
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Computational Prediction of Amino Acid Preferences of Potentially Multispecific Peptide-Binding Domains Involved in Protein-Protein Interactions
In This Article
Summary
We describe a methodology based on sequence diversification to estimate the amino acid preferences of multispecific binding sites in protein-protein interactions (PPIs). In this strategy, thousands of potential peptide ligands are generated and screened in silico, thus overcoming some limitations of available experimental methods.
Abstract
Many protein-protein interactions involve the binding of short protein segments to peptide-binding domains. Usually, such interactions require the recognition of linear motifs with variable conservation. The combination of highly conserved and more variable regions in the same ligands often contributes to the multispecificity of binding, a common property of enzymes and cell signaling proteins. Characterization of amino acid preferences of peptide-binding domains is important for the design of mediators of protein-protein interactions (PPIs). Computational methods are an efficient alternative to the often costly and cumbersome experimental techniques, enabling the design of potential mediators that can be later validated in downstream experiments. Here, we described a methodology using the Pepspec application of the Rosetta molecular modeling package to predict the amino acid preferences of peptide-binding domains. This methodology is useful when the structure of the receptor protein and the nature of the peptide ligand are both known or can be inferred. The methodology starts with a well-characterized anchor from the ligand, which is extended by randomly adding amino acid residues. The binding affinity of peptides generated this way is then evaluated by flexible-backbone peptide docking in order to select the peptides with the best predicted binding scores. These peptides are then used to calculate amino acid preferences and to optionally compute a position-weight matrix (PWM) that can be used in further studies. To illustrate the application of this methodology, we used the interaction between subunits of human interferon regulatory factor 5 (IRF5), previously known to be multispecific but globally guided by a short conserved motif called pLxIS. The estimated amino acid preferences were consistent with previous knowledge about the IRF5 binding surface. Positions occupied by phosphorylatable serine residues exhibited a high frequency of aspartate and glutamate, likely because their negatively charged side chains are similar to phosphoserine.
Introduction
Interaction between two proteins often involves the binding of short segments of amino acids to peptide-binding domains, resembling protein-peptide interfaces. Receptor proteins involved in such protein-protein interactions (PPI) often have the ability to recognize a certain set of overlapping but divergent ligand sequences, a property known as multispecificity1,2. Multispecific recognition is a feature of many cellular proteins, but it is particularly remarkable in enzymes and cell signaling proteins3. Proteins interacting with multispecific binding sites often have a combination of more and less conserved regions in their sequence4,5,6. In this scenario, the more conserved sequence motifs are involved in stringent molecular interactions. Conversely, the more variable sequences interact with somehow permissive surfaces in the receptor binding site. Usually, these less conserved but still functionally relevant segments are loops lacking defined secondary structure patterns or have even more dynamic conformations, such as those typical of intrinsically disordered proteins7.
Identification of potential peptide ligands of binding sites is usually the first step in the design of mediators able to interfere with the corresponding PPIs8. However, it is often unlikely to find a single most frequent amino acid residue at most sequence positions in ligands of multispecific binding sites. Instead, these sites may have particular preferences for a specific class of amino acids according to their chemical properties, e.g., acidic and negatively charged amino acids such as aspartate or glutamate, bulky aromatic amino acids such as phenylalanine or more hydrophobic residues such as aliphatic amino acids alanine, valine, leucine or isoleucine3. Several experimental methods can provide insights about amino acid preferences of protein binding sites, including directed evolution9, multi-codon scanning mutagenesis10, and deep mutational scanning11. All of these methods follow the approach of sequence diversification, which is based on introducing mutations to original ligands and further analyzing their effect on the function of the receptor protein (see Bratulic and Badran12 for a comprehensive review). However, these methods often require the survey of large sequence libraries, which makes them more cumbersome, costly, and time-consuming.
Computational methods to infer the amino acid preferences of multispecific binding sites have the potential to circumvent the limitations of wet lab methods. Among these, the in silico sequence diversification approach evaluates the energetic impact of a wide range of amino acid replacements in the ligand sequence as a way to characterize the structural plasticity of the PPI13. This method begins with the structure or model of the peptide ligand bound to the receptor binding site and subsequently introduces mutations to the ligand sequence. Statistical and energy-scoring functions are then used to evaluate the impact of these mutations on stability and binding affinity. The set of best-scoring ligand sequences resulting from the evaluation phase can then be used to compute the amino acid preferences. This strategy has the potential to process a very high number of ligand sequences in an efficient manner. Therefore, it can provide a more complete and consistent inference of amino acid preferences compared to those computed from the more limited number of sequences that can usually be processed in wet lab approaches.
The Pepspec application of the Rosetta molecular modeling suite14 is a tool that performs sequence diversification as a key step of its peptide design mode. This application requires a structure or model of the receptor protein with a bound peptide down to a single amino acid residue in length, which is used as an anchor for the next steps. The sequence of the bound peptide is then extended (if necessary) and diversified to generate a large number of putative peptide ligands. The binding affinity of these peptides is then evaluated by flexible-backbone peptide docking in order to select those with the best predicted binding scores. Although the main output of this application is the best peptide candidates selected at the end of the design phase, the much larger set of peptides accepted during this phase can also be used to compute the amino acid preferences of the target binding site. Amino acid preferences are computed as the frequency of each amino acid residue per position of the ligand sequence represented either as a position weight matrix (PWM) or as a more visual sequence logo.
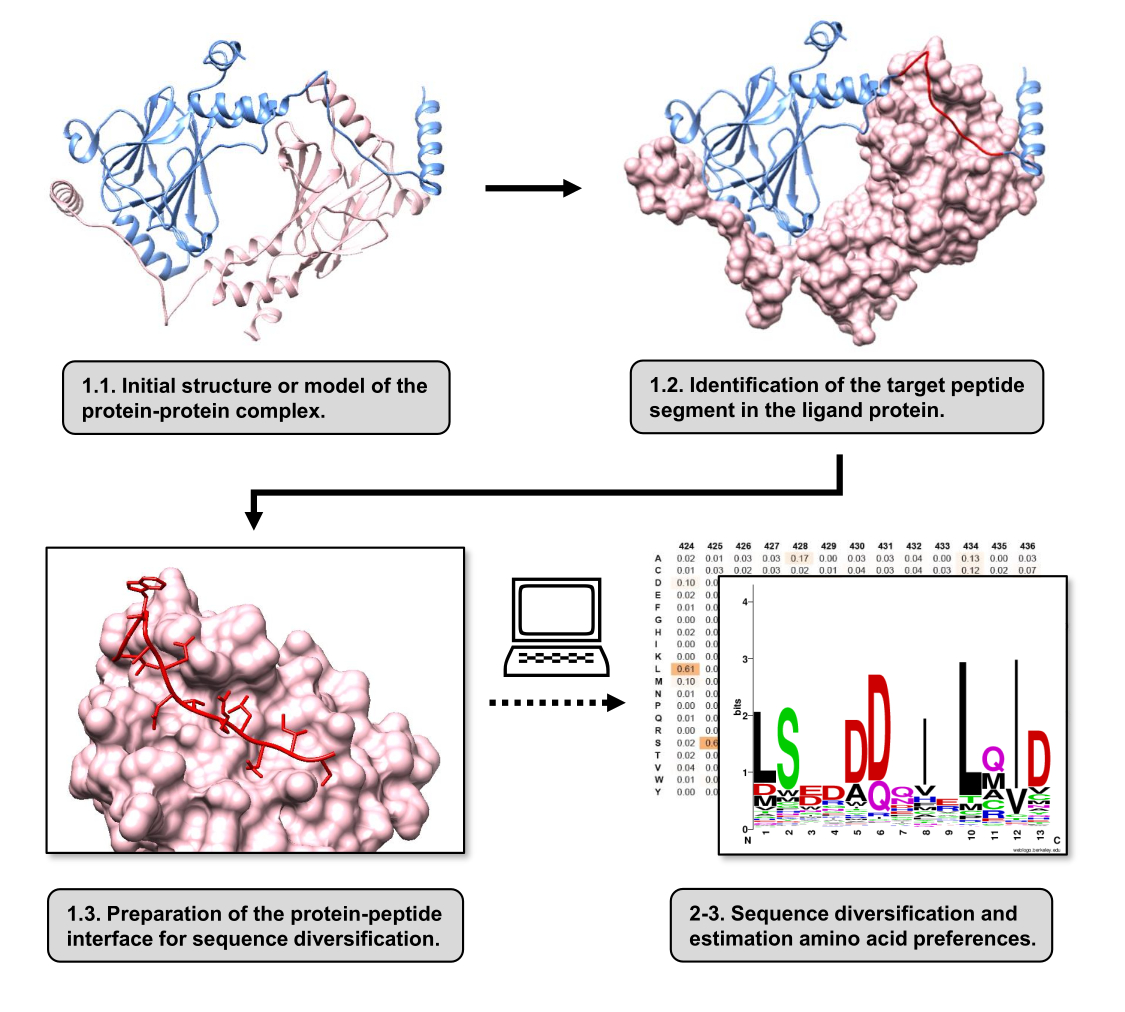
In this article, we describe a protocol to estimate the amino acid preferences of the binding surface of a receptor protein involved in a PPI. The protocol is focused on PPIs in which a linear segment of the protein-ligand is known to bind to the receptor protein, so the scenario can be modeled as a protein-peptide interface. In this scenario, conserved motifs from the ligand typically interact with defined pockets in the receptor binding site, although the entire ligand segment involved in the PPI may contain less conserved regions. A flowchart summarizing the major steps of the protocol is shown in Figure 1. The protocol starts with the 3D structure of the protein-protein complex and further reduces the ligand protein to the potential best-interacting segment, leaving the receptor protein intact. The best-interacting segment is inferred by using the BUDE Alanine Scan server15, which conducts computational alanine scanning mutagenesis to identify hot-spot residues between the two interacting proteins. In this approach, residues from the ligand are individually replaced by alanine, and the estimated change in free energy or stability of the complex (ΔΔG) is then used to infer the relevance of the corresponding residue for the target PPI. Once the best-interacting segment is inferred, its complex with the receptor protein is used as the base structure submitted to Pepspec to perform sequence diversification.

Figure 1: Overview of the main steps of the protocol proposed in this work. Numbers match step numbers in the protocol section. Figures were made with the protein-protein complex used as the example described in the text. In this complex, the protein chain considered as the receptor is shown in pink, while the chain considered as the ligand is shown in light blue with its predicted best-interacting segment highlighted in red. Please click here to view a larger version of this figure.
{kind=link}
One of the limitations of the suggested protocol is the requirement for a resolved structure of the protein-peptide interface. The protocol may alternatively begin with a model of the target protein-peptide interface, although the specific modeling steps are not described herein. Moreover, although the protocol can be conducted on a personal computer running any operating system, a Linux environment is required for the steps involving the Rosetta applications. A computer cluster is also highly recommended for the sequence diversification step due to the large number of iterations typically performed by Pepspec.
Application of the suggested protocol is illustrated with the estimation of amino acid preferences of the biding surface of IRF5, a member of the human interferon regulatory factor (IRF) family. We chose this protein as an example because, during its activation, two subunits bind to form a dimer whose structure is well characterized16. In IRF dimers, binding can be modeled as a protein-peptide interface in which one subunit provides the binding surface and the other one interacts through a region containing a short conserved motif called pLxIS17,18. In addition, binding to IRF subunits is multispecific; therefore, they can form homodimers, heterodimers, and complexes with other cellular proteins known as coactivators18.
Access restricted. Please log in or start a trial to view this content.
Protocol
1. Initial preparation of the protein-peptide interface
- Downloading the structure of the protein-protein complex
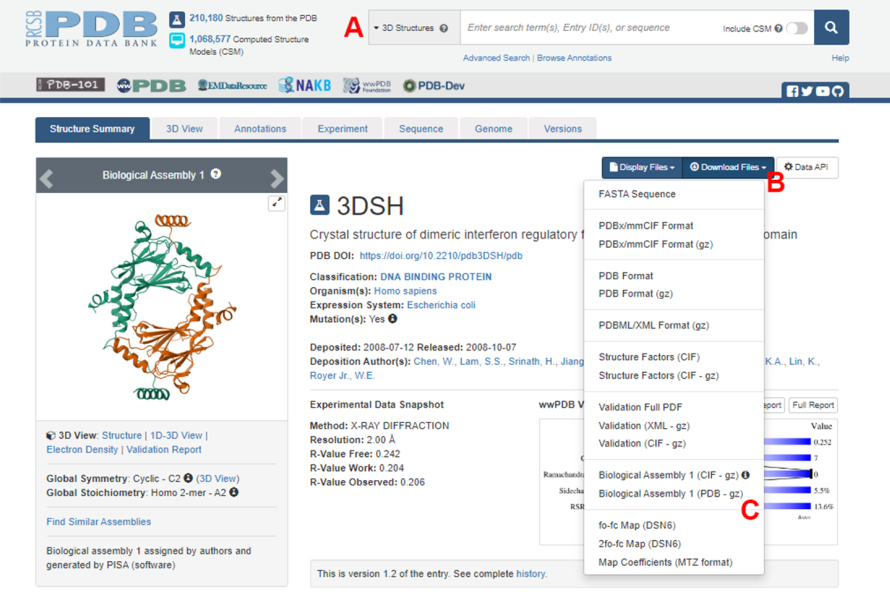
- Navigate to the Protein Data Bank (PDB) homepage (https://www.rcsb.org/) and type the PDB ID for the structure of the protein-protein complex in the main search box (Figure 2A). The PDB ID for the structure of the IRF5 dimer, used as an example in this work, is 3DSH19.
- In the main page for the desired structure, click on Download Files (Figure 2B) and then on Biological Assembly 1 (PDB - gz) (Figure 2C).
NOTE: In the PDB database, structures of many protein complexes formed by identical monomers are represented as biological assemblies, in which only the structure of one monomer (asymmetric unit) is stored in the PDB file. The structure of the multimer, in this case, the IRF5 dimer, must be downloaded as the biological assembly containing two instances of the asymmetric unit. To facilitate the next steps of this protocol, the two monomers are first separated, and different chain IDs are assigned to them. - Open the downloaded structure in UCSF Chimera20 and click on Tools > Structure Editing > Change Chain IDs. In this example, both chains in the biological assembly are named A. Rename the second chain (labeled #0.2) to B and click OK.
- Click on Favorites > Model Panel and then select the model containing the two chains. Click on the Group/Ungroup button to separate each chain into a different model. Then, select the two models and click on the Copy/Combine button. Enter a new name for the combined model, check Close Source Models, and click OK.
- Click on Select > Chain and confirm that each chain in the dimer is now identified by a different letter, namely, A and B.
- Use File > Save PDB to save the edited structure to a different PDB file, which will be used in the next steps of the protocol (here, the name IRF5_dimer.pdb was used).

Figure 2: The Protein Data Bank (PDB) page for the structure used as a representative example in this work. (A) Search box to introduce the PDB accession code of the target structure. (B) Menu to download the structure in several formats. (C) Options to download biological assemblies when the structure has been saved as an asymmetric unit (see step 1.1.2 for more details). Please click here to view a larger version of this figure.
{kind=link}
- Identifying the target segment in the ligand protein
- Navigate to the BUDE Alanine Scan server (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Click on the Choose File button under Structure Upload and select the PDB file saved in step 1.1.6.
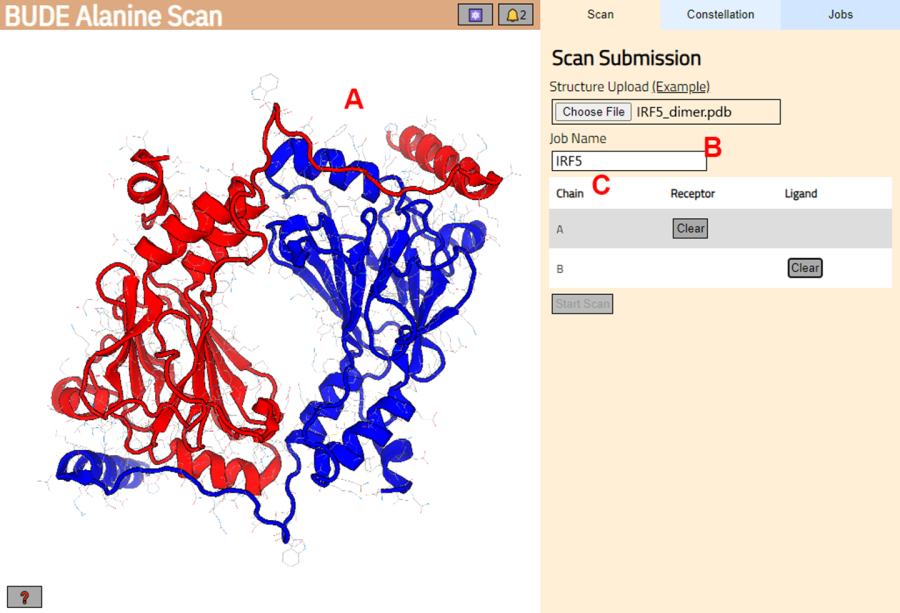
- On the next page, check that the structure was correctly loaded (Figure 3A) and enter a name for the job in the server (Figure 3B).
- Set the chains from the PDB that will be treated as receptor (A) and ligand (B) (Figure 3C). Then, click on the Start Scan button to submit the job.
- Once the job is finished, click on Show Results to open the results page (Figure 4).
NOTE: In the results page, residues from the ligand structure are colored according to their estimated change in free energy (ΔΔG), and those with higher values are colored in red. - From the residue list, select the stretch of residues predicted to better interact with the target binding surface. Ensure these residues cluster the higher values for the difference in free energy (ΔΔG). In this example, the segment between residues Leu424 and Ser436 was selected (highlighted with a red box in the right panel of Figure 4).
- Preparing the protein-peptide interface for sequence diversification
- Open the PDB file saved in step 1.1.6 in Chimera and check that there are no missing atoms or bonds in the structure of target subunits.
- Delete all small molecules, ions, and solvents that were co-crystalized with the original structure. To do this, click on Select > Residues and then select all molecules other than standard amino acids. Then, click on Actions > Atoms/Bonds and Delete.
- Crop the ligand chain to the best-interacting segment chosen in step 1.2.5. To do this, click on Favorites and Sequence and then click on the chain considered as the ligand (B). In the Sequence panel, drag the mouse to select all residues except those between positions 424 and 436. To delete these residues, click on Actions > Atoms/Bonds and Delete.
- Use File > Save PDB to save the edited structure to a different PDB file, which is used in the next steps of the protocol (here, the name IRF5_interface.pdb was used).

Figure 3: Selection of receptor and ligand in the BUDE Alanine Scan server. (A) Graphic representation of the protein-protein complex. (B) Text box to enter the name of the job in the server. (C) Panel to interactively select the chains that will be considered as receptor and ligand (see step 1.2 for more details). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Results page of the BUDE Alanine Scan server. The potential best-interacting segment in the ligand sequence is indicated with a red box. In the left panel, the residue with the higher predicted energy contribution (Leu433) is highlighted in green. Please click here to view a larger version of this figure.
{kind=link}
2. Sequence diversification
NOTE: In the following steps, rosetta_main refers to the main Rosetta installation directory, which is typically located at /opt/rosetta_src_<version>_bundle/main/, where <version> indicates the installed Rosetta version. Also, it is assumed that Rosetta applications are accessible system-wide; if this is not the case, the full path to the executables has to be provided. When compiled from the source, these executables are located in the /rosetta_main/source/bin/ directory.
- Initial optimization of amino acid side chains
- Copy the edited structure saved in step 1.3.4 to a Linux location accessible by the Rosetta applications.
- Use Rosetta's FixBB application to perform a repack of all the amino acid side chains of the base structure before sequence diversification. In this operation, the orientation of all amino acid side chains is optimized to minimize energy and improve the stability of the complex. To do this, run the following command:

NOTE: This command outputs a PDB file named after the original structure with an additional numerical suffix (IRF5_interface_0001.pdb in this example). - To facilitate the next step of the protocol, rename the repacked PDB file with the _repack suffix using the following command:
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- Sequence diversification
- Run Pepspec in design mode to perform the actual sequence diversification step using the following command:

The following are general options:- -s indicates the input file (the repacked PDB file generated in step 2.1.3).
- -o indicates the prefix to name output files.
- - database indicates the path to the main Rosetta 3 database.
- -ex1, -ex2, and extrachi_cutoff are rotamer library options (see Pepspec documentation for more details).
- -overwrite tells the application to overwrite possible pre-existing outputs generated by previous iterations.
- -pepspec:pep_chain indicates the PDB chains considered as ligand ('b' in this example).
- -pepspec:native_pep_anchor indicates the amino acid residue used as an anchor (in this example, the Leu residue at position 10 of the ligand peptide).
- -pepspec:n_peptides indicates the number of peptide structures to output.
- -pepspec:no_prepack_prot tells the application to skip repacking in the input base structure (since this was previously performed in step 2.1).
NOTE: The main Pepspec output is a directory containing the PDB files for peptides resulting from the design phase, named using the output prefix with the .pdbs suffix (IRF5.pdbs in the example). Additionally, Pepspec outputs all the accepted peptide sequences tested as part of the sequence diversification step and their corresponding Rosetta energy scores in a tab-delimited text file named after the output prefix, with the .spec suffix (IRF5.spec in the example). Since the protocol described in this work aims to estimate amino acid preferences rather than the actual peptide design, the next steps use IRF5.spec instead of the PDB structures in the .pdbs directory.
- Run Pepspec in design mode to perform the actual sequence diversification step using the following command:
3. Estimation of amino acid preferences
- Computing a PWM
- To generate a PWM, use the gen_pepspec_pwm.py script included in the Rosetta suite. To run this script, use the following command:

where:- IRF5.spec is the Pepspec output file generated in step 2.2.
- -1 indicates that there are not additional N-terminal residues in the sequence and, therefore, positions in the PWM are 1-based.
- 0.2 tells the script to only consider the top 20% best-scoring peptides from the Pepspec output (the default value is 0.1, corresponding to the 10%)
- interface_score tells the script to rank peptides based on the interface score, which is one of the various Rosetta scores included in the Pepspec output file.
NOTE: This script generates two output files, one for the computed PWM (with the .pwm suffix) and the other for the sequences of the subset of peptides used to compute the PWM (with the .seq suffix). The names of these files also include the score and the fraction of peptides used for ranking. In this example, these files are respectively named IRF5_interface_score_0.2.pwm and IRF5_interface_score_0.2.seq.
- To generate a PWM, use the gen_pepspec_pwm.py script included in the Rosetta suite. To run this script, use the following command:
- Generating a sequence logo
- Navigate to the WebLogo server (https://weblogo.berkeley.edu/logo.cgi)21 and click on the Choose File button next to Upload Sequence Data. Upload the file with peptide sequences generated in step 3.1.1 (IRF5_interface_score_0.2.seq in this example).
- Choose the desired format and size of the logo according to the input length. The example uses PDF format and a size of 15 cm x 12 cm. Click on Create Logo.
Access restricted. Please log in or start a trial to view this content.
Results
In this article, we described a protocol to predict the amino acid preferences of the binding surface of IRF5, a member of a family of transcription factors known as human interferon regulatory factors. These proteins are regulators of innate and adaptive immune responses and participate in the differentiation and activation of several immune cells. IRF subunits have highly plastic and multispecific binding surfaces, being capable of forming homodimers, heterodimers, and complexes with other cellular proteins
Access restricted. Please log in or start a trial to view this content.
Discussion
The present article describes a protocol to estimate the amino acid preferences of potentially multispecific binding sites based on in silico sequence diversification. Few computational tools have been developed to estimate amino acid preferences of protein-peptide interfaces14,25,26. These tools have a predictive nature, but they differ in the computational algorithms used to perform their predictions and the corrections they i...
Access restricted. Please log in or start a trial to view this content.
Disclosures
Authors have nothing to disclose.
Acknowledgements
Financial support by Sistema Nacional de Investigación (SNI) (grant numbers SNI-043-2023 and SNI-170-2021), Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) of Panama and Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) are gratefully acknowledged. Authors would like to thank Dr. Miguel Rodríguez for carefully reviewing the manuscript.
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
References
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627(2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), London, England. 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267(2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33(2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999(2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451(2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614(2017).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved