
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Determinação do genoma de sincronismo Replication Mammalian por DNA Medição conteúdo
Neste Artigo
Resumo
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Resumo
A replicação do genoma ocorre durante a fase S do ciclo celular em um processo altamente regulado que assegura a fidelidade de duplicação de ADN. Cada região genómico é replicada em um momento diferente durante a fase S através da activação simultânea de múltiplas origens de replicação. Tempo de replicação (ToR) correlaciona-se com muitas características genômicas e epigenéticos e está ligada a taxas de mutação e câncer. Compreender o ponto de vista genômica completa do programa de replicação, na saúde e na doença é uma importante meta para o futuro e desafio.
Este artigo descreve em detalhes o "Número Taxa de cópia de S / G1 para mapear Tempo genômico de replicação" método (aqui chamado: CNR-ToR), uma abordagem simples para mapear o genoma ampla ToR de células de mamíferos. O método baseia-se nas diferenças no número de cópias entre as células em fase S e células em fase G1. O método CNR-TdR é realizado em 6 passos: 1. Preparação de células e a coloração com iodeto de propídio (PI); 2. SorTing G1 e células em fase S utilizando células activadas por fluorescência (FACS); 3. purificação de ADN; 4. sonicação; 5. preparação e sequenciamento da biblioteca; e 6. A análise bioinformática. O método CNR-Tor é uma abordagem fácil e rápido que resulta em mapas de replicação detalhadas.
Introdução
replicação de ADN de mamífero é estritamente regulada para assegurar a replicação precisa de cada cromossoma apenas uma vez durante o ciclo celular. A replicação ocorre de acordo com uma ordem altamente regulado - várias regiões genômicas grandes (~ Mb) replicar no início da fase S (início de replicar domínios), enquanto que outras regiões genômicas replicar mais tarde em (domínios médio e replicar tarde) médio ou fase S tardia 1. A maior parte do genoma replica ao mesmo tempo em todos os tecidos (domínios ToR constitutivos), enquanto que 30% - 50% do genoma, muda a sua TdR entre tecidos 2, durante a diferenciação 3, 4 e, em menor medida, também durante a transformação do cancro 5 . Além disso, certas regiões genómicas replicar de forma assíncrona 6, 7, 8, ou seja, existe uma diferençana especificação técnica entre os dois alelos.
ToR correlaciona com muitas características genômicas e epigenômicos incluindo os níveis de transcrição, conteúdo GC, estado de cromatina, a densidade gene, etc. 1, 9. Tor também está associada a taxas de mutação e tipos 10, 11 e, portanto, sem surpresa, perturbações do programa de replicação estão ligados ao câncer 12, 13. A relação causal entre ToR e estrutura da cromatina ainda não é compreendido. É possível que a cromatina aberta facilita a replicação inicial. No entanto, um modelo alternativo sugere que a cromatina é montada durante a replicação e os diferentes reguladores da cromatina presentes no início e no fim do avanço de fase S para embalagem diferencial de regiões de replicação precoce e tardio 1, 14 . Mostrámos recentemente que o TdR molda o teor de GC ao afectar o tipo de mutações que ocorrem em diferentes regiões genómicas 11.
Hibridização fluorescente in situ (FISH) é o método principal para a medição TdR em loci individuais. Ela é realizada simplesmente por meio da contagem da percentagem de células em fase S que exibem sinais de FISH individuais contra a percentagem de dupletos para um determinado alelo 15, 16. Um método alternativo, é constituído por impulsos marcação do ADN com BrdU, as células de apartação de acordo com o seu conteúdo em ADN para vários pontos de tempo ao longo S, imunoprecipitação ADN contendo BrdU, e verificando a abundância de ADN precipitado com 17 qPCR.
TdR mapeamento genómico pode ser alcançada através de dois métodos. O primeiro método é uma versão genómica do método baseado em IP BrdU descrito acima, em que a quantificação da quantidadede ADN foi precipitado em cada fracção é feito, simultaneamente, para a totalidade do genoma por meio de hibridação com micromatrizes ou por sequenciação de profundidade. O segundo método, CNR-TdR, baseia-se na medição do número de cópias de cada região genómica de células em fase S e normalizando pelo conteúdo em ADN em células G1. Neste método, as células são classificadas por FACS em não-replicante (fase G1) e replicação (fase S) grupos (Figura 1). As células em G1 tem o mesmo número de cópias em todas as regiões genómicas e, assim, o seu conteúdo de ADN deve ser o mesmo. Por outro lado, o número de cópias de ADN em S depende da TdR, uma vez que regiões replicadoras iniciais foram submetidos a replicação na maioria das células e, por conseguinte, o seu conteúdo de ADN é duplicada, enquanto que as regiões de replicação final ainda não replicado na maioria das células e, por conseguinte, o seu conteúdo de ADN serão ser semelhante ao de células G1. Daí a S à relação de G1 de conteúdo de DNA é indicativo do ToR. A quantidade de ADN para cada região genómica é medido quer por hibridação commicroarrays ou por uma profunda sequenciamento 2, 8. As vantagens do método CNR-TdR será discutido.
Este artigo descreve o método CNR-ToR para mapeamento genômico ToR como descrito na Figura 2. O artigo discute os detalhes de todo o processo a partir de células coleta até a análise básica dos resultados ea criação de mapas ToR genômicos. O protocolo descrito no presente trabalho foi realizado com sucesso em vários tipos de células cultivadas em cultura. Futuras melhorias deste protocolo pode levar ao mapeamento dos TdR in vivo e em tipos de células raras.
Protocolo
Nota: ToR pode ser avaliada apenas com o crescimento de células, não sincronizadas. O procedimento deve começar com pelo menos 1 - 2 x 10 6 células de crescimento rápido, que geralmente resultam na ~ 1 x 10 5 células em fase S (o passo limitante da velocidade). Recomenda-se para conduzir cada experiência utilizando-se dois ou três repetições. Todo o processo de CNR-ToR pode ser concluída dentro de uma semana - dois dias deve ser dedicado a todas as etapas até a preparação biblioteca, um a dois dias são necessários para sequenciamento e um dia adicional é necessário para a análise de dados inicial.
1. Recolha de Células de Cultura
NOTA: O protocolo é escrito para as células que crescem em cultura em placas de 10 cm (contendo cerca de 2 - 5 x 10 6 células), mas pode ser facilmente ajustado para outras plataformas.
- Para as células que foram cultivadas em suspensão, proceder à fixação (seção 2).
- Para células aderentes, aspirar e lavar o plcomeu com 3 mL PBS, sem Ca 2+ e Mg 2+.
- Descartar o PBS e incubar as células durante 5 min a 37 ° C com 1 ml de tripsina-EDTA comercial até separar células.
NOTA: A duração do tratamento com tripsina deve ser ajustado para cada tipo de célula. - Adicionar 3 mL meios de cultura para neutralizar a tripsina e recolher as células num tubo cónico de 15 mL ou um tubo de poliestireno de 5 ml. Manter em gelo.
2. Fixação
NOTA: Para esta parte, todas as etapas devem ser feitas a 4 ° C.
- Centrifugar as células a 300 xg durante 5 min a 4 ° C.
- Aspirar e lavar as células duas vezes com 1 mL de PBS frio.
- Ressuspender as células em 250 mL (total) PBS frio.
- Enquanto o tubo suavemente vortex, adiciona-se lentamente gota a gota 800 uL de -20 ° C, 100% de etanol. Isto leva a uma concentração final de etanol de 70 - 80%.
NOTA: alta pureza etanol é recomendado nesta etapa. - Incubar as células em gelo foR 30 min.
Nota: Neste células estágio pode ser mantido por alguns dias a 4 ° C ou durante alguns meses a -20 ° C.
3. PI Coloração
- Centrifugar as células a 500 xg durante 10 min a 4 ° C.
- Aspirar o sobrenadante cuidadosamente e lavar as células duas vezes com 1 mL de PBS frio.
- Aspirar e ressuspender cada amostra com a seguinte mistura: 1 mL de PBS, 5 uL de 10 mg / ml de ARNase, 50 uL de 1 mg / mL de iodeto de propídio (PI; misturar frasco antes de usar). Ajustar a concentração até aproximadamente 2 x 10 6 células / ml).
NOTA: Mantenha fora da luz - PI é sensível à luz. - Filtrar através de 35 mm de malha a um tubo de poliestireno 5 mL, e fechar com Parafilm.
- Incubar à temperatura ambiente no escuro, durante 15 - 30 min.
NOTA: As células estão prontas para análise FACS. Se necessário, as células coradas podem ser armazenados durante pelo menos 24 h a 4 ° C no escuro.
4. Organizar
- Ordenação de células utilizando uma máquina FACS. NosÊ O laser de 561 nm para diferenciar células com base na sua intensidade PI. Outros lasers perto do máximo de excitação de 535 nm, tal como 488 nm ou 532 pode ser utilizado, dependendo da configuração da máquina de FACS.
- Para obter resultados óptimos, utilize o menor bocal recomendado para o tamanho de célula específico (para a maioria das células 85 uM). Priorizar modos de pureza mais rendimento. Use um fluxo lento constante, geralmente até 300-500 eventos / s, com uma pressão do invólucro de 45 psi.
- Usando gating, discriminar as células mortas e detritos subcelular (baixa FCS e alto SSC) traçando FCS vs SSC. A partir das células viáveis discriminar dupletos locando SSC-largura (W) vs SSC-altura (h), seguido por um FSC-W vs lote FSC-H e por PI-W vs PI-H (dupletos terá o mesmo valor H- mas maior W-valor). Para as células individuais viáveis desenhar um histograma da intensidade de PI-área (A), que representa o teor de ADN das células.
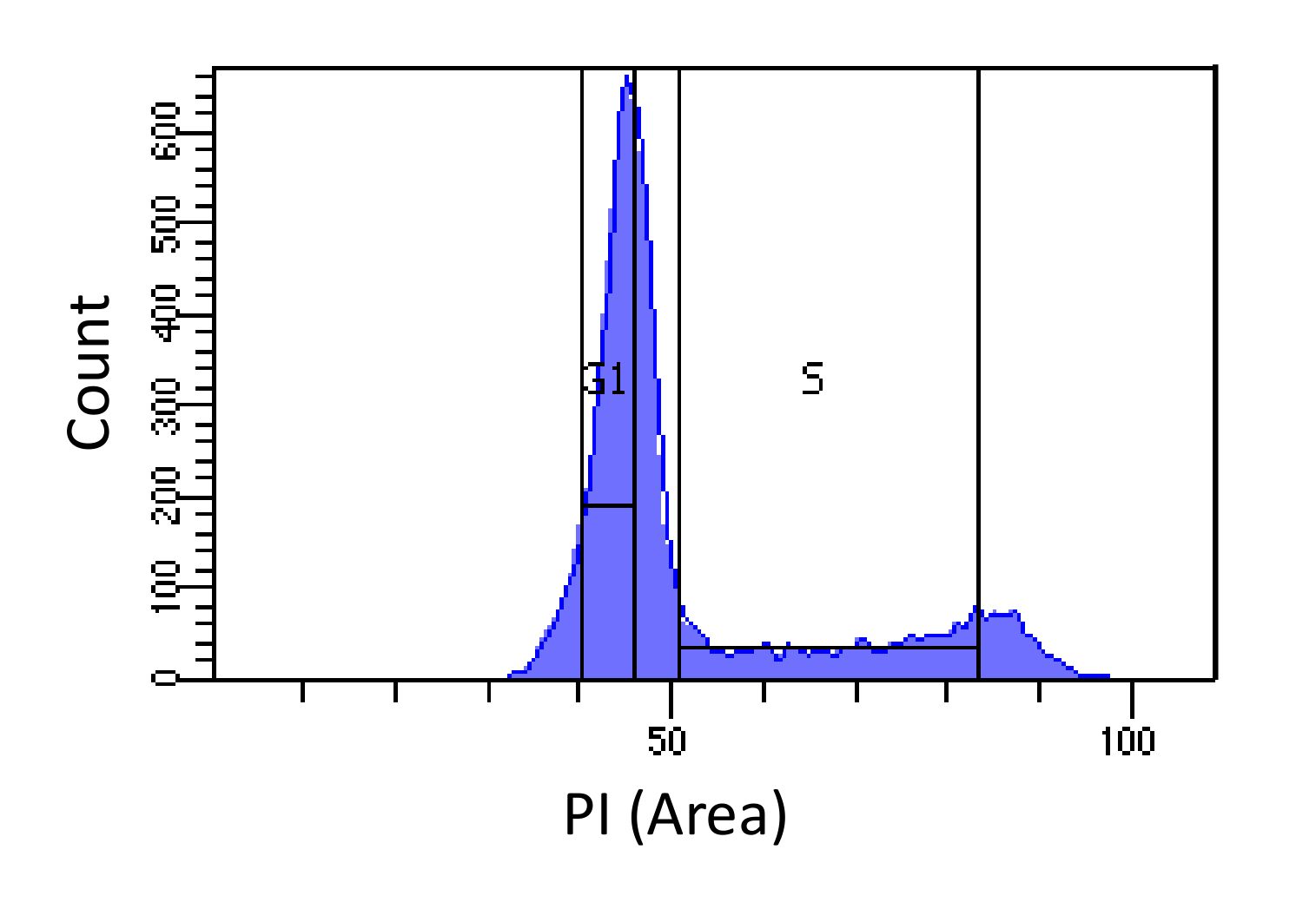
- Ordenação de células em fases G1 e S, como mostrado na Figura 1. Gating para S devem ser largas e invadir as fases G1 e G2, G1 enquanto gating deve ser estreita e, na medida do possível S.

Figura determinação fase do ciclo 1. celular baseado na intensidade PI. Histograma que mostra a distribuição do conteúdo em ADN celular (medido por PI-Área) de rato embrionário de fibroblastos (MEF) população. O conteúdo de DNA é usada para classificar a população em duas sub-populações de i) células G1 (conteúdo de DNA 2N) e ii) as células em fase S (2N - conteúdo de DNA 4N), usando as regiões marcadas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
NOTA: A finalidade da recolha de células G1 é para explicar desvios na eficiência sequenciamento entre diferentes regiões genômicas. Um aplicativo alternativabarata é a utilização de células paradas G1 a partir do mesmo tipo de célula. Esta abordagem dá resultados mais limpos (uma vez que minimiza a contaminação fase S), mas podem introduzir vieses decorrentes de diferenças genéticas entre as células presas e as células medidos.
- Classificar em condições de frio e recolher células ordenadas em 1,5 / 2 tubos mL. Mantenha os tubos no gelo após a ordenação.
NOTA: De modo a melhorar a recuperação de ADN, é preferível utilizar tubos de ligação baixa ou o uso de tubos revestidos com 4-5% de BSA durante 1-3 horas a 4 ° C 18.
5. Purificação de DNA
- Para cada amostra (G1 e S) purificar o ADN utilizando um kit de purificação de ADN.
NOTA: Quando utilizando o kit comercial, elui-se com 400 ul de tampão de eluição para um tubo de 2 mL para um rendimento elevado, tal como recomendado pelo fabricante. - Verificar concentração de DNA usando fluorómetro.
NOTA: A partir de 100.000 células de mamíferos coletados pelo FACS, deve-se obter ~ 1 mg de DNA. UMAt esta fase ADN pode ser mantido durante alguns dias a 4 ° C ou a -20 ° C para armazenamento a longo.
6. sonicação
- Transferência de ADN para um tubo de 1,7 ml, que é compatível com o suporte magnético utilizado.
- Concentra-se o ADN utilizando pérolas 2x SPRI de acordo com as instruções do fabricante, utilizando um suporte magnético, e elui-se em 50 ul de tampão de eluição.
- DNA de cisalhamento com um ultrasonicator focada para um tamanho médio de pico alvo de 250 pb. Utilize as seguintes definições para uma amostra de DNA de 50 mL: 50 W, factor de utilização de 20%, 200 ciclos por explosão, 20 ° C, 120 s.
- Verificar o tamanho do ADN cortado por electroforese. A distribuição de tamanho recomendado é de 200-700 bp com um pico em ~ 250 pb.
NOTA: Nesta fase o ADN pode ser mantido durante alguns dias a 4 ° C ou a -20 ° C para armazenamento a longo.
7. Biblioteca Preparação e Sequencing
NOTA: Muitos kits de preparação biblioteca e diferemplataformas de sequenciamento ent deve funcionar de forma semelhante aos utilizados por nós e mencionados na seção materiais. Na realidade, no passado, foram gerados mapas TdR usando um método muito semelhante com plataformas de microarray 2.
- Prepare bibliotecas usando qualquer kit de preparação biblioteca comercial.
- No final de preparação da biblioteca, selecionar o tamanho usando esferas magnéticas para 300-800 pb.
- Depois de preparar bibliotecas, medir a concentração de DNA usando fluorômetro.
- Medir o Tamanho do ADN utilizando electroforese.
- Execute sequenciamento em qualquer plataforma.
NOTA: Sequencing pelo menos 10 M leituras por amostra é recomendado. Esta profundidade é equivalente a uma leitura aproximadamente em cada 300 pb (para ~ 3 genomas de tamanho Gb), e é suficiente para medições ToR com uma resolução de 50 - 100 kb. Aumentando a profundidade irá resultar numa diminuição no tamanho das janelas e, assim, vai permitir aumentar a resolução com maior certeza. Emparelhado sequenciamento final não é necessáriaeste protocolo já que a informação única cobertura é coletado. É, no entanto, pode ajudar a resolver o local de leituras que contêm sequências repetitivas.
8. Análise
NOTA: A análise dos dados baseia-se no método usado por A. Koren et ai. 19.
- Os dados do mapa de sequenciamento para o genoma correspondente utilizando bowtie2 ou qualquer curto-alinhador de leitura confiável. Definir variando-size, janelas cromossômicas igual de cobertura como segmentos cobertos por 200 lê na fração G1 e contar fase S lê nas mesmas janelas.
- Calcula-se a relação S / G1 para cada janela. Isto deve gerar um mapa com grandes flutuações da razão S / G1 ao longo do genoma (Figura 3). Um bom controle para a confiabilidade das medidas ToR é comparar este mapa para a relação G1 / G1 (de duas medidas separadas do G1), que deve ser muito mais plana.
- Normalizar os dados para 0 média e 1 SD subtraindo cada value o valor médio de todas as janelas (excluindo o cromossoma X), e dividindo o resultado pelo desvio padrão do S / G1 de todas as janelas. Isto é feito, a fim de converter-se ao escore z e permitir a comparação entre os diferentes experimentos.
- Remover todas as regiões constantes de hiato pelo navegador genoma do UCSC, bem como entre cada fragmento diferença remanescente contendo menos de 15 janelas de dados.
- Suavizar os fragmentos restantes com uma spline suavização cúbico através da função csaps Matlab com um parâmetro de 10 - 16 e interpolar em pontos fixos cada 100 kb.
NOTA: Parâmetros de alisamento e a interpolação deve ser ajustada com base na profundidade dos dados. Existem outros métodos de suavização adequados e as funções e pode ser usado. - Depois de confirmar visualmente a fiabilidade de cada réplica, mesclar todas as leituras e calcular um perfil mais profunda resolução realizando o mesmo processo descrito acima nesta dados.
Resultados
Um mapa típico TdR é mostrado na Figura 3 para fibroblastos de rato embrionários (MEFs). Esta figura demonstra o processo de análise, uma vez que mostra ambos os pontos, que são a razão E / S para G1 normalizada janelas individuais (passo 8.3), bem como a linha que resulta da interpolação e alisamento cúbico (passo 8.5).
Tais mapas capturar a organização do programa de replicação, que é uma colch...
Discussão
CNR-TdR pode ser realizado, em princípio, em qualquer população de células em proliferação eucariótica que pode ser dividida por FACS para fases G1 e S (revisto por Rhind N. e Gilbert 20 DM). O método aqui descrito foi adaptado para células de mamífero com um tamanho de genoma de ~ 3 Gb, tais como humano e de ratinho. Pequenas mudanças no protocolo CNR-ToR (em preparação celular e profundidade sequenciamento) são necessários, a fim de ajustá-lo para outros eucariotas. Deve ser dad...
Divulgações
No conflicts of interest declared.
Agradecimentos
Agradecemos Oriya Vardi de assistência na geração de números. Trabalho no grupo IS foi apoiado pelo Israel Science Foundation (concessão No. 567/10) e do Conselho Europeu de Investigação Começando Grant (# 281306).
Materiais
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Referências
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados