
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
通过DNA含量测定的哺乳动物复制时序的全基因组的测定
摘要
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
摘要
基因组的复制过程中在一个高度调节的过程,以确保DNA复制的保真度在细胞周期的S期时发生。每个基因组区域是在不同的时间在S期通过复制多个起源的同时激活复制。复制(TOR)的时间与许多基因组和表观遗传特征相关,并链接到突变率和癌症。解读复制程序的完整基因组认为,在健康和疾病是未来的一个主要目标和挑战。
本文详细介绍了该法的"S / G1的用于映射复制基因组时代拷贝数比率"(以下称为:CNR-TOR),一个简单的方法来绘制哺乳动物细胞的基因组范围的职责范围。该方法是基于S期细胞和G1期细胞的拷贝数的差异。在6个步骤进行CNR-TOR方法:1,细胞和染色与碘化丙啶(PI)的制备; 2.索尔廷G1和使用荧光激活细胞分选S期细胞(FACS); 3. DNA纯化; 4.超声; 5.文库制备和测序; (六)生物信息学分析。该CNR-TOR法是一种快速简便的办法,导致了详细的复制地图。
引言
哺乳动物DNA复制是严格调节细胞周期过程中,以确保精确地一次每个染色体的精确复制。复制根据一个高度管制的次序发生-多个大型基因组区域(〜兆)S期的开始复制(早期复制域),而其他的基因组区域的中后期S期(中晚期复制域)后重复1。大多数基因组的复制在所有组织中(组成职责范围域)的同时,而30% -在基因组的50%,也期间癌症转化5改变组织2之间它的职责范围,分化3,4期间和在较小程度上。此外,某些基因组区域复制异步6,7,8,即是有区别的在两个等位基因之间的职责范围。
职责范围与许多基因组和表观基因的功能,包括转录水平,GC含量,染色质的状态,基因密度等 1,9相关。职责范围也与突变率和类型10,11,因此毫不奇怪相关联,则复制程序的扰动与癌症12,13。职责范围和染色质结构之间的因果关系尚不清楚。这可能是开放的染色质促进早期复制。然而,另一种模型表明,在复制过程中的染色质组装和存在于开头的不同染色质调节和S期引线端至差分早期和晚期复制区域1的包装,14 。我们最近表明,职责范围形状通过影响发生在不同的基因组区域11突变型GC含量。
荧光原位杂交(FISH)是在各个基因检测职责范围的主要方法。它是通过计算显示出单一的FISH信号与双重对于给定的等位基因15,16百分比S期细胞的百分比简单地进行。一种替代方法,由脉冲的标记用BrdU的DNA,根据它们的DNA含量,以多个时间点沿小号分拣细胞,免疫沉淀含有BrdU的DNA,并检查沉淀的DNA的丰度与定量PCR 17。
基因组职责范围映射可以通过两种方法来实现。第一种方法是上述的的BrdU-IP为基础的方法的基因组的版本,其中,所述量的量化在每个级分沉淀的DNA被用于通过杂交整个基因组,以微阵列或通过深度测序同时进行。第二种方法,CNR-TOR,基于由在G1期细胞的DNA含量测定S期细胞和正火的每个基因组区域的拷贝数。在该方法中,细胞通过FACS分类成非复制(G1期),并复制(S期)基团( 图1)。在G1期细胞在所有的基因组区域的相同拷贝数和因此它们的DNA含量应该是相同的。另一方面,在S中的DNA拷贝数依赖于职责范围,因为早期复制区域中大多数细胞进行复制,因此,它们的DNA含量增加一倍,而晚复制区域没有在大多数细胞尚未复制,因此,它们的DNA含量将是类似的G1期细胞。因此,S键DNA含量的G1比例指示职责范围。 DNA的每个基因组区域的量测得的通过杂交要么芯片或深度测序2,8。的CNR-TOR方法的优点将被进一步讨论。
本文介绍在图2描述了基因组职责范围映射CNR-TOR方法。本文从收集细胞,直到结果的基本分析和基因组职责范围的地图的创建讨论的全过程的细节。在本文中描述的协议已经在培养中生长的各种细胞类型被成功执行。这个协议的未来的改进可导致职责范围体内的映射和在罕见的细胞类型。
研究方案
注意:职责范围只能上生长,不同步的细胞来测量。 2×10 6个快速生长的细胞,这通常会导致在〜1×10 5个细胞在S期(速率限制步骤) -一个程序应当至少有1开始。它建议进行使用两个或三个重复各实验。 CNR-TOR的整个过程可以在一个星期内完成 - 两个天应专用于所有步骤达文库制备,需要用于测序一至两天,另外天是必要的初始数据分析。
1.培养细胞的采集
注:该协议是为于10cm板(含有约2 - 5×10 6个细胞)中培养生长的细胞编写的,但可以很容易地调整到其他平台。
- 对于悬浮生长的细胞,进行固定(第2节)。
- 对于贴壁细胞,吸洗了PL用3毫升的PBS吃不含Ca 2+和镁离子 。
- 丢弃PBS中,并用1mL商业胰蛋白酶-EDTA孵育细胞5分钟,在37℃下直到细胞分离。
注:胰蛋白酶处理的持续时间应调整到不同的细胞类型。 - 加入3毫升培养基中和胰蛋白酶,并收集在15毫升锥形管或一个5毫升聚苯乙烯管细胞。置于冰上。
2.固定
注:对于这部分,所有的步骤应在4℃下进行。
- 离心细胞,在300×g离心在4℃下5分钟。
- 洗板细胞两次用1毫升冷PBS。
- 悬浮细胞在250微升(总)冷PBS。
- 同时轻轻涡旋管,慢慢°C 100%的乙醇添加-20滴加800微升。这导致70的最终乙醇浓度 - 80%。
注意:高纯度乙醇建议在此步骤。 - 孵育细胞在冰上FO-R 30分钟。
注:在此阶段细胞可以保持几天在4℃或在-20℃下几个月。
3. PI染色
- 离心细胞,在500×g离心在4℃下10分钟。
- 小心吸出上清液并用1mL冷PBS洗涤细胞两次。
- 吸液和悬浮用下列混合物各样品:1毫升的PBS,5微升10毫克/毫升RNA酶A,50微升1mg / mL的碘化丙啶(PI;使用前混合瓶)。调整浓度可达〜2×10 6个细胞/ mL)。
注意:远离光 - PI对光敏感。 - 通过35微米网状到5毫升聚苯乙烯管过滤器,并关闭封口膜。
- 孵育在室温在黑暗中,15 - 30分钟。
注:细胞现在已经准备好FACS分析。如果需要,染色的细胞可以在4℃保存至少24小时在黑暗中。
4.排序
- 使用FACS机器排序细胞。我们e为561纳米的激光来区分基于其PI强度细胞。靠近535 nm激发最大像488纳米或532其它激光器可以根据FACS机器的配置中使用。
- 为获得最佳结果,使用推荐用于特定小区的大小(对于大多数细胞85微米)最小的喷嘴。优先于产量纯度模式。使用稳定的缓慢流动,通常高达300-500事件/秒,以45 psi的鞘压力。
- 使用选通,通过绘制FCS VS SSC区分死细胞和亚细胞碎片(低FCS和高SSC)。从活细胞通过绘制SSC-宽度(W) 与后跟一个FSC-W VS FSC-H曲线图,并通过PI-W¯¯SSC-高度(H)的区别双峰VS的PI-H(双双将具有相同的H值但更大的W值)。的活单细胞绘制其表示细胞的DNA含量的PI-区(A)的强度的直方图。
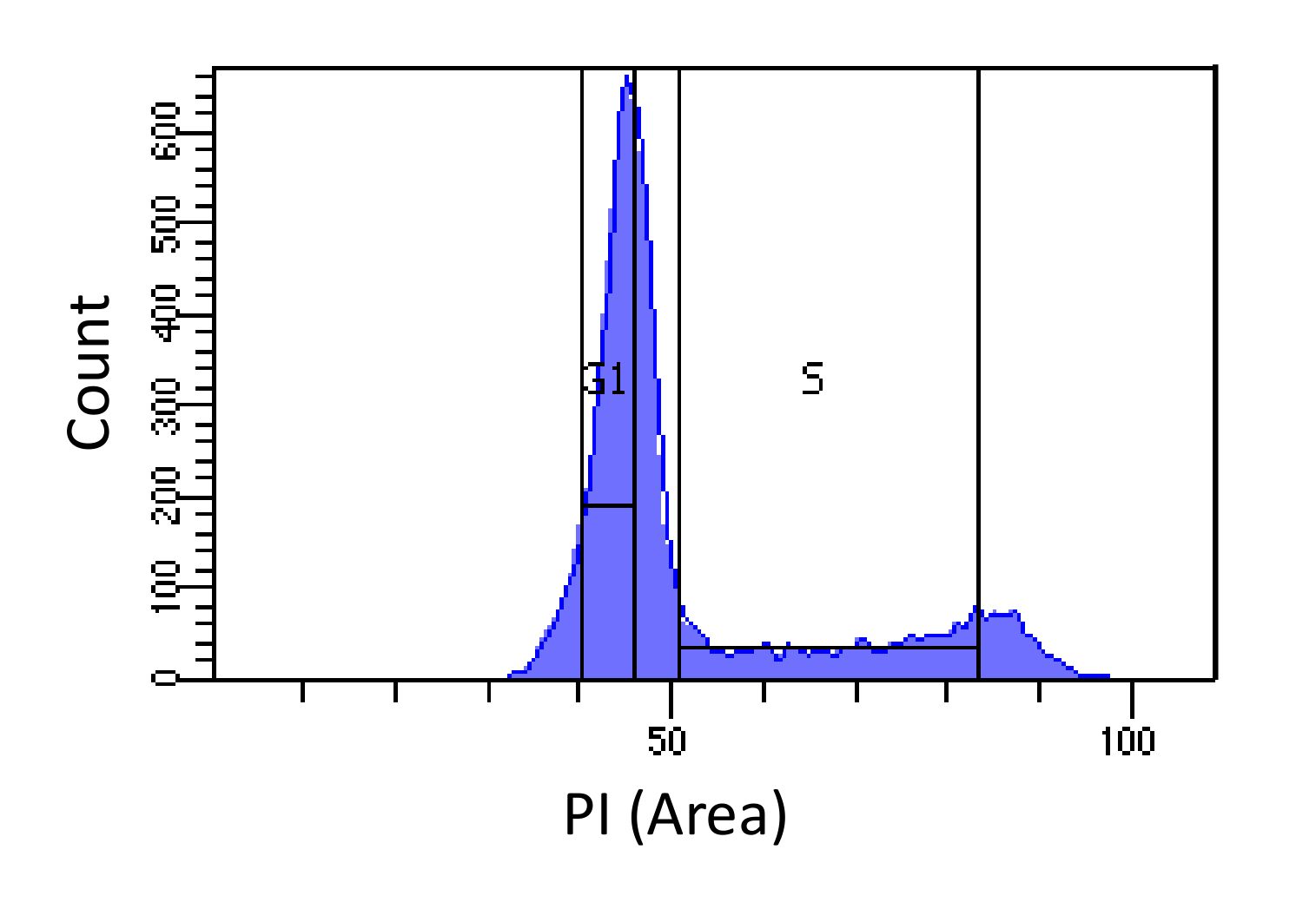
- 排序细胞进入G1和S期, 如图1。门控对于s应宽和侵入G1和G2期,而G1门应该是狭窄,尽量选自S越好。

图1.细胞周期的阶段决定基于PI的强度。柱状图表示细胞DNA含量的小鼠胚胎成纤维细胞(MEF)总体的分布(由PI-区测量)。 DNA含量用于人口排序成两个亚群ⅰ)G1期细胞(2N DNA含量)和ii)S期细胞(2N - 4N DNA含量),使用标记的区域。 请点击此处查看该图的放大版本。
{kind=link}
注:G1期细胞的集合的目的是考虑到在不同的基因组区域之间的测序效率偏差。另一种应用蟑螂是使用G1停滞细胞来自相同细胞类型。这种方法使清洁剂的结果(因为它最大限度地减少S期的污染),但它可能会引入偏见从被捕细胞和细胞测量之间的遗传差异所产生的。
- 排序在寒冷条件下,收集排序细胞到1.5 / 2 mL管。保持排序在冰上管。
注意:为了提高DNA回收率,最好是使用低的结合管或使用涂有4个试管-在4℃下18 3小时- 5%BSA的1。
5. DNA纯化
- 对于每个样本(G1和S)用DNA纯化试剂盒纯化DNA。
注:当使用市售的试剂盒,洗脱用400微升的洗脱缓冲液到2mL管产量高,如制造商推荐的。 - 使用荧光检查DNA浓度。
注:从由FACS收集100000哺乳动物细胞中,人们应该得到的DNA〜1微克。一个吨该阶段DNA可在4℃或在-20℃长期储存保持几天。
6.超声
- 转移DNA导入1.7毫升管,其是与所使用的磁性底座兼容。
- 集中的DNA根据制造商的说明使用2倍SPRI珠,用磁性支架,并在50微升洗脱缓冲液洗脱。
- 剪切DNA用聚焦超声发生器到250基点的平均目标峰大小。使用以下设置为50μLDNA样本:50 W,20%占空比系数,每爆200个周期,20℃,120秒。
- 验证通过电泳的剪切的DNA的大小。推荐大小分布为200 - 700个碱基,在〜250 bp的高峰。
注:在此阶段的DNA可以保持几天,在4℃或在-20℃长期保存。
7.文库制备和测序
注意:许多文库制备试剂盒和不同耳鼻喉科测序平台应的工作方式类似于我们所使用,并提到在材料部分的人。实际上,在过去,职责范围的地图用与微阵列平台2非常相似的方法产生的。
- 准备使用任何商业库制备试剂盒库。
- 800 bp的 - 在图书馆准备结束时,利用磁珠300选择大小。
- 准备库后,使用荧光测定DNA浓度。
- 使用电泳测量DNA大小。
- 在任何平台上进行测序。
注意:每个样品测序至少10μM的读取建议。该深度相当于一个读大约每300bp的(为〜3千兆大小的基因组),并足以为Tor测量在50,分辨率 - 100 kb的。增加深度将导致在窗口的尺寸的减小,从而使增加与更高的确定性的分辨率。配对末端测序并非必要因为只有覆盖信息这个协议被收集。然而,它可能会帮助解决的位置读取包含重复序列。
8.分析
注意:数据分析是基于由A.科伦等人所使用的方法。 19。
- 地图测序数据使用bowtie2或任何可靠的短读取对准相应的基因组。定义不同的尺寸,相当于覆盖染色体窗口由200段涵盖在G1分数读取和计数S期读取同一个窗口。
- 计算每个窗口在S / G1比。这应该产生与沿基因组中( 图3)在S / G1比大的波动的地图。良好控制职责范围测量的可靠性是在该地图进行比较,以在G1 / G1比(从G1的两个独立测量值),其应该是更平坦。
- 数据标准化为0均值和1个SD通过各V减去ALUE所有窗口(不包括X染色体)和由所有窗口的S / G1的标准偏差除以结果的平均值。这样做是为了将其转换为z值和使不同实验之间的比较完成的。
- 除去由UCSC基因组浏览器以及包含少于15个数据窗每个剩余间间隙片段列出的所有的间隙区域。
- 通过与10个参数的MATLAB函数csaps一个三次样条函数平滑剩余的碎片- 16,并在设定点插每100 KB。
注:平滑和内插的参数应该根据数据的深度来调节。其它合适的平滑化方法和功能存在,并且可以使用。 - 视觉上确认的每个复制的可靠性后,合并所有的读取和通过对这个数据与上述相同的过程计算更深分辨率轮廓。
结果
一个典型的职责范围的地图显示在图3为小鼠胚胎成纤维细胞(MEFs)。该图演示了分析过程,因为它同时显示了点,这是各个窗口(步骤8.3)的归一化的S / G1期的比例,以及从该立方平滑和内插(步骤8.5)产生的线。
这类地图捕获复制程序的组织,这是两种类型的职责范围的域的拼凑:ⅰ)大的区域(在一个兆碱基...
讨论
CNR-TOR可以在原则上任何真核增殖细胞群,可以通过FACS对S和G1期(由莱因德N。和吉尔伯特DM 20中综述)被划分来执行。这里所描述的方法已被调整到哺乳动物细胞用〜3千兆的基因组大小如人和小鼠。在CNR-TOR协议的微小变化(在细胞制剂和测序深度)都需要,以便将其调整到其它真核生物。因为它是限速步骤必须注意到的足够量的S期的细胞的集合。因此,对于细胞周期的初步FACS...
披露声明
No conflicts of interest declared.
致谢
我们感谢奥里亚瓦迪对生成的数字援助。在IS组的工作是由以色列科学基金会(批准号:十分之五百六十七)和欧洲研究理事会资助启动(#281306)的支持。
材料
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
参考文献
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。