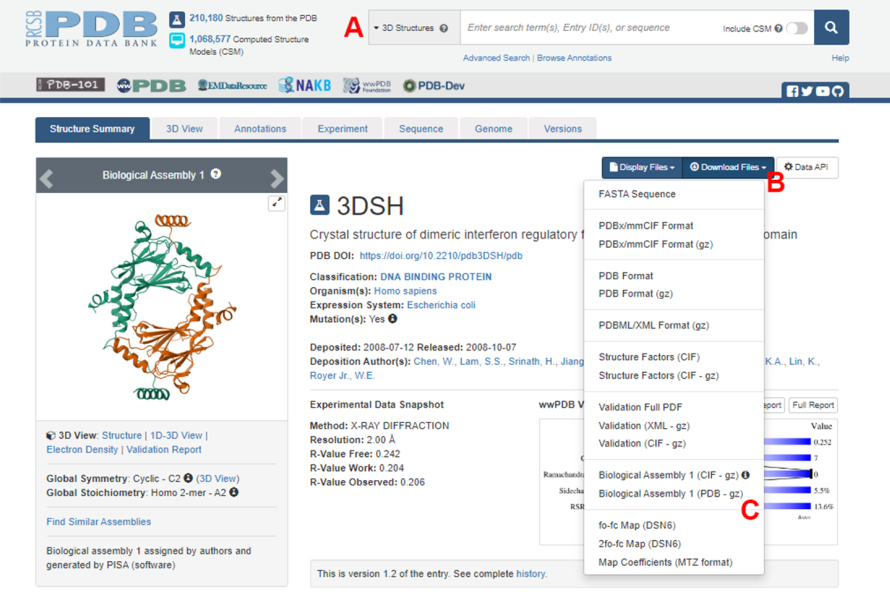
- נווטו אל דף הבית של בנק נתוני החלבונים (PDB) (https://www.rcsb.org/) והקלידו את מזהה PDB עבור המבנה של קומפלקס החלבון-חלבון בתיבת החיפוש הראשית (איור 2A). מזהה PDB עבור המבנה של דימר IRF5, המשמש כדוגמה בעבודה זו, הוא 3DSH19.
- בעמוד הראשי של המבנה הרצוי, לחצו על Download Files (איור 2B) ולאחר מכן על Biological Assembly 1 (PDB – gz) (איור 2C).
הערה: במסד הנתונים PDB, מבנים של קומפלקסים חלבוניים רבים שנוצרו על ידי מונומרים זהים מיוצגים כמכלולים ביולוגיים, שבהם רק המבנה של מונומר אחד (יחידה אסימטרית) מאוחסן בקובץ PDB. יש להוריד את מבנה הרבב, במקרה זה, הדימר IRF5, כמכלול ביולוגי המכיל שני מופעים של היחידה הא-סימטרית. כדי להקל על השלבים הבאים של פרוטוקול זה, שני המונומרים מופרדים תחילה, ומזהי שרשרת שונים מוקצים להם. - פתח את המבנה שהורדת ב- UCSF Chimera20 ולחץ על כלים > עריכת מבנה > שינוי מזהי שרשרת. בדוגמה זו, שתי השרשראות בהרכבה הביולוגית נקראות A. שנה את שם השרשרת השנייה (המסומנת #0.2) ל - B ולחץ על אישור.
- לחצו על Favorites > Model Panel ובחרו בדגם הכולל את שתי השרשראות. לחץ על כפתור Group/Ungroup כדי להפריד כל שרשרת למודל אחר. לאחר מכן, בחר את שני הדגמים ולחץ על העתק / שלב לחצן. הזינו שם חדש לדגם המשולב, בדקו את האפשרות 'סגור דגמי מקור' ולחצו על הלחצן 'אשר'.
- לחץ על בחר שרשרת > ואשר שכל שרשרת בדימר מזוהה כעת על ידי אות אחרת, כלומר A ו - B.
- השתמש ב - File > Save PDB כדי לשמור את המבנה הערוך בקובץ PDB אחר, שישמש בשלבים הבאים של הפרוטוקול (כאן, נעשה שימוש בשם IRF5_dimer.pdb ).

איור 2: דף בנק נתוני החלבונים (PDB) עבור המבנה המשמש כדוגמה מייצגת בעבודה זו. (A) תיבת חיפוש להצגת קוד ההצטרפות PDB של מבנה היעד. (B) תפריט להורדת המבנה במספר פורמטים. (C) אפשרויות להורדת מכלולים ביולוגיים כאשר המבנה נשמר כיחידה אסימטרית (ראה שלב 1.1.2 לפרטים נוספים). אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
- זיהוי מקטע המטרה בחלבון הליגנד
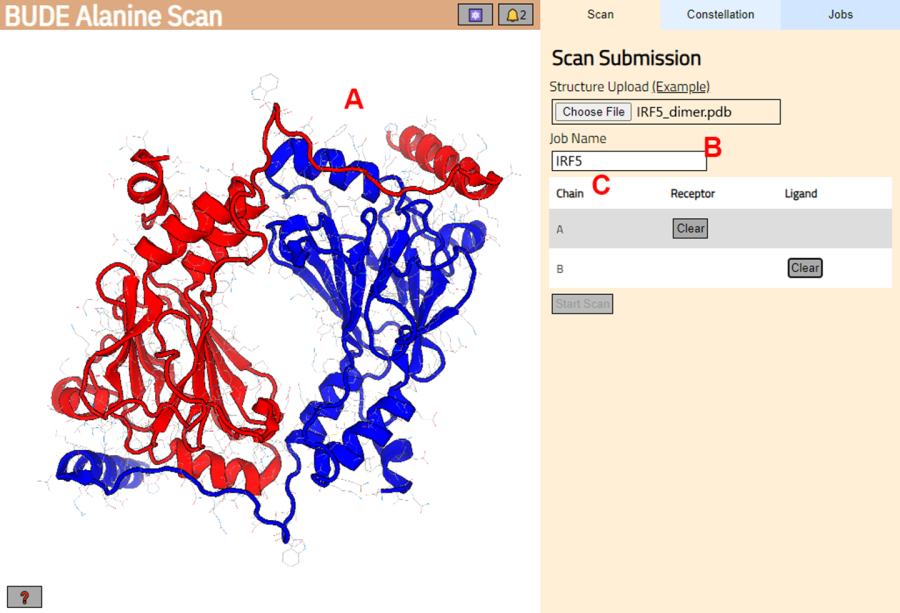
- נווט אל שרת סריקת אלנין BUDE (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). לחץ על בחר קובץ כפתור תחת מבנה העלאה ובחר את קובץ PDB שנשמר בשלב 1.1.6.
- בעמוד הבא, בדקו שהמבנה נטען כהלכה (איור 3A) והזינו שם למשימה בשרת (איור 3B).
- קבעו את השרשראות מה-PDB שיטופלו כקולטן (A) וליגנד (B) (איור 3C). לאחר מכן, לחץ על התחל סריקה כפתור כדי לשלוח את העבודה.
- לאחר סיום העבודה, לחץ על הצג תוצאות כדי לפתוח את דף התוצאות (איור 4).
הערה: בדף התוצאות, שאריות ממבנה הליגנד צבועות בהתאם לשינוי המשוער שלהן באנרגיה חופשית (ΔΔG), ואלה עם ערכים גבוהים יותר צבועות באדום. - מרשימת השאריות, בחר את רצף השאריות הצפויות לקיים אינטראקציה טובה יותר עם משטח הקישור של המטרה. ודא ששאריות אלה מקבצות את הערכים הגבוהים יותר עבור ההבדל באנרגיה חופשית (ΔΔG). בדוגמה זו נבחר המקטע בין השאריות Leu424 ו-Ser436 (מסומן בתיבה אדומה בלוח הימני של איור 4).
- הכנת ממשק חלבון-פפטיד לגיוון רצפים
- פתח את קובץ PDB שנשמר בשלב 1.1.6 בכימרה ובדוק שאין אטומים או קשרים חסרים במבנה של תת-יחידות המטרה.
- מחק את כל המולקולות, היונים והממסים הקטנים שהתגבשו יחד עם המבנה המקורי. לשם כך, לחץ על בחר > שאריות ולאחר מכן בחר את כל המולקולות מלבד חומצות אמינו סטנדרטיות. לאחר מכן, לחץ על פעולות > אטומים/אג"ח ומחק.
- חתוך את שרשרת הליגנד למקטע בעל האינטראקציה הטובה ביותר שנבחר בשלב 1.2.5. לשם כך, לחץ על מועדפים ורצף ולאחר מכן לחץ על השרשרת הנחשבת לליגנד (B). בחלונית 'רצף', גרור את העכבר לבחירת כל השאריות, פרט לשאריות שבין מיקומים 424 ו- 436. כדי למחוק שאריות אלה, לחץ על פעולות > אטומים/אג"ח ומחק.
- השתמש ב - File > Save PDB כדי לשמור את המבנה הערוך בקובץ PDB אחר, המשמש בשלבים הבאים של הפרוטוקול (כאן, נעשה שימוש בשם IRF5_interface.pdb ).

איור 3: בחירת קולטן וליגנד בשרת סריקת אלנין BUDE. (A) ייצוג גרפי של קומפלקס חלבון-חלבון. (B) תיבת טקסט להזנת שם המשימה בשרת. (C) פאנל לבחירה אינטראקטיבית של השרשראות שייחשבו כקולטן וליגנד (ראה שלב 1.2 לפרטים נוספים). אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}

איור 4: דף התוצאות של שרת סריקת BUDE Alanine. המקטע הפוטנציאלי בעל האינטראקציה הטובה ביותר ברצף הליגנד מסומן בתיבה אדומה. בפאנל השמאלי, השאריות עם תרומת האנרגיה החזויה הגבוהה יותר (Leu433) מודגשות בירוק. אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
2. גיוון רצף
הערה: בשלבים הבאים, rosetta_main מתייחס לספריית ההתקנה הראשית של Rosetta, הממוקמת בדרך כלל ב - /opt/rosetta_src__bundle/main/, כאשר מציין את גירסת Rosetta המותקנת. כמו כן, ההנחה היא כי יישומי רוזטה נגישים בכל המערכת; אם זה לא המקרה, יש לספק את הנתיב המלא לקבצי ההפעלה. כאשר הם מהודרים מהמקור, קבצי הפעלה אלה ממוקמים בספריה /rosetta_main/source/bin/ .
- אופטימיזציה ראשונית של שרשראות צד חומצות אמינו
- העתק את המבנה הערוך שנשמר בשלב 1.3.4 למיקום Linux הנגיש ליישומי Rosetta.
- השתמש ביישום FixBB של Rosetta כדי לבצע אריזה מחדש של כל שרשראות הצד של חומצות האמינו של מבנה הבסיס לפני גיוון הרצף. בפעולה זו, הכיוון של כל שרשראות הצד של חומצות אמינו מותאם כדי למזער את האנרגיה ולשפר את יציבות המתחם. לשם כך, הפעל את הפקודה הבאה:

הערה: פקודה זו מפיקה קובץ PDB שנקרא על שם המבנה המקורי עם סיומת מספרית נוספת (IRF5_interface_0001.pdb בדוגמה זו). - כדי להקל על השלב הבא של הפרוטוקול, שנה את שם קובץ PDB שנארז מחדש בסיומת _repack באמצעות הפקודה הבאה:
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- גיוון רצף
- הפעל את Pepspec במצב עיצוב כדי לבצע את שלב גיוון הרצף בפועל באמצעות הפקודה הבאה:

להלן אפשרויות כלליות:- -s מציין את קובץ הקלט (קובץ PDB שנארז מחדש שנוצר בשלב 2.1.3).
- -o מציין את הקידומת למתן שם לקובצי פלט.
- - מסד נתונים מציין את הנתיב למסד הנתונים הראשי של רוזטה 3.
- -ex1, -ex2 ו- extrachi_cutoff הן אפשרויות ספריית rotamer (ראה תיעוד Pepspec לפרטים נוספים).
- -overwrite מורה ליישום להחליף פלטים אפשריים קיימים מראש שנוצרו על-ידי חזרות קודמות.
- - pepspec:pep_chain מציין את שרשראות PDB הנחשבות לליגנד ('b' בדוגמה זו).
- -pepspec:native_pep_anchor מציין את שאריות חומצת האמינו המשמשות כעוגן (בדוגמה זו, שאריות Leu במיקום 10 של פפטיד הליגנד).
- -pepspec:n_peptides מציין את מספר המבנים הפפטידים שיש להפיק.
- -pepspec:no_prepack_prot מורה ליישום לדלג על אריזה מחדש במבנה בסיס הקלט (מכיוון שפעולה זו בוצעה בעבר בשלב 2.1).
הערה: פלט Pepspec הראשי הוא ספרייה המכילה את קובצי PDB עבור פפטידים הנובעים משלב העיצוב, הנקראת באמצעות קידומת הפלט עם הסיומת .pdbs (IRF5.pdbs בדוגמה). בנוסף, Pepspec מפיק את כל רצפי הפפטידים המקובלים שנבדקו כחלק משלב גיוון הרצף ואת ציוני אנרגיית רוזטה המתאימים להם בקובץ טקסט מופרד בטאבים הקרוי על שם קידומת הפלט, עם . סיומת מפרט (IRF5.spec בדוגמה). מכיוון שהפרוטוקול המתואר בעבודה זו נועד להעריך את העדפות חומצות האמינו ולא את תכנון הפפטיד בפועל, השלבים הבאים משתמשים ב- IRF5.spec במקום במבני PDB בספריית .pdbs .
- הפעל את Pepspec במצב עיצוב כדי לבצע את שלב גיוון הרצף בפועל באמצעות הפקודה הבאה:
3. הערכת העדפות חומצות אמינו
- חישוב PWM
- כדי ליצור PWM, השתמש בסקריפט gen_pepspec_pwm.py הכלול בחבילת Rosetta. כדי להפעיל קובץ script זה, השתמש בפקודה הבאה:

איפה:- IRF5.spec הוא קובץ הפלט של Pepspec שנוצר בשלב 2.2.
- -1 מציין שאין שאריות N-terminal נוספות ברצף, ולכן המיקומים ב-PWM הם מבוססי 1.
- 0.2 מורה לסקריפט לשקול רק את 20% הפפטידים בעלי הניקוד הטוב ביותר מפלט Pepspec (ערך ברירת המחדל הוא 0.1, המתאים ל- 10%)
- interface_score מורה לסקריפט לדרג פפטידים בהתבסס על ציון הממשק, שהוא אחד מציוני רוזטה השונים הכלולים בקובץ הפלט של Pepspec.
הערה: סקריפט זה יוצר שני קובצי פלט, אחד עבור PWM המחושב (עם הסיומת .pwm ) והשני עבור הרצפים של תת-קבוצת הפפטידים המשמשת לחישוב PWM (עם הסיומת .seq ). שמות קבצים אלה כוללים גם את הניקוד ואת חלק הפפטידים המשמשים לדירוג. בדוגמה זו, קבצים אלה נקראים בהתאמה IRF5_interface_score_0.2.pwm ו- IRF5_interface_score_0.2.seq.
- כדי ליצור PWM, השתמש בסקריפט gen_pepspec_pwm.py הכלול בחבילת Rosetta. כדי להפעיל קובץ script זה, השתמש בפקודה הבאה:
- יצירת לוגו רצף
- נווט אל שרת WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 ולחץ על הלחצן בחר קובץ לצד העלה נתוני רצף. העלה את הקובץ עם רצפי פפטידים שנוצרו בשלב 3.1.1 (IRF5_interface_score_0.2.seq בדוגמה זו).
- בחר את הפורמט והגודל הרצויים של הלוגו בהתאם לאורך הקלט. הדוגמה משתמשת בתבנית PDF בגודל 15 ס"מ x 12 ס"מ. לחץ על צור לוגו.
תוצאות
במאמר זה תיארנו פרוטוקול לחיזוי העדפות חומצות האמינו של משטח הקישור של IRF5, חבר במשפחה של גורמי שעתוק הידועים כגורמי ויסות אינטרפרון אנושיים. חלבונים אלה מווסתים תגובות חיסוניות מולדות ונרכשות ומשתתפים בהתמיינות ובהפעלה של מספר תאי חיסון. תת-יחידות IRF הן בעלות משטחי קיש?...
Discussion
המאמר הנוכחי מתאר פרוטוקול להערכת העדפות חומצות האמינו של אתרי קישור רב-ספציפיים פוטנציאליים בהתבסס על גיוון רצף סיליקו. כלים חישוביים מעטים פותחו כדי להעריך את העדפות חומצות האמינו של ממשקי חלבון-פפטיד 14,25,26. לכלים אל?...
Disclosures
למחברים אין מה לחשוף.
Acknowledgements
תמיכה כספית על ידי Sistema Nacional de Investigación (SNI) (מספרי מענקים SNI-043-2023 ו- SNI-170-2021), Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) של פנמה ו- Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) מוכרים בהכרת תודה. המחברים רוצים להודות לד"ר מיגל רודריגז על שבחן בקפידה את כתב היד.
Materials
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
References
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved