
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Determinación de todo el genoma de los mamíferos sincronización de replicación de ADN por medición del contenido
En este artículo
Resumen
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Resumen
La replicación del genoma se produce durante la fase S del ciclo celular en un proceso altamente regulado que asegura la fidelidad de la duplicación del ADN. Cada región genómica se replica en un momento distinto durante la fase S a través de la activación simultánea de múltiples orígenes de replicación. Tiempo de replicación (TdR) se correlaciona con muchas características genómicas y epigenéticos y está vinculada a las tasas de mutación y cáncer. La comprensión de la vista genómico completo del programa de réplica, en la salud y la enfermedad es un importante objetivo futuro y desafío.
En este artículo se describe en detalle la "Relación de número de copia S / G1 para el mapeo genómico Tiempo de replicación" método (denominado en el presente documento: CNR-TdR), un enfoque simple para mapear el genoma amplia TdR de células de mamíferos. El método se basa en las diferencias de número de copias entre las células de fase S y las células en fase G1. El método CNR-TdR se lleva a cabo en 6 pasos: 1. Preparación de las células y la tinción con yoduro de propidio (PI); 2. Sorting G1 y células en fase S utilizando células activadas por fluorescencia (FACS); 3. purificación de ADN; 4. La sonicación; 5. Preparación y secuenciación Biblioteca; y 6. El análisis bioinformático. El método CNR-TdR es un método rápido y fácil que resulta en mapas detallados de replicación.
Introducción
la replicación del ADN de los mamíferos está estrechamente regulada para asegurar la replicación precisa de cada cromosoma exactamente una vez durante el ciclo celular. La replicación se realiza de acuerdo con un orden altamente regulado - varias grandes regiones genómicas (~ Mb) se replican en el inicio de la fase S (a principios de replicar dominios), mientras que otras regiones genómicas replican luego en (dominios medias y replicar tarde) medio o fase S tardía 1. La mayor parte del genoma se replica al mismo tiempo en todos los tejidos (dominios TdR constitutivo), mientras que 30% - 50% del genoma, cambia su TdR entre los tejidos 2, durante la diferenciación de 3, 4 y en menor medida también durante la transformación del cáncer 5 . Por otra parte, ciertas regiones genómicas se replican de forma asíncrona 6, 7, 8, es decir, hay una diferenciaen los términos de referencia entre los dos alelos.
TdR se correlaciona con muchas características genómicas y epigenómicos incluyendo los niveles de transcripción, contenido de GC, el estado de la cromatina, la densidad de genes, etc. 1, 9. TdR también se asocia con tasas de mutación y los tipos 10, 11 y por lo tanto era de esperar, perturbaciones del programa de réplica están ligados al cáncer 12, 13. La relación causal entre TdR y la estructura de la cromatina todavía no se entiende. Es posible que la cromatina abierta facilita la replicación temprana. Sin embargo, un modelo alternativo sugiere que la cromatina se monta durante la replicación y los diferentes reguladores de la cromatina presentes en el inicio y final de adelanto de fase S a diferencial de embalaje de las regiones de replicación temprana y tardía 1, 14 . Recientemente hemos demostrado que los TdR da forma al contenido de GC al afectar el tipo de mutaciones que se producen en diferentes regiones genómicas 11.
Hibridación in situ fluorescente (FISH) es el principal método para la medición de TdR en loci individual. Se lleva a cabo simplemente contando el porcentaje de células en fase S que exhiben las señales de FISH individuales vs. el porcentaje de dobletes para un determinado alelo 15, 16. Un método alternativo, se compone de pulso etiquetado de ADN con BrdU, las células de clasificación de acuerdo con su contenido de ADN a múltiples puntos de tiempo a lo largo de S, la inmunoprecipitación de ADN que contiene BrdU, y la comprobación de la abundancia de ADN precipitado con qPCR 17.
mapeo TdR genómico se puede lograr mediante dos métodos. El primer método es una versión genómica del método basado en BrdU-IP se ha descrito anteriormente, en el que la cuantificación de la cantidadde ADN precipitado en cada fracción se realiza de forma simultánea para todo el genoma a través de la hibridación de microarrays o por secuenciación profunda. El segundo método, CNR-TdR, se basa en medir el número de copias de cada región genómica de células en fase S y la normalización por el contenido de ADN en las células G1. En este método, las células están ordenados por FACS en no replicante (fase G1) y replicar (fase S) grupos (Figura 1). Las células en G1 tienen el mismo número de copias de todas las regiones genómicas y por lo tanto su contenido de ADN deben ser los mismos. Por otro lado, el número de copias de ADN en S depende de la TdR, ya que las regiones de replicación primeros se sometieron a la replicación en la mayoría de las células y por lo tanto su contenido de ADN se duplica, mientras que las regiones de replicación finales no se han replicado todavía en la mayoría de las células y por lo tanto su contenido de ADN se ser similar a la de las células G1. Por lo tanto la S a la proporción de G1 del contenido de ADN es indicativa de la TdR. La cantidad de ADN para cada región genómica se mide por hibridación amicroarrays o mediante secuenciación profunda 2, 8. Se discutirán además las ventajas del método CNR-TdR.
Este artículo describe el método de CNR-TdR para el mapeo genómico TdR como se describe en la Figura 2. El documento analiza los detalles finos de todo el proceso desde la recolección de células hasta que el análisis básico de los resultados y la creación de mapas genómicos Tor. El protocolo descrito en este documento se ha realizado con éxito en diversos tipos de células que crecen en cultivo. Mejoras futuras de este protocolo pueden dar lugar a la asignación de los TdR in vivo e in tipos de células raras.
Protocolo
Nota: TdR se puede medir sólo en cultivo, las células no sincronizadas. El procedimiento debe comenzar con al menos 1 - 2 x 10 6 células de crecimiento rápido, que dará lugar generalmente a ~ 1 x 10 5 células en fase S (el paso limitante de la velocidad). Se recomienda llevar a cabo cada experimento usando dos o tres repeticiones. Todo el proceso de CNR-TdR se puede completar en una semana - dos días deben estar dedicados a todos los pasos hasta la preparación de la biblioteca, se necesitan uno o dos días para la secuenciación y es necesario un día adicional para el análisis de datos inicial.
1. Recolección de células de Cultura
NOTA: El protocolo está escrito para las células que crecen en cultivo en placas de 10 cm (que contenían aproximadamente 2 - 5 x 10 6 células), pero se puede ajustar fácilmente a otras plataformas.
- Para las células que se cultivaron en suspensión, proceder a la fijación (sección 2).
- Para células adherentes, aspirar y lavar la plcomió con 3 ml de PBS sin Ca2 + y Mg2 +.
- Desechar el PBS y se incuban las células durante 5 min a 37 ° C con 1 ml de tripsina-EDTA comercial hasta que las células se separan.
NOTA: La duración del tratamiento con tripsina se debe ajustar a cada tipo de célula. - Añadir 3 ml de medios de cultivo para neutralizar la tripsina y recoger las células en un tubo cónico de 15 ml o un tubo de poliestireno de 5 ml. Mantener en hielo.
2. Fijación
NOTA: Para esta parte, todas las medidas se debe hacer a 4 ° C.
- Centrifugar las células a 300 xg durante 5 min a 4 ° C.
- Aspirar y lavar las células dos veces con 1 ml de PBS frío.
- Resuspender las células en 250 l (total) de PBS frío.
- Mientras vórtex suavemente el tubo, añadir lentamente gota a gota 800 l de -20 ° C 100% de etanol. Esto conduce a una concentración de etanol final de 70 - 80%.
NOTA: alta pureza etanol se recomienda en esta etapa. - Se incuban las células en hielo for 30 min.
NOTA: En esta etapa las células se pueden mantener durante unos días a 4 ° C, o durante unos meses a -20 ° C.
3. La tinción PI
- Centrifugar las células a 500 xg durante 10 min a 4 ° C.
- Aspirar el sobrenadante con cuidado y lavar las células dos veces con 1 ml de PBS frío.
- Aspirar y resuspender cada muestra con la siguiente mezcla: 1 ml de PBS, 5 l 10 mg / ml ARNasa A, 50 l 1 mg / ml de yoduro de propidio (PI; botella de la mezcla antes de su uso). Ajuste la concentración hasta ~ 2 x 10 6 células / ml).
NOTA: Mantener fuera de la luz - PI es sensible a la luz. - Filtrar a través de 35 micras de malla a un tubo de poliestireno de 5 ml, y se cierra con Parafilm.
- Se incuba a RT en la oscuridad, por 15 - 30 min.
NOTA: Las células están ya listos para el análisis FACS. Si es necesario, las células teñidas se pueden almacenar durante al menos 24 horas a 4 ° C en la oscuridad.
4. Ordenar
- Ordenar las células utilizando una máquina de FACS. Nose el láser 561 nm para diferenciar las células en función de su intensidad PI. Otros láser cerca del 535 nm de excitación máxima como 488 nm o 532 se pueden utilizar dependiendo de la configuración de la máquina FACS.
- Para obtener resultados óptimos, utilice la boquilla más pequeña recomendado para el tamaño específico de célula (para la mayoría de las células de 85 micras). Dar prioridad a los modos de pureza más de rendimiento. Utilice un flujo lento constante, por lo general hasta 300-500 eventos / s, con una presión de funda de 45 psi.
- El uso de compuerta, discriminar las células muertas y los desechos subcelular (FCS bajo y alto SSC) mediante el trazado de FCS frente al SSC. A partir de las células viables discriminar dobletes trazando SSC-Ancho (W) frente al SSC-Altura (H) seguido de una FSC-W vs parcela FSC-H y por PI-W vs PI-H (dobletes tendrá el mismo valor H pero más grande W-valor). Para las células individuales viables dibujar un histograma de la intensidad de PI-área (A) que representa el contenido de ADN de las células.
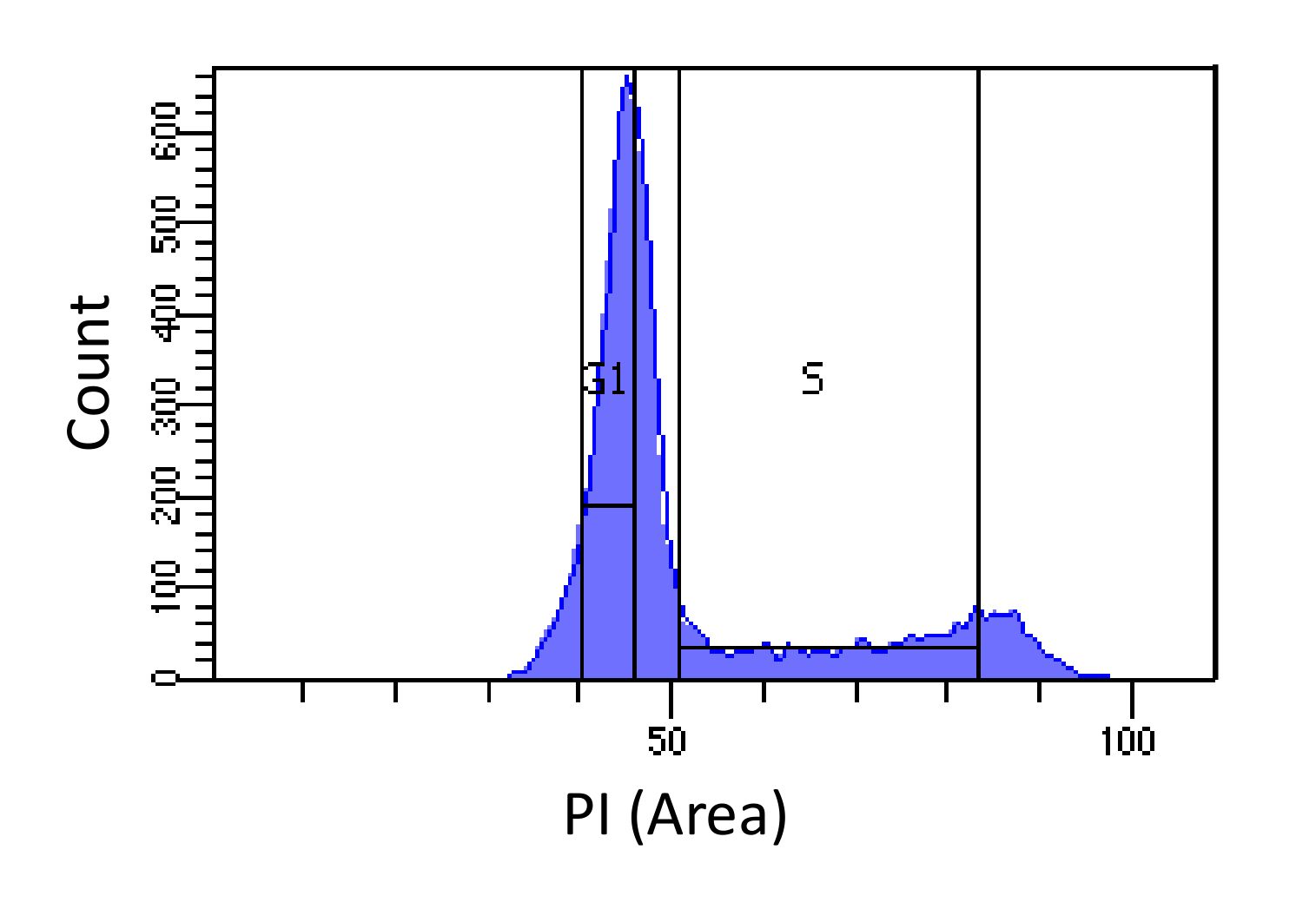
- Células Ordenar en fases G1 y S, como se muestra en la Figura 1. Compuerta de S debe ser amplia y entrometerse en las fases G1 y G2, G1, mientras compuerta debe ser estrecha y tan lejos de S como sea posible.

Figura 1. determinación de la fase del ciclo celular basada en la intensidad de PI. Histograma que muestra la distribución del contenido de ADN celular (medida por PI-Area) de ratón fibroblastos de embriones de población (MEF). El contenido de ADN se utiliza para clasificar la población en dos subpoblaciones i) células G1 (contenido de ADN 2N) y ii) las células en fase S (2N - contenido de ADN 4N), usando las regiones marcadas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
NOTA: La finalidad de la recogida de las células G1 es dar cuenta de sesgos en la eficiencia de la secuenciación entre diferentes regiones genómicas. Una aplicación alternativaRoach es utilizar células detenidas G1 del mismo tipo de células. Este enfoque da resultados más limpios (ya que minimiza la contaminación de la fase S), pero puede introducir sesgos derivados de las diferencias genéticas entre las células detenidas y las células de medición.
- Ordenar en condiciones de frío y recoger células clasificadas en 1,5 / 2 ml tubos. Mantenga los tubos en hielo después de la especie.
NOTA: Con el fin de mejorar la recuperación de ADN, es mejor utilizar tubos de unión de baja o de utilizar tubos recubiertos con 4-5% de BSA durante 1-3 horas a 4 ° C 18.
5. Purificación de ADN
- Para cada muestra (G1 y S) purificar ADN utilizando un kit de purificación de ADN.
NOTA: Cuando se utiliza el kit comercial, eluir con 400 l de tampón de elución en un tubo de 2 ml de alto rendimiento, según lo recomendado por el fabricante. - Compruebe la concentración de ADN utilizando fluorómetro.
NOTA: A partir de 100.000 células de mamíferos recogidos por la FACS, se debe obtener ~ 1 g de ADN. UNt este ADN etapa se puede mantener por unos días a 4 ° C oa -20 ° C durante un almacenamiento prolongado.
6. La sonicación
- La transferencia de ADN en un tubo de 1,7 ml que es compatible con el soporte magnético utilizado.
- Concentrado de ADN usando perlas 2x SPRI de acuerdo con las instrucciones del fabricante, utilizando un soporte magnético, y se eluye en tampón de elución de 50 l.
- ADN cizalla con una de ultrasonidos enfocados a un tamaño medio de pico blanco de 250 pb. Utilice los siguientes parámetros para una muestra de ADN 50 l: 50 W, 20% Factor de servicio, 200 ciclos por ráfaga, 20 ° C, 120 s.
- Verificar el tamaño del ADN cortado por electroforesis. La distribución del tamaño recomendado es de 200 a 700 pb con un pico a ~ 250 pb.
NOTA: En esta etapa el ADN se puede mantener por unos días a 4 ° C oa -20 ° C durante un almacenamiento prolongado.
7. Biblioteca Preparación y secuenciación
NOTA: Muchos kits de preparación de la biblioteca y difierenplataformas de secuenciación tes deben funcionar de manera similar a los utilizados por nosotros y que se mencionan en la sección de materiales. En realidad, en el pasado, mapas TdR se generaron utilizando un método muy similar con plataformas de microarrays 2.
- Preparar las bibliotecas utilizando cualquier kit de preparación de bibliotecas de tipo comercial.
- Al final de la preparación de la biblioteca, seleccione el tamaño usando perlas magnéticas para 300 personas - 800 pb.
- Después de preparar bibliotecas, medir la concentración de ADN utilizando fluorómetro.
- Medir el tamaño de ADN mediante electroforesis.
- Realizar la secuenciación en cualquier plataforma.
NOTA: La secuenciación al menos 10 M lee por muestra se recomienda. Esta profundidad es equivalente a una lectura aproximadamente cada 300 pb (para ~ 3 genomas de tamaño GB), y es suficiente para las mediciones de TdR a una resolución de 50 - 100 kb. El aumento de la profundidad dará lugar a una disminución en el tamaño de las ventanas y por lo tanto permitirá aumento de la resolución con mayor certeza. secuenciación final emparejado no es necesario eneste protocolo ya que la información se recoge solamente la cobertura. Es, sin embargo, puede ayudar en la resolución de la ubicación de lecturas que contienen secuencias repetitivas.
8. Análisis
NOTA: El análisis de datos se basa en el método utilizado por A. Koren et al. 19.
- Los datos del mapa de secuenciación del genoma a la correspondiente usando bowtie2 o cualquier alineador de lectura corta fiable. Definir la variación de tamaño, de igual cobertura ventanas cromosómicas como segmentos cubiertos por 200 lee en la fracción G1 y cuentan fase S se lee en las mismas ventanas.
- Calcular la relación S / G1 para cada ventana. Esto debería generar un mapa con grandes fluctuaciones en la relación S / G1 a lo largo del genoma (Figura 3). Un buen control de la fiabilidad de las mediciones TdR es comparar este mapa a la relación de G1 / G1 (a partir de dos mediciones separadas de G1) que debe ser mucho más plana.
- Normalizar los datos a 0 y 1 significan SD restando de cada value el valor medio de todas las ventanas (excluyendo el cromosoma X) y dividiendo el resultado por la desviación estándar de la S / G1 de todas las ventanas. Esto se hace con el fin de convertir a las puntuaciones z y permitir la comparación entre los diferentes experimentos.
- Retirar todas las regiones brecha enumerados por la UCSC genoma navegador, así como entre cada fragmento diferencia restante que contiene menos de 15 ventanas de datos.
- Suavizar los fragmentos restantes con una spline cúbica de suavizado a través de la función de Matlab CSAPs con un parámetro de 10 - 16 e interpolar los puntos de ajuste en cada 100 kb.
NOTA: Los parámetros de suavizado y la interpolación se deben ajustar en base a la profundidad de los datos. Otros métodos de suavizado adecuados y funciones existen y pueden ser utilizados. - Después de confirmar visualmente la fiabilidad de cada repetición, fusionar todas las lecturas y calcular un perfil más profundo resolución llevando a cabo el mismo proceso descrito anteriormente en estos datos.
Resultados
Un mapa típico TdR se muestra en la Figura 3 para los fibroblastos embrionarios de ratón (MEFs). Esta figura demuestra el proceso de análisis, ya que muestra tanto los puntos, que son la razón normalizada / G1 S para las ventanas individuales (paso 8.3), así como la línea que resulta de la suavización cúbico y la interpolación (paso 8.5).
Tales mapas capturan la organización del programa de replicaci...
Discusión
CNR-TdR se puede realizar, en principio, en cualquier población de células en proliferación eucariota que puede ser dividido por FACS a S y fases G1 (revisado por Rhind N. y Gilbert DM 20). El método descrito aquí se ha ajustado a células de mamífero con un tamaño de genoma de ~ 3 Gb tales como humanos y de ratón. se necesitan pequeños cambios en el protocolo CNR-TdR (en preparación de células y la profundidad de secuenciación), con el fin de ajustarla a otros eucariotas. Se debe pre...
Divulgaciones
No conflicts of interest declared.
Agradecimientos
Agradecemos a Oriya Vardi de asistencia en la producción de las figuras. El trabajo del grupo se fue apoyado por la Fundación de Ciencias de Israel (subvención Nº 567/10) y el Consejo Europeo de Investigación subvenciones de inicio (# 281306).
Materiales
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Referencias
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados