Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Вычислительное прогнозирование аминокислотных предпочтений потенциально мультиспецифических пептид-связывающих доменов, участвующих в белок-белковых взаимодействиях
В этой статье
Резюме
Мы описываем методологию, основанную на диверсификации последовательностей, для оценки аминокислотных предпочтений мультиспецифических сайтов связывания в белок-белковых взаимодействиях (ИПП). В рамках этой стратегии тысячи потенциальных пептидных лигандов генерируются и экранируются in silico, тем самым преодолевая некоторые ограничения доступных экспериментальных методов.
Аннотация
Многие белок-белковые взаимодействия включают связывание коротких белковых сегментов с пептид-связывающими доменами. Обычно такие взаимодействия требуют распознавания линейных мотивов с переменным сохранением. Комбинация высококонсервативных и более вариабельных областей в одних и тех же лигандах часто способствует мультиспецифичности связывания, что является общим свойством ферментов и клеточных сигнальных белков. Характеристика аминокислотных предпочтений пептид-связывающих доменов важна для разработки медиаторов белок-белковых взаимодействий (ИПП). Вычислительные методы являются эффективной альтернативой часто дорогостоящим и громоздким экспериментальным методам, позволяя разрабатывать потенциальные медиаторы, которые впоследствии могут быть проверены в последующих экспериментах. В данной работе мы описали методологию с использованием приложения Pepspec пакета молекулярного моделирования Rosetta для прогнозирования аминокислотных предпочтений пептид-связывающих доменов. Эта методология полезна, когда структура рецепторного белка и природа пептидного лиганда известны или могут быть выведены. Методология начинается с хорошо охарактеризованного якоря из лиганда, который расширяется за счет случайного добавления аминокислотных остатков. Аффинность связывания полученных таким образом пептидов затем оценивается с помощью докинга пептидов гибкого каркаса с целью выбора пептидов с наилучшими прогнозируемыми показателями связывания. Эти пептиды затем используются для расчета аминокислотных предпочтений и для опционального вычисления матрицы позицион-вес (ШИМ), которая может быть использована в дальнейших исследованиях. Чтобы проиллюстрировать применение этой методологии, мы использовали взаимодействие между субъединицами регуляторного фактора интерферона человека 5 (IRF5), ранее известного как мультиспецифичный, но глобально управляемого коротким консервативным мотивом под названием pLxIS. Оцененные предпочтения аминокислот согласуются с предыдущими знаниями о поверхности связывания IRF5. Позиции, занимаемые фосфорилируемыми сериновыми остатками, демонстрировали высокую частоту аспартата и глутамата, вероятно, потому, что их отрицательно заряженные боковые цепи подобны фосфосерину.
Введение
Взаимодействие между двумя белками часто включает связывание коротких сегментов аминокислот с пептид-связывающими доменами, напоминающими белок-пептидные границы. Рецепторные белки, участвующие в таких белок-белковых взаимодействиях (ИПП), часто обладают способностью распознавать определенный набор перекрывающихся, но расходящихся последовательностей лигандов, свойство, известное как мультиспецифичность 1,2. Мультиспецифическое распознавание является особенностью многих клеточных белков, но особенно заметно оно проявляется в ферментах и клеточных сигнальных белках3. Белки, взаимодействующие с мультиспецифическими сайтами связывания, часто имеют комбинацию более и менее консервативных областей в своей последовательности 4,5,6. В этом сценарии более консервативные мотивы последовательности вовлечены в строгие молекулярные взаимодействия. И наоборот, более вариабельные последовательности взаимодействуют с каким-то образом разрешающими поверхностями в месте связывания рецептора. Как правило, эти менее консервативные, но все же функционально значимые сегменты представляют собой петли, лишенные определенных структурных паттернов или имеющие еще более динамичные конформации, такие как типичные для внутренне неупорядоченныхбелков.
Идентификация потенциальных пептидных лигандов сайтов связывания обычно является первым шагом в разработке медиаторов, способных интерферировать с соответствующими ИПП8. Тем не менее, часто маловероятно найти один наиболее часто встречающийся аминокислотный остаток в большинстве позиций последовательности в лигандах мультиспецифических сайтов связывания. Вместо этого эти сайты могут иметь особые предпочтения в отношении определенного класса аминокислот в соответствии с их химическими свойствами, например, кислых и отрицательно заряженных аминокислот, таких как аспартат или глутамат, объемных ароматических аминокислот, таких как фенилаланин, или более гидрофобных остатков, таких как алифатические аминокислоты аланин, валин, лейцин или изолейцин3. Несколько экспериментальных методов могут дать представление о аминокислотных предпочтениях сайтов связывания белков, включая направленную эволюцию9, мутагенез мультикодонового сканирования10 и глубокое мутационное сканирование11. Все эти методы основаны на подходе диверсификации последовательностей, который основан на введении мутаций в исходные лиганды и дальнейшем анализе их влияния на функцию рецепторного белка (см. Bratulic and Badran12 для всестороннего обзора). Однако эти методы часто требуют изучения больших библиотек последовательностей, что делает их более громоздкими, дорогостоящими и трудоемкими.
Вычислительные методы для вывода аминокислотных предпочтений мультиспецифических сайтов связывания могут обойти ограничения методов мокрой лаборатории. Среди них подход к диверсификации последовательностей in silico оценивает энергетическое воздействие широкого спектра заменителей аминокислот в последовательности лигандов как способ характеристики структурной пластичности PPI13. Этот метод начинается со структуры или модели пептидного лиганда, связанного с рецепторным сайтом связывания, и впоследствии вводит мутации в последовательность лиганда. Затем статистические функции и функции оценки энергии используются для оценки влияния этих мутаций на стабильность и аффинность связывания. Набор последовательностей лигандов с наилучшей оценкой, полученных в результате фазы оценки, может быть затем использован для вычисления предпочтений аминокислот. Эта стратегия обладает потенциалом для эффективной обработки очень большого числа последовательностей лигандов. Таким образом, он может обеспечить более полный и последовательный вывод о предпочтениях аминокислот по сравнению с теми, которые были вычислены из более ограниченного числа последовательностей, которые обычно могут быть обработаны в подходах к мокрой лаборатории.
Приложение Pepspec пакета молекулярного моделированияRosetta 14 представляет собой инструмент, который выполняет диверсификацию последовательностей в качестве ключевого шага в режиме пептидного проектирования. Для этого требуется структура или модель рецепторного белка со связанным пептидом вплоть до одного аминокислотного остатка в длину, который используется в качестве якоря для следующих шагов. Затем последовательность связанного пептида удлиняют (при необходимости) и диверсифицируют для получения большого числа предполагаемых пептидных лигандов. Аффинность связывания этих пептидов затем оценивается с помощью докинга пептидов гибкого каркаса, чтобы выбрать те из них с наилучшими прогнозируемыми показателями связывания. Несмотря на то, что основным результатом данного применения являются наилучшие пептидные кандидаты, выбранные в конце фазы проектирования, гораздо больший набор пептидов, принятых во время этой фазы, также может быть использован для вычисления аминокислотных предпочтений целевого сайта связывания. Аминокислотные предпочтения вычисляются как частота каждого аминокислотного остатка на позицию лигандной последовательности, представленной либо в виде матрицы веса положения (ШИМ), либо в виде более визуального логотипа последовательности.
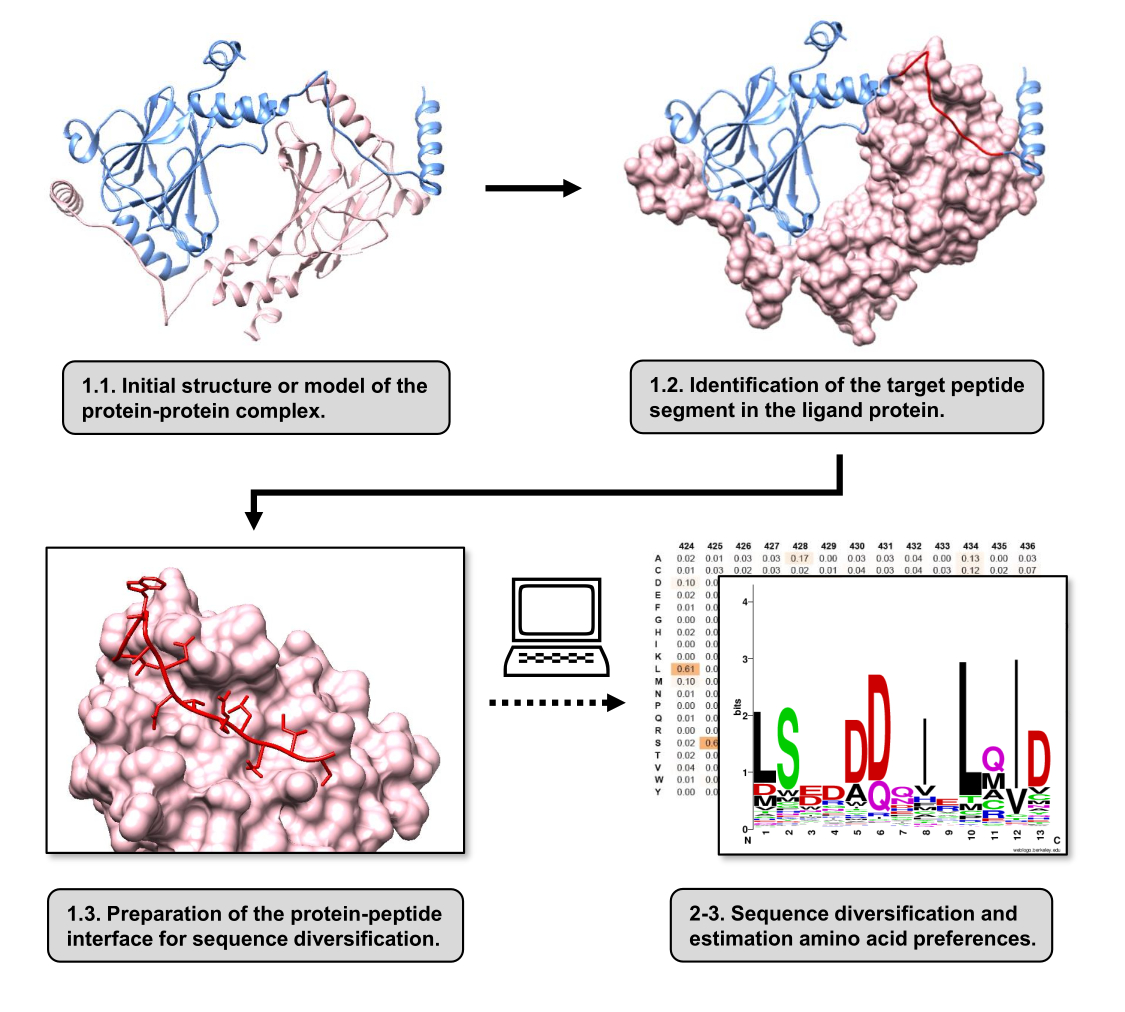
В этой статье мы описываем протокол для оценки аминокислотных предпочтений на поверхности связывания рецепторного белка, участвующего в ИПП. Протокол ориентирован на ИПП, в которых линейный сегмент белок-лиганда, как известно, связывается с рецепторным белком, поэтому сценарий может быть смоделирован как граница белок-пептид. В этом сценарии консервативные мотивы лиганда обычно взаимодействуют с определенными карманами в сайте связывания рецептора, хотя весь сегмент лиганда, участвующий в ИПП, может содержать менее консервативные области. Блок-схема, обобщающая основные этапы протокола, показана на рисунке 1. Протокол начинается с 3D-структуры белок-белкового комплекса и далее восстанавливает лигандный белок до потенциально наиболее взаимодействующего сегмента, оставляя рецепторный белок нетронутым. Наиболее взаимодействующий сегмент определяют с помощью сервера15 сканирования аланина BUDE, который проводит компьютерный мутагенез сканирования аланина для идентификации остатков горячих точек между двумя взаимодействующими белками. При таком подходе остатки лиганда по отдельности замещаются аланином, а расчетное изменение свободной энергии или стабильности комплекса (ΔΔG) затем используется для вывода о значимости соответствующего остатка для целевого PPI. После того, как выведен наиболее взаимодействующий сегмент, его комплекс с рецепторным белком используется в качестве базовой структуры, представленной в Pepspec для выполнения диверсификации последовательностей.

Рисунок 1: Обзор основных этапов протокола, предложенных в данной работе. Номера совпадают с номерами шагов в разделе протокола. Рисунки были выполнены с использованием белок-белкового комплекса, используемого в качестве примера, описанного в тексте. В этом комплексе белковая цепь, рассматриваемая как рецептор, показана розовым цветом, в то время как цепь, рассматриваемая как лиганд, показана светло-голубым цветом, а ее прогнозируемый наиболее взаимодействующий сегмент выделен красным. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
Одним из ограничений предлагаемого протокола является требование к разрешенной структуре белок-пептидного интерфейса. В качестве альтернативы протокол может начинаться с моделирования интерфейса белок-пептид-мишень, хотя конкретные этапы моделирования в настоящем документе не описаны. Более того, хотя протокол может быть реализован на персональном компьютере под управлением любой операционной системы, для выполнения шагов, связанных с приложениями Rosetta, требуется среда Linux. Компьютерный кластер также настоятельно рекомендуется для этапа диверсификации последовательностей из-за большого количества итераций, обычно выполняемых Pepspec.
Применение предложенного протокола проиллюстрировано оценкой аминокислотных предпочтений бидинговой поверхности IRF5, входящего в семейство фактора регуляции интерферона человека (IRF). Мы выбрали этот белок в качестве примера, потому что во время его активации две субъединицы связываются, образуя димер, структура которого хорошо охарактеризована16. В димерах IRF связывание может быть смоделировано как граница белок-пептид, в которой одна субъединица обеспечивает поверхность связывания, а другая взаимодействует через область, содержащую короткий консервативный мотив, называемый pLxIS17,18. Кроме того, связывание с субъединицами IRF является мультиспецифичным; Таким образом, они могут образовывать гомодимеры, гетеродимеры и комплексы с другими клеточными белками, известными как коактиваторы18.
протокол
1. Начальная подготовка белок-пептидной границы раздела
- Скачивание структуры белок-белкового комплекса
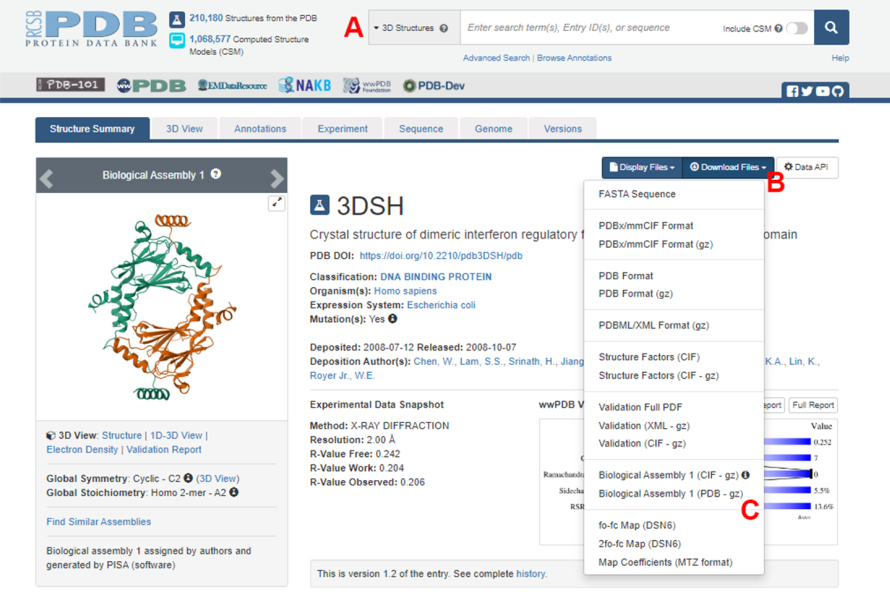
- Перейдите на домашнюю страницу Protein Data Bank (PDB) (https://www.rcsb.org/) и введите PDB ID для структуры белок-белкового комплекса в главном окне поиска (рис. 2A). Идентификатор PDB для структуры димера IRF5, используемый в качестве примера в данной работе, — 3DSH19.
- На главной странице нужной структуры нажмите на Download Files (рисунок 2B), а затем на Biological Assembly 1 (PDB - gz) (рисунок 2C).
Примечание: В базе данных PDB структуры многих белковых комплексов, образованных идентичными мономерами, представлены в виде биологических сборок, в которых в PDB-файле хранится только структура одного мономера (асимметричной единицы). Структура мультимера, в данном случае димера IRF5, должна быть загружена в виде биологической сборки, содержащей два экземпляра асимметричной единицы. Чтобы облегчить следующие шаги этого протокола, два мономера сначала разделяются, и им присваиваются различные идентификаторы цепей. - Откройте загруженную структуру в UCSF Chimera20 и нажмите «Инструменты» > «Редактирование структуры> «Изменить идентификаторы цепочки». В этом примере обе цепочки в биологической сборке называются A. Переименуйте вторую цепь (помеченную #0.2) в B и нажмите OK.
- Нажмите «Избранное» > панели модели, а затем выберите модель, содержащую две цепочки. Нажмите кнопку «Сгруппировать/Разгруппировать», чтобы разделить каждую цепочку в отдельную модель. Затем выберите две модели и нажмите кнопку «Копировать/Объединить». Введите новое имя для объединенной модели, установите флажок «Закрыть исходные модели» и нажмите кнопку «ОК».
- Нажмите на Select > Chain и подтвердите, что каждая цепочка в димере теперь идентифицируется разными буквами, а именно A и B.
- Используйте File > Save PDB , чтобы сохранить отредактированную структуру в другой PDB-файл, который будет использоваться на следующих этапах протокола (здесь использовалось имя IRF5_dimer.pdb ).

Рисунок 2: Страница банка данных белков (PDB) для структуры, используемой в качестве репрезентативного примера в данной работе. (A) Окно поиска для введения кода присоединения PDB целевой структуры. (B) Меню для скачивания структуры в нескольких форматах. (C) Опции для загрузки биологических сборок, когда структура была сохранена как асимметричная единица (подробнее см. шаг 1.1.2). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
- Идентификация целевого сегмента в белке лиганда
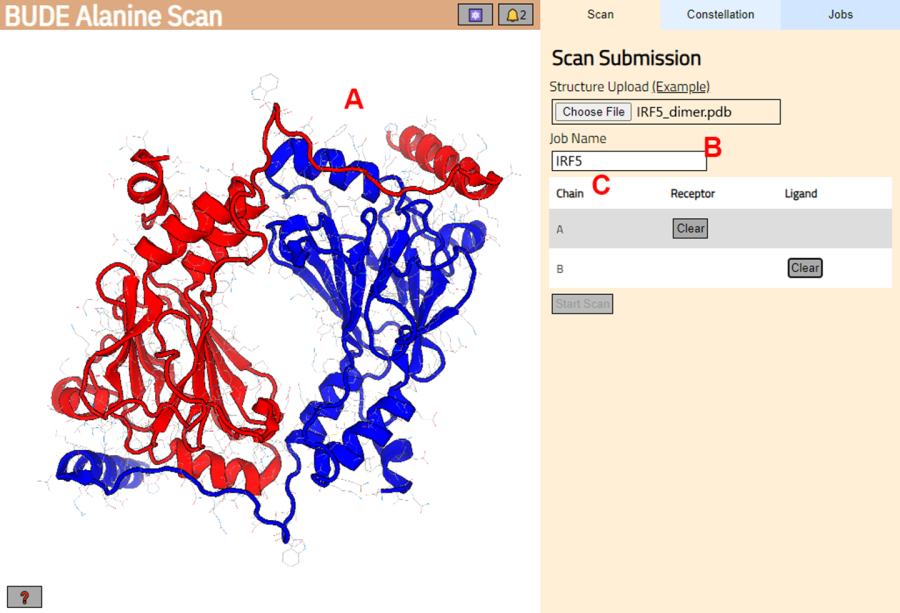
- Перейдите к серверу сканирования аланина BUDE (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Нажмите кнопку « Выбрать файл » в разделе «Загрузка структуры » и выберите PDB-файл, сохраненный на шаге 1.1.6.
- На следующей странице убедитесь, что структура была правильно загружена (рисунок 3A), и введите имя задания на сервере (рисунок 3B).
- Установите цепи из PDB, которые будут рассматриваться как рецептор (А) и лиганд (В) (рис. 3В). Затем нажмите кнопку « Начать сканирование », чтобы отправить задание.
- После завершения задания нажмите кнопку "Показать результаты ", чтобы открыть страницу результатов (рисунок 4).
ПРИМЕЧАНИЕ: На странице результатов остатки лигандной структуры окрашены в соответствии с их расчетным изменением свободной энергии (ΔΔG), а остатки с более высокими значениями окрашены в красный цвет. - В списке остатков выберите прогнозируемый участок остатков для лучшего взаимодействия с целевой поверхностью связывания. Убедитесь, что эти остатки кластеризуются с более высокими значениями разницы в свободной энергии (ΔΔG). В этом примере был выбран сегмент между остатками Leu424 и Ser436 (выделен красным прямоугольником на правой панели рисунка 4).
- Подготовка белок-пептидного интерфейса к диверсификации последовательностей
- Откройте PDB-файл, сохраненный на шаге 1.1.6 в Chimera, и проверьте, что в структуре целевых субъединиц нет недостающих атомов или связей.
- Удалите все мелкие молекулы, ионы и растворители, которые были сокристаллизованы с исходной структурой. Для этого нажмите « Выбрать > остатки », а затем выберите все молекулы, кроме стандартных аминокислот. Затем нажмите на Действия > Атомы/Связи и Удалить.
- Обрежьте цепь лиганда до наиболее взаимодействующего сегмента, выбранного на шаге 1.2.5. Для этого нажмите на «Избранное» и «Последовательность », а затем нажмите на цепь, которая считается лигандом (B). На панели «Последовательность » перетащите мышь, чтобы выделить все остатки, кроме тех, которые находятся между позициями 424 и 436. Чтобы удалить эти остатки, нажмите на Действия > Атомы/Связи и Удалить.
- Используйте File > Save PDB , чтобы сохранить отредактированную структуру в другой PDB-файл, который используется на следующих этапах протокола (здесь использовалось имя IRF5_interface.pdb ).

Рисунок 3: Выбор рецептора и лиганда на сервере BUDE Alanine Scan. (A) Графическое представление комплекса белок-белок. (B) Текстовое поле для ввода имени задания на сервере. (C) Панель для интерактивного выбора цепей, которые будут рассматриваться как рецепторы и лиганды (подробнее см. шаг 1.2). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 4: Страница результатов работы сервера сканирования аланина BUDE. Потенциальный наиболее взаимодействующий сегмент в последовательности лигандов обозначен красным прямоугольником. На левой панели остаток с более высоким прогнозируемым вкладом энергии (Leu433) выделен зеленым цветом. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
2. Диверсификация последовательностей
ПРИМЕЧАНИЕ: На следующих шагах rosetta_main ссылается на основной каталог установки Rosetta, который обычно находится по адресу /opt/rosetta_src__bundle/main/, где указывает на установленную версию Rosetta. Кроме того, предполагается, что приложения Rosetta доступны в масштабах всей системы; Если это не так, необходимо предоставить полный путь к исполняемым файлам. При компиляции из исходного кода эти исполняемые файлы находятся в каталоге /rosetta_main/source/bin/ .
- Первичная оптимизация боковых цепей аминокислот
- Скопируйте отредактированную структуру, сохраненную на шаге 1.3.4, в папку Linux, доступную для приложений Rosetta.
- Используйте приложение FixBB от Rosetta для выполнения переупаковки всех боковых цепей аминокислот базовой структуры перед диверсификацией последовательности. В этой операции ориентация всех боковых цепей аминокислот оптимизируется для минимизации энергии и повышения стабильности комплекса. Для этого выполните следующую команду:

ПРИМЕЧАНИЕ: Эта команда выводит PDB-файл, названный в честь исходной структуры с дополнительным числовым суффиксом (IRF5_interface_0001.pdb в данном примере). - Чтобы облегчить следующий шаг протокола, переименуйте переупакованный PDB-файл с суффиксом _repack с помощью следующей команды:
MV IRF5_interface_0001.pdb IRF5_repack.pdb
- Диверсификация последовательностей
- Запустите Pepspec в режиме проектирования, чтобы выполнить фактический шаг диверсификации последовательности с помощью следующей команды:

Ниже приведены общие варианты:- -s указывает на входной файл (переупакованный PDB-файл, сгенерированный на шаге 2.1.3).
- -o указывает префикс для именования выходных файлов.
- - база данных указывает путь к основной базе данных Rosetta 3.
- -ex1, -ex2 и extrachi_cutoff являются опциями библиотеки rotamer (подробнее см. документацию Pepspec).
- -overwrite указывает приложению перезаписать возможные ранее существовавшие выходные данные, сгенерированные предыдущими итерациями.
- -pepspec:pep_chain указывает на PDB-цепи, рассматриваемые как лиганды (в этом примере 'b').
- -pepspec:native_pep_anchor указывает на аминокислотный остаток, используемый в качестве якоря (в данном примере остаток Leu в положении 10 пептида лиганда).
- -pepspec:n_peptides указывает количество пептидных структур для вывода.
- -pepspec:no_prepack_prot указывает приложению пропустить переупаковку в входной базовой структуре (поскольку это ранее было выполнено на шаге 2.1).
ПРИМЕЧАНИЕ: Основным выходом Pepspec является каталог, содержащий PDB-файлы для пептидов, полученных на этапе проектирования, названные с использованием выходного префикса с суффиксом .pdbs (в примере IRF5.pdbs ). Кроме того, Pepspec выводит все принятые пептидные последовательности, протестированные в рамках этапа диверсификации последовательностей, и соответствующие им оценки калорий Rosetta в текстовом файле, разделенном табуляцией, названном по выходному префиксу, с . spec (IRF5.spec в примере). Поскольку протокол, описанный в этой работе, направлен на оценку предпочтений аминокислот, а не фактического пептидного дизайна, на следующих этапах будет использоваться IRF5.spec вместо PDB-структур в каталоге .pdbs .
- Запустите Pepspec в режиме проектирования, чтобы выполнить фактический шаг диверсификации последовательности с помощью следующей команды:
3. Оценка аминокислотных предпочтений
- Вычисление ШИМ
- Чтобы сгенерировать ШИМ, используйте сценарий gen_pepspec_pwm.py , входящий в состав пакета Rosetta. Чтобы запустить этот скрипт, используйте следующую команду:

где:- IRF5.spec - это выходной файл Pepspec, сгенерированный на шаге 2.2.
- -1 указывает на то, что в последовательности нет дополнительных N-концевых остатков и, следовательно, позиции в ШИМ основаны на 1.
- 0.2 указывает скрипту учитывать только 20% наиболее результативных пептидов из выходных данных Pepspec (значение по умолчанию — 0.1, что соответствует 10%)
- interface_score указывает скрипту ранжировать пептиды на основе оценки интерфейса, которая является одной из различных оценок Rosetta, включенных в выходной файл Pepspec.
ПРИМЕЧАНИЕ: Этот скрипт генерирует два выходных файла, один для вычисленной ШИМ (с суффиксом .pwm ), а другой для последовательностей подмножества пептидов, используемых для вычисления ШИМ (с суффиксом .seq ). Названия этих файлов также включают оценку и фракцию пептидов, используемых для ранжирования. В этом примере эти файлы называются соответственно IRF5_interface_score_0.2.pwm и IRF5_interface_score_0.2.seq.
- Чтобы сгенерировать ШИМ, используйте сценарий gen_pepspec_pwm.py , входящий в состав пакета Rosetta. Чтобы запустить этот скрипт, используйте следующую команду:
- Создание логотипа последовательности
- Перейдите на сервер WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 и нажмите кнопку «Выбрать файл » рядом с пунктом «Загрузить данные последовательности». Загрузите файл с пептидными последовательностями, сгенерированными на шаге 3.1.1 (в данном примере IRF5_interface_score_0.2.seq ).
- Выберите желаемый формат и размер логотипа в соответствии с длиной ввода. В примере используется формат PDF и размер 15 см x 12 см. Нажмите на кнопку «Создать логотип».
Результаты
В этой статье мы описали протокол для прогнозирования аминокислотных предпочтений на поверхности связывания IRF5, члена семейства транскрипционных факторов, известных как регуляторные факторы интерферона человека. Эти белки являются регуляторами врожденных и адапт?...
Обсуждение
В настоящей статье описан протокол оценки аминокислотных предпочтений потенциально мультиспецифических сайтов связывания на основе диверсификации последовательностей in silico. Разработано несколько вычислительных инструментов для оценки аминокислотных предпочтен...
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Выражаем благодарность за финансовую поддержку со стороны Sistema Nacional de Investigación (SNI) (номера грантов SNI-043-2023 и SNI-170-2021), Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) Панамы и Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU). Авторы хотели бы поблагодарить доктора Мигеля Родригеса за тщательное рецензирование рукописи.
Материалы
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Ссылки
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены