
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Détermination de l'échelle du génome de Timing de réplication mammalienne par l'ADN Mesure Contenu
Dans cet article
Résumé
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Résumé
La réplication du génome se produit pendant la phase S du cycle cellulaire dans un processus hautement régulé qui assure la fidélité de la duplication de l'ADN. Chaque région génomique est répliqué à un moment distinct lors de la phase S par l'activation simultanée de multiples origines de réplication. Temps de réplication (TDR) est en corrélation avec de nombreuses caractéristiques génomiques et épigénétiques et est liée à des taux de mutation et le cancer. Comprendre la vue complète du génome du programme de réplication, de la santé et de la maladie est un objectif de l'avenir et un défi.
Cet article décrit en détail le "Rapport de copie Nombre de S / G1 pour la cartographie Temps génomique de réplication" méthode (appelée ici: CNR-TdR), une approche simple pour cartographier le génome large TdR de cellules de mammifères. La méthode est basée sur les différences de nombre de copies entre les cellules en phase S et les cellules en phase G1. La méthode CNR-TdR est effectué en 6 étapes: 1. Préparation des cellules et la coloration avec l'iodure de propidium (PI); 2. Sørting G1 et des cellules en phase S à l'aide de cellules activées par fluorescence (FACS); 3. purification d'ADN; 4. sonication; 5. préparation et séquençage Bibliothèque; et 6. L'analyse bioinformatique. La méthode CNR-TdR est une approche simple et rapide qui se traduit par des cartes de réplication détaillées.
Introduction
la réplication de l'ADN de mammifère est étroitement régulée pour garantir la réplication précise de chaque chromosome exactement une fois au cours du cycle cellulaire. La réplication se produit selon un ordre très réglementé - plusieurs grandes régions génomiques (~ Mb) répliquent au début de la phase S (réplication précoce des domaines) , tandis que d' autres régions génomiques reproduisent plus tard , au milieu ou en phase S tardive (milieu et fin répliquant les domaines) 1. La majeure partie du génome réplicats en même temps dans tous les tissus (domaines TdR constitutifs), tandis que 30% - 50% du génome, change TdR entre les tissus 2, au cours de la différenciation 3, 4 et , dans une moindre mesure , au cours de la transformation cancéreuse 5 . Par ailleurs, certaines régions génomiques se répliquent de manière asynchrone 6, 7, 8, à savoir il y a une différencedans les TdR entre les deux allèles.
TdR est en corrélation avec de nombreuses caractéristiques génomiques et épigénomiques y compris les niveaux de transcription, le contenu GC, l' état de la chromatine, la densité de gènes, etc. 1, 9. TdR est également associée à des taux et des types 10, 11 et donc sans surprise la mutation, des perturbations du programme de réplication sont liés au cancer 12, 13. La relation de cause à effet entre les TdR et la structure de la chromatine est pas encore compris. Il est possible que la chromatine ouverte facilite la réplication précoce. Cependant, un autre modèle suggère que la chromatine est assemblé lors de la réplication et les différents régulateurs de chromatine présents au début et à la fin de la phase S conduisent à différentiel emballage des régions de réplication précoce et tardive 1, 14 . Nous avons récemment montré que le TdR façonne la teneur en GC en affectant le type de mutations qui se produisent dans différentes régions du génome 11.
Fluorescence hybridation in situ (FISH) est la principale méthode de mesure TDR à des loci particuliers. Elle est réalisée simplement en comptant le pourcentage de cellules en phase S qui présentent des signaux de poissons isolés par rapport au pourcentage de doublets pour un allèle donné 15, 16. Une autre méthode est constitué d'impulsions marquage de l'ADN avec du BrdU, le tri des cellules en fonction de leur teneur en ADN à plusieurs points temporels le long de S, immunoprécipitation ADN contenant BrdU, et la vérification de l'abondance de l' ADN précipité avec 17 qPCR.
TdR cartographie génomique peut être réalisée par deux méthodes. La première méthode est une version génomique de la méthode basée sur la BrdU IP décrite ci-dessus, dans lequel la quantification de la quantitéde l'ADN précipité dans chaque fraction est effectuée simultanément pour l'ensemble du génome par hybridation à microarrays ou par séquençage profond. La deuxième méthode, CNR-TdR, est basé sur la mesure du nombre de copies de chaque région génomique de cellules en phase S et en normalisant par la teneur en ADN dans les cellules G1. Dans cette méthode, les cellules sont triées par FACS dans la non-répliquant (phase G1) et la reproduction (phase S) des groupes (figure 1). Les cellules en G1 ont le même nombre de copies dans toutes les régions du génome et par conséquent leur teneur en ADN devraient être les mêmes. D'autre part, le nombre de copies d'ADN dans S dépend de la TdR, puisque les régions de réplication premières ont subi la réplication dans la plupart des cellules et donc leur teneur en ADN est doublé, tandis que les régions de réplication en retard ne sont pas encore repris dans la plupart des cellules et donc leur teneur en ADN similaire à celui des cellules G1. D'où le rapport S G1 du contenu ADN est indicatif du TdR. La quantité d'ADN pour chaque région génomique est mesurée soit par hybridation àmicroarrays ou par séquençage profond 2, 8. Les avantages de la méthode CNR-TdR seront discutés.
Le présent document décrit la méthode CNR-TdR pour la cartographie génomique TdR comme décrit dans la figure 2. Le document traite des détails fins de l'ensemble du processus de collecte de cellules jusqu'à ce que l'analyse de base des résultats et la création de cartes TdR génomiques. Le protocole décrit dans le présent document a été réalisé avec succès sur différents types de cellules en culture. Les améliorations futures de ce protocole peuvent conduire à la cartographie des TdR in vivo et dans des types cellulaires rares.
Protocole
Remarque: TdR peut être mesurée uniquement sur la croissance, les cellules non synchronisées. La procédure devrait commencer avec au moins 1 - 2 x 10 6 cellules à croissance rapide, ce qui se traduira généralement en ~ 1 x 10 5 cellules en phase S (l'étape de limitation du débit). Il est recommandé d'effectuer chaque expérience en utilisant deux ou trois répétitions. L'ensemble du processus CNR-TdR peut être complété en une semaine - deux jours devraient être consacrées à toutes les étapes jusqu'à la préparation bibliothèque, un à deux jours sont nécessaires pour le séquençage et une journée supplémentaire est nécessaire pour l'analyse initiale des données.
1. Collecte de cellules provenant de la culture
NOTE: Le protocole est écrit pour la croissance des cellules en culture dans des plaques de 10 cm (contenant environ 2 - 5 x 10 6 cellules), mais peut être facilement ajustée à d' autres plates - formes.
- Pour les cellules qui ont été cultivées en suspension, procéder à la fixation (section 2).
- Pour les cellules adhérentes, aspirer et laver le plmangé avec 3 ml de PBS sans Ca 2+ et Mg 2+.
- Jeter le PBS et on incube les cellules pendant 5 min à 37 ° C avec 1 ml de trypsine-EDTA commercial jusqu'à ce que les cellules se détachent.
REMARQUE: La durée du traitement à la trypsine doit être ajustée pour chaque type de cellule. - Ajouter 3 ml de milieux de culture pour neutraliser la trypsine et de recueillir des cellules dans un tube de 15 ml conique ou un tube 5 ml de polystyrène. Gardez sur la glace.
2. Fixation
NOTE: Pour cette partie, toutes les étapes devrait être fait à 4 ° C.
- Centrifuger les cellules à 300 xg pendant 5 min à 4 ° C.
- Aspirer et laver les cellules deux fois avec 1 ml de PBS froid.
- Resuspendre les cellules dans 250 pi (total) PBS froid.
- Alors que tourbillonner doucement le tube, ajouter lentement goutte à goutte 800 pi de -20 ° C à 100% d'éthanol. Cela conduit à une concentration finale d'éthanol de 70-80%.
NOTE: l'éthanol de haute pureté est recommandé à cette étape. - Incuber les cellules sur la glace for 30 min.
NOTE: A ce stade de cellules peuvent être conservées pendant quelques jours à 4 ° C ou pendant quelques mois à -20 ° C.
3. PI Staining
- Centrifuger les cellules à 500 xg pendant 10 min à 4 ° C.
- Aspirer le surnageant avec précaution et laver les cellules deux fois avec 1 ml de PBS froid.
- Aspirer et remettre en suspension chaque échantillon avec le mélange suivant: 1 ml de PBS, 5 ul de 10 mg / ml de RNase, 50 ul de 1 mg / ml d'iodure de propidium (PI; bouteille mélanger avant utilisation). Ajuster la concentration jusqu'à ~ 2 x 10 6 cellules / ml).
NOTE: Conserver hors de la lumière - PI est sensible à la lumière. - Filtrer à travers 35 pm maille à un tube 5 ml de polystyrène, et à proximité avec Parafilm.
- Incubation à température ambiante dans l'obscurité, pendant 15 - 30 min.
NOTE: Les cellules sont maintenant prêtes pour l'analyse FACS. Si nécessaire, les cellules colorées peuvent être conservées pendant au moins 24 heures à 4 ° C dans l'obscurité.
4. Trier
- Trier les cellules en utilisant une machine FACS. Nouse le laser 561 nm pour différencier les cellules en fonction de leur intensité de PI. D'autres lasers proches de l'excitation maximale de 535 nm, 488 nm ou comme 532 peuvent être utilisés en fonction de la configuration de la machine FACS.
- Pour des résultats optimaux, utilisez la plus petite buse recommandé pour la taille de la cellule spécifique (pour la plupart des cellules de 85 pm). Prioriser modes de pureté plus le rendement. Utiliser un débit lent stable, habituellement jusqu'à 300-500 événements / s, avec une pression de la gaine de 45 psi.
- Utilisation de gating, discriminer les cellules mortes et les débris subcellulaire (faible FCS et de haute SSC) par Traçage FCS vs SSC. A partir des cellules viables discriminer doublets en traçant SSC-Largeur (W) vs SSC-Hauteur (H) suivie d'une FSC-W vs FSC-H parcelle et par PI- W vs PI-H (doublets aura la même valeur H mais plus grande valeur W). Pour les cellules individuelles viables dessiner un histogramme de l'intensité PI-zone (A) qui représente la teneur en ADN des cellules.
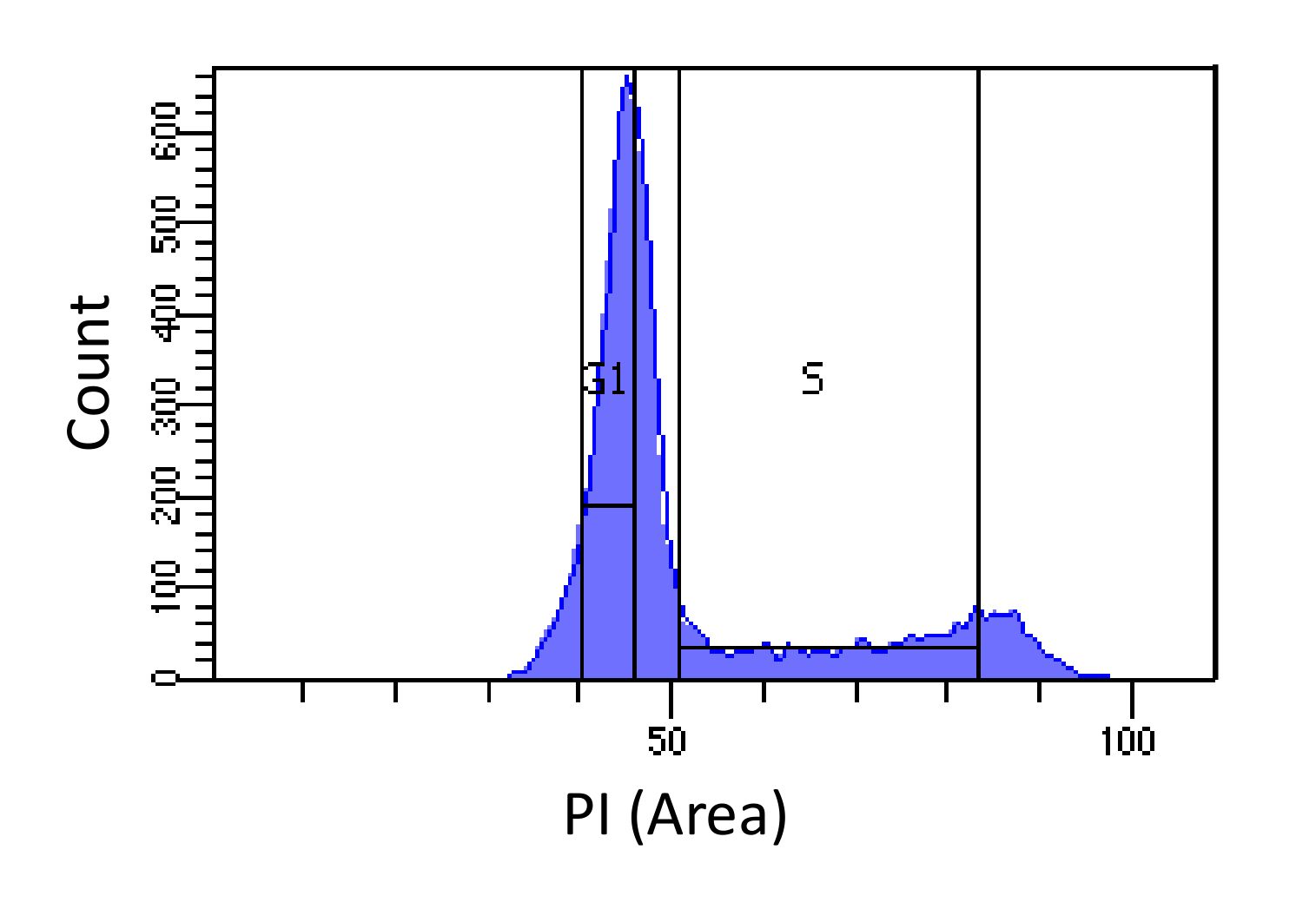
- Trier les cellules en G1 et S phases, comme le montre la figure 1. Gating S devrait être large et intrusion dans les phases G1 et G2, tandis que G1 gating devrait être étroite et aussi loin de S que possible.

Figure 1. Cellule détermination de phase de cycle basée sur l' intensité de la PI. Histogramme montrant la distribution de la teneur en ADN cellulaire (mesurée par PI-Area) de souris fibroblaste embryonnaire (MEF) population. La teneur en ADN est utilisé pour trier la population en deux sous-populations i) les cellules G1 (teneur en ADN de 2N) et ii) des cellules en phase S (2N - 4N teneur en ADN), en utilisant les régions marquées. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
NOTE: L'objet de la collecte des cellules G1 pour tenir compte des distorsions dans l'efficacité de séquençage entre les différentes régions génomiques. Une application de remplacementroach est d'utiliser des cellules arrêtées G1 du même type de cellule. Cette approche donne des résultats plus propres (car il minimise la contamination de la phase S), mais il peut introduire des biais résultant de différences génétiques entre les cellules arrêtées et les cellules mesurées.
- Trier dans des conditions froides et recueillir des cellules triées dans 1,5 / 2 tubes ml. Garder les tubes sur la glace après le tri.
NOTE: Afin d'améliorer la récupération de l' ADN, il est préférable d'utiliser des tubes de liaison faible ou d'utiliser des tubes revêtus de 4-5% de BSA pendant 1 à 3 h à 4 ° C 18.
5. Purification d'ADN
- Pour chaque échantillon (G1 et S) purifier l'ADN en utilisant un kit de purification d'ADN.
REMARQUE: Lorsque vous utilisez le kit commercial, éluer avec un tampon d'élution de 400 pi dans un tube de 2 mL pour un rendement élevé, tel que recommandé par le fabricant. - Vérifiez la concentration de l'ADN en utilisant fluoromètre.
NOTE: De 100.000 cellules de mammifères recueillies par le FACS, on devrait obtenir ~ 1 ug d'ADN. UNEt cet ADN de scène peut être conservé pendant quelques jours à 4 ° C ou à -20 ° C pour un stockage.
6. sonication
- Transférer de l'ADN dans un tube de 1,7 ml qui est compatible avec le support magnétique utilisé.
- Concentrer l'ADN en utilisant des billes 2x SPRI selon les instructions du fabricant, en utilisant un support magnétique, et éluer dans un tampon d'élution de 50 ul.
- ADN cisaillement avec un appareil à ultrasons focalisé à une taille cible maximale moyenne de 250 pb. Utilisez les paramètres suivants pour un échantillon d'ADN de 50 ul: 50 W, 20% Facteur de service, 200 cycles par rafale, 20 ° C, 120 s.
- Vérifier la taille de l'ADN cisaillée par électrophorèse. La distribution de la taille recommandée est de 200 - 700 pb avec un pic à ~ 250 pb.
NOTE: A ce stade de l'ADN peut être conservé pendant quelques jours à 4 ° C ou à -20 ° C pour un stockage.
7. Bibliothèque Préparation et séquençage
NOTE: De nombreux kits de préparation de la bibliothèque et diffèrentplates-formes de séquençage ent devraient fonctionner de façon similaire à ceux utilisés par nous et mentionnés dans la section des matériaux. En fait , dans le passé, les cartes TdR ont été générés en utilisant une méthode très similaire avec les plates - formes de biopuces 2.
- Préparer les bibliothèques en utilisant toute trousse de préparation de la bibliothèque commerciale.
- A la fin de la préparation de bibliothèque de sélectionner la taille en utilisant des billes magnétiques pour 3-800 pb.
- Après la préparation de bibliothèques, mesurer la concentration de l'ADN en utilisant fluoromètre.
- Mesurer la taille de l'ADN par électrophorèse.
- Effectuer le séquençage sur toute plate-forme.
NOTE: Le séquençage d'au moins 10 M lit par échantillon est recommandée. Cette profondeur correspond à une lecture d'environ tous les 300 pb (pour ~ 3 Gb génomes de taille), et est suffisante pour les mesures de TdR à une résolution de 50 - 100 kb. L'augmentation de la profondeur se traduira par une diminution de la taille des fenêtres et ainsi permettra à augmenter la résolution avec une certitude élevée. séquençage final Couplé est pas nécessairece protocole puisque l'information ne de couverture sont collectées. Il, cependant, peut aider à résoudre l'emplacement de lectures qui contiennent des séquences répétitives.
8. Analyse
NOTE: L' analyse des données est basée sur la méthode utilisée par A. Koren et al. 19.
- données Plan de séquençage du génome correspondant en utilisant bowtie2 ou tout alignement fiable de lecture courte. Définir variables de taille, égale-couverture fenêtres chromosomiques comme segments couverts par 200 lectures dans la fraction G1 et compter la phase S se lit dans les mêmes fenêtres.
- Calculer le rapport S / G1 pour chaque fenêtre. Cela devrait générer une carte avec de grandes fluctuations dans le rapport S / G1 long du génome (figure 3). Un bon contrôle de la fiabilité des mesures TdR est de comparer cette carte pour le rapport G1 / G1 (à partir de deux mesures distinctes de G1) qui devrait être beaucoup plus plat.
- Normaliser les données à 0 moyenne et 1 SD en soustrayant de chaque value la valeur moyenne de toutes les fenêtres (à l'exclusion du chromosome X), et en divisant le résultat par l'écart type de l'indice S / G1 de toutes les fenêtres. Ceci est fait dans le but de se convertir à z scores et de permettre la comparaison entre les différentes expériences.
- Retirer toutes les régions de l'écart énumérés par le navigateur de génome UCSC ainsi que chaque fragment d'espace inter restante contenant moins de 15 fenêtres de données.
- Lisser les fragments restants avec une spline de lissage cubique via la fonction csaps Matlab avec un paramètre de 10 - 16 et interpoler à des points de consigne 100 kb.
REMARQUE: Les paramètres de lissage et d'interpolation doit être ajustée en fonction de la profondeur des données. D'autres procédés et fonctions de lissage appropriés existent et peuvent être utilisés. - Après avoir confirmé visuellement la fiabilité de chaque répétition, fusionner toutes les lectures et calculer un profil de résolution plus profonde en effectuant le même processus décrit ci-dessus sur ces données.
Résultats
Une carte typique TdR est représenté sur la figure 3 pour les fibroblastes embryonnaires de souris (MEF). Cette figure montre le processus d'analyse, car il montre à la fois les points, qui sont le rapport E / G1 normalisé S pour les fenêtres individuelles (étape 8.3), ainsi que la ligne qui résulte du lissage cubique et d'interpolation (étape 8.5).
Ces cartes capturent l'organisation du pr...
Discussion
CNR-TdR peut être effectuée , en principe , sur une population de cellules proliférantes eucaryote qui peut être divisé par FACS à S et les phases G1 (examinées par Rhind N. et Gilbert 20 DM). La méthode décrite ici a été ajusté pour les cellules de mammifères avec une taille de génome de ~ 3 Gb tels que l'homme et la souris. De petits changements dans le protocole CNR-TdR (en préparation cellulaire et de la profondeur de séquençage) sont nécessaires, afin de l'adapter ?...
Déclarations de divulgation
No conflicts of interest declared.
Remerciements
Nous remercions Oriya Vardi pour l'aide à générer des chiffres. Le travail dans le groupe IS a été soutenu par la Fondation Israël Science (subvention n ° 567/10 du) et le Conseil européen de la recherche Starting Grant (# 281306).
matériels
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Références
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.