
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Генома Определение МЛЕКОПИТАЮЩИХ репликации ДНК с помощью Timing Content измерения
В этой статье
Резюме
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Аннотация
Репликация генома происходит во время S-фазе клеточного цикла в сильно регулируемом процессе, который обеспечивает точность дублирования ДНК. Каждый геномная область реплицируется в отдельное время во время фазы S через одновременной активации нескольких начала репликации. Время репликации (ПКВ) коррелирует со многими геномных и эпигенетических особенностей и связан с частоты мутаций и рака. Осмысливая полную геномную вид программы репликации, в здоровье и болезни является одной из основных целей будущего и вызов.
В данной статье подробно описывается "Copy Number Коэффициент S / G1 для отображения геномную времени репликации" метод (в данном документе под названием: CNR-TOR), простой подход к карте генома широкий ToR клеток млекопитающих. Метод основан на количестве копий различий между S фазы клеток и фазы G1 клеток. Метод CNR-TOR выполняется в 6 этапов: 1. Подготовка клеток и окрашивание пропидиума йодида (PI); 2. Сортин G1 и фазы S клетки с помощью флуоресцентной активированный сортировки клеток (FACS); 3. Очистка ДНК; 4. Ультразвуком; 5. Подготовка библиотеки и последовательности; и 6. Биоинформатический анализ. Метод CNR-TOR является быстрый и простой подход, который приводит к репликации подробных карт.
Введение
Млекопитающим репликации ДНК жестко регулируется, чтобы обеспечить точную репликацию каждой хромосомы только один раз в течение клеточного цикла. Репликация происходит в соответствии с жестко регламентированной порядке - несколько больших геномных областей (~ Мб) реплицировать в начале фазы S (начало репликации доменов) , тогда как другие геномные области репликации позже в средней или поздней фазе S (средние и поздние реплицирующихся доменов) 1. Большая часть генома реплицируются в то же время во всех тканях (конститутивные домены ПКВ), в то время как 30% - 50% генома, меняет свое техническое задание между тканями 2, во время дифференцировки 3, 4 , и в меньшей степени , также во время раковой трансформации 5 , Кроме того, некоторые геномные области репликации в асинхронном режиме 6, 7, 8, а именно есть разницав ПКВ между двумя аллелями.
ToR коррелирует со многими геномных и эпигеномном функций , включая уровни транскрипции, содержание GC, хроматина состояние, плотность генов и т.д. 1, 9. ToR также связан с частоты мутаций и типов 10, 11 и , следовательно , неудивительно, что возмущения программы репликации связаны с раком 12, 13. Причинно-следственная связь между ПКВ и структуры хроматина пока не понял. Вполне возможно, что открытая хроматина способствует раннему репликации. Тем не менее, альтернативная модель предполагает , что хроматин собирается во время репликации и различные регуляторы хроматина , присутствующие в начале и в конце S фазы приводят к дифференциальной упаковки ранних и поздних воспроизводящихся областей 1, 14 . Недавно мы показали , что формирует техническое задание содержание GC, влияя на тип мутаций , которые происходят в различных геномных областей 11.
Флуоресцентная гибридизация (FISH) в основной метод для измерения ПКВ на отдельных локусов. Она осуществляется просто путем подсчета процента клеток фазы S , которые обладают одной рыбы сигналы vs. процент дублетов для заданного аллель 15, 16. Альтернативный способ, состоит из последовательностей импульсов мечения ДНК , с помощью BrdU, сортировка клеток в соответствии с их содержанием ДНК в различные моменты времени по S, иммунопреципитации ДНК , содержащей BrdU, и проверять обилие осажденной ДНК с кПЦР 17.
Геномные отображение ТЗ может быть достигнуто двумя способами. Первый способ представляет собой геномную вариант метода, основанного BrdU-IP описанный выше, в котором количественное определение суммыосажденной ДНК в каждой фракции осуществляется одновременно в течение всего генома путем гибридизации с микрочипов или глубокой последовательности. Второй метод, CNR-ToR, основан на измерении числа копий каждой геномной области фазовых клеток S и нормализацию по содержанию ДНК в G1 клетках. В этом способе клетки сортируются по FACS в не-тиражирование (G1 фаза) и тиражирование (фаза S) группы (рисунок 1). Клетки в G1 имеют один и тот же номер копии всех геномных областей и, следовательно, их содержание ДНК должно быть одинаковым. С другой стороны, ДНК, число копий в S зависит от технического задания, так как ранние регионы Дублирование подверглись репликации в большинстве клеток и, следовательно, их содержание ДНК удваивается, в то время как поздние регионы Дублирование еще не реплицируются в большинстве клеток и, следовательно, их содержание ДНК будет быть похож на G1 клеток. Следовательно, S соотношение G1 содержания ДНК свидетельствует о ПКВ. Количество ДНК для каждой области генома измеряется либо путем гибридизациимикрочипов или глубокой последовательности 2, 8. будут дополнительно обсуждены преимущества метода CNR-TOR.
В данной статье описывается метод CNR-TOR для геномного картирования ToR , как описано на рисунке 2. В статье рассматриваются мелкие детали всего процесса от момента сбора клеток до базового анализа результатов и создания геномных ToR карт. Протокол, описанный в этой статье была успешно выполнена на различных типах клеток, выращенных в культуре. Дальнейшее совершенствование этого протокола может привести к отображению ПКВ в естественных условиях и в редких типов клеток.
протокол
Примечание: TOR может быть измерена только на растущих, несинхронизированных клетках. Эта процедура должна начинаться с по крайней мере , 1 - 2 х 10 6 быстро растущих клеток, которые обычно приводят к ~ 1 × 10 5 клеток в фазе S (шаг ограничение скорости). Рекомендуется проводить каждый эксперимент с использованием двух или трех повторах. Весь процесс CNR-TOR может быть завершена в течение одной недели - через два дня должен быть посвящен всем шагам до подготовки библиотеки, от одного до двух дней, необходимых для секвенирования и дополнительный день необходимо для первоначального анализа данных.
1. Сбор клеток из культуры
Примечание: Протокол написан для клеток , растущих в культуре в 10 см пластинах (содержащих примерно 2 - 5 × 10 6 клеток), но может быть легко отрегулирована на другие платформы.
- Для клеток, выращенных в суспензии, перейти к фиксации (раздел 2).
- Для прилипшие клетки, аспирация и мыть плел с 3 мл PBS без Са 2+ и Mg 2+.
- Выбросите PBS и инкубируют клетки в течение 5 мин при 37 ° С в 1 мл трипсина-коммерческого ЭДТА, пока клетки отделяться.
Примечание: Продолжительность обработки трипсином должна быть отрегулирована для каждого типа клеток. - Добавьте 3 мл культуральной носитель для нейтрализации трипсина и собирают клетки в 15 мл коническую пробирку или 5 мл полистирола трубки. Держите на льду.
2. Закрепление
Примечание: В этой части все шаги должны быть сделано при 4 ° С.
- Центрифуга клетки при 300 мкг в течение 5 мин при температуре 4 ° С.
- Отберите и мыть клетки дважды 1 мл холодного PBS.
- Ресуспендируют клеток в 250 мкл (общий) холодной PBS.
- В то время как осторожно встряхивая пробирку, медленно по каплям добавляют 800 мкл 20 ° C 100% этанола. Это приводит к конечной концентрации этанола 70 - 80%.
Примечание: Высокий этанол чистота рекомендуется на этом этапе. - Инкубируйте клетки на льду Foг 30 мин.
Примечание: На этом этапе клетки могут храниться в течение нескольких дней при температуре 4 ° С или в течение нескольких месяцев при -20 ° C.
3. PI Окрашивание
- Центрифуга клетки при 500 мкг в течение 10 мин при температуре 4 ° С.
- Аспирируйте супернатант тщательно и мыть клетки дважды 1 мл холодного PBS.
- Отберите и ресуспендирования каждый образец со следующей смеси: 1 мл PBS, 5 мкл 10 мг / мл РНКазы А, йодид пропидия 50 мкл 1 мг / мл (PI; перемешать флакон перед использованием). Отрегулируйте концентрацию до ~ 2 · 10 6 клеток / мл).
ПРИМЕЧАНИЕ: Хранить света - PI чувствителен к свету. - Фильтруют через 35 мкм меш в 5 мл полистирола трубки и близко парафильмом.
- Выдержите при комнатной температуре в темноте в течение 15 - 30 мин.
Примечание: Клетки теперь готовы для анализа FACS. При необходимости, Окрашенные клетки могут храниться в течение не менее 24 ч при температуре 4 ° С в темноте.
4. Сортировка
- Сортировка клеток с использованием машины FACS. НасЕ 561 нм лазер дифференцироваться клетки в зависимости от их интенсивности ПИ. Другие лазеры вблизи максимума возбуждения на 535 нм, как 488 нм или 532 могут использоваться в зависимости от конфигурации FACS машины.
- Для получения оптимальных результатов используйте наименьшее сопло рекомендованного для конкретного размера ячейки (для большинства клеток 85 мкм). Приоритетность режимы чистоты по сравнению с выходом. Используйте постоянный медленный поток, как правило, до 300-500 событий / с, при давлении оболочки 45 фунтов на квадратный дюйм.
- Использование стробирования, различать мертвые клетки и субклеточных мусора (FCS низкие и высокие SSC), откладывая FCS против SSC. Из жизнеспособных клеток дискриминировать дублеты путем построения графика SSC-Ширина (W) против SSC-высота (H) , а затем FSC-W против FSC-H участка и по PI- W против PI-H (дублеты будет иметь тот же H-значение но больше W-значение). Для получения жизнеспособных одиночных клеток начертить гистограмму интенсивности ПИ-зона (А), которая представляет содержание ДНК в клетках.
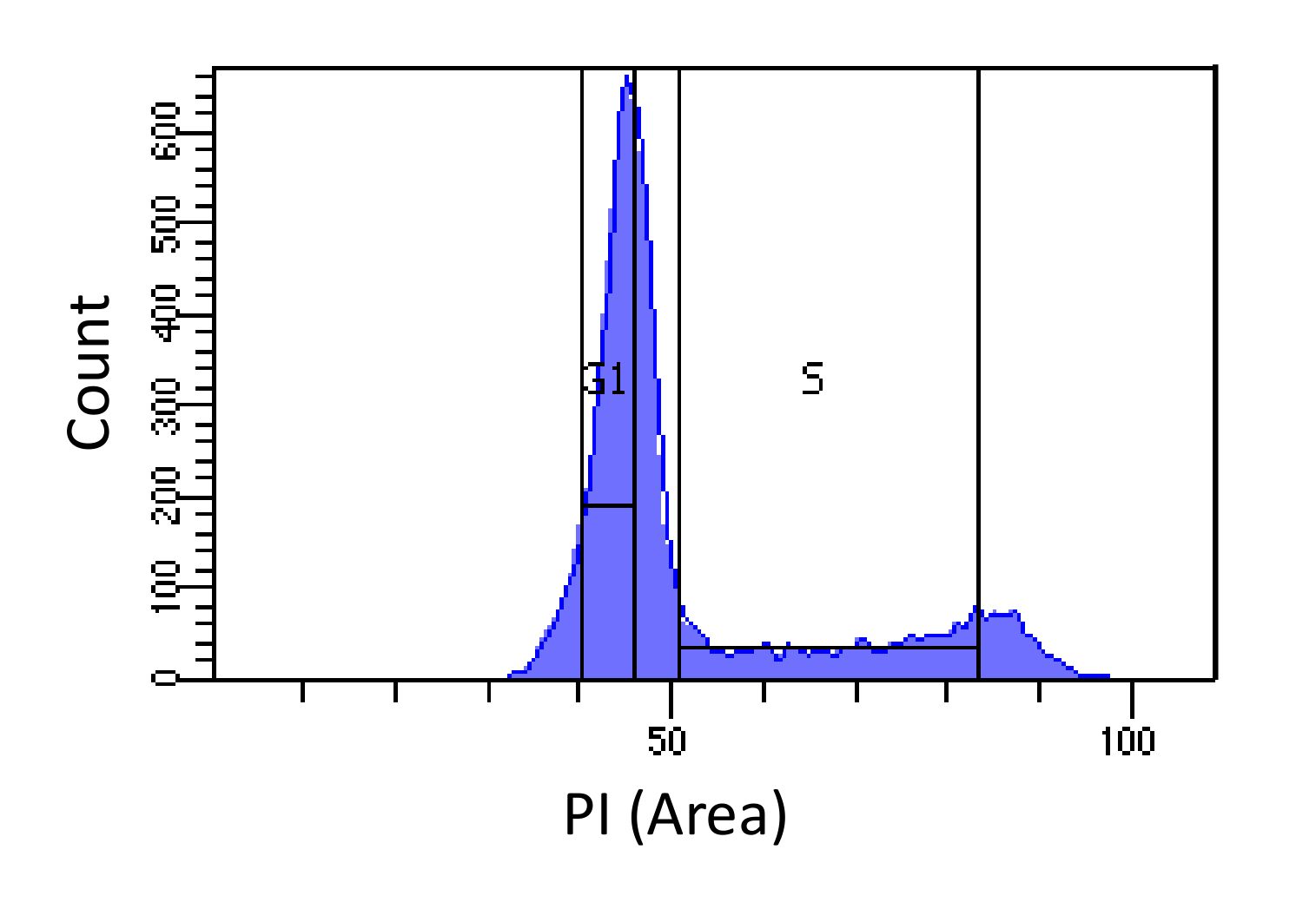
- Сортировка клеток в фазах G1 и S, как показано на рисунке 1, Gating для S должны быть широкими и вторгаться в G1 и G2 фаз, в то время как G1 стробирования должна быть узкой и как можно дальше от S, насколько это возможно.

Рисунок определение фазы цикла 1. Ячейка на основе интенсивности ПИ. Гистограмма, показывающая распределение содержания клеточной ДНК (измеряется PI-площадь) мышиных эмбриональных фибробластов (MEF) населения. Содержание ДНК используется для сортировки население на две подгруппы населения I) G1 клетки (содержание 2н ДНК) и II) S фазы клетки (2N - 4N содержание ДНК), используя отмеченные регионы. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
Примечание: Цель коллекции G1 клеток для учета погрешностей в эффективности между различными секвенирования геномных областей. Альтернативное приложениеРоач является использование G1 клеток, захваченных из того же самого типа клеток. Такой подход дает более чистые результаты (так как это сводит к минимуму загрязнение фазы S), но это может ввести предвзятости, вытекающие из генетических различий между арестованными клеток и измеренных клеток.
- Сортировка в холодных условиях и сбора отсортированных клеток в 1,5 / 2 мл пробирки. Хранить трубы на льду следующие сорта.
Примечание: Для того , чтобы улучшить восстановление ДНК, то лучше использовать низким связыванием трубы или использовать трубы с покрытием с 4 - 5% БСА в течение 1 - 3 ч при 4 ° С 18.
5. ДНК Очистка
- Для каждого образца (G1 и S) очищают ДНК с использованием набора для очистки ДНК.
Примечание: При использовании коммерческого набора, элюции 400 мкл буфера для элюции в 2 мл пробирку для высокого выхода, в соответствии с рекомендациями производителя. - Проверьте концентрацию ДНК с помощью флуорометр.
Примечание: Из 100000 клеток млекопитающих, собранных FACS, следует получить ~ 1 мкг ДНК.т этот этап ДНК может сохраняться в течение нескольких дней при температуре 4 ° С или при -20 ° С в течение длительного хранения.
6. Ультразвуком
- Перенос ДНК в 1,7 мл пробирку, который совместим с магнитной подставкой, используемой.
- Концентрат ДНК с использованием 2х SPRI бусин в соответствии с инструкциями производителя, используя магнитную стойку, и элюирования в 50 мкл буфера для элюции.
- Shear ДНК с сфокусированного ультразвукового дезинтегратора до среднего размера целевого пика 250 пар оснований. Используйте следующие параметры для образца ДНК в 50 мкл: 50 Вт, 20% коэффициент заполнения, 200 циклов в порыве, 20 ° C, 120 сек.
- Проверьте размер фрагментированной ДНК с помощью электрофореза. Рекомендуемый размер дистрибутива составляет 200 - 700 пар оснований с максимумом при ~ 250 пар оснований.
Примечание: На этом этапе ДНК может сохраняться в течение нескольких дней при температуре 4 ° С или при -20 ° С в течение длительного хранения.
7. Библиотека Подготовка и секвенирование
Примечание: Многие наборы для подготовки библиотеки и различающиесялор секвенирование платформы должны работать так же, как те, которые используются нами и упомянутые в разделе материалов. На самом деле в прошлом, ToR карты были получены с использованием очень похожий метод с микрочипов платформ 2.
- Подготовка библиотеки с использованием любого набора коммерческой подготовки библиотеки.
- По окончании подготовки библиотеки, выберите размер с помощью магнитных шариков 300 - 800 пар оснований.
- После подготовки библиотек, измерения концентрации ДНК с помощью флуорометр.
- Измерьте размер ДНК с помощью электрофореза.
- Выполните последовательность на любой платформе.
Примечание: Секвенирование по меньшей мере, 10 М читает на пробу рекомендуется. Эта глубина эквивалентно прочитанного приблизительно каждые 300 пар оснований (за ~ 3 Гб размер геномов), и является достаточным для измерения ToR при разрешении 50 - 100 кб. Увеличение глубины приведет к уменьшению размера окна и, таким образом, позволит увеличить разрешение с более высокой достоверностью. Соединенный конец последовательности не является необходимым вэтот протокол, поскольку информация только покрытия собирается. Это, однако, может помочь в решении местоположения прочтений, которые содержат повторяющиеся последовательности.
8. Анализ
Примечание: анализ данных на основе метода , используемого А. Корен и соавт. 19.
- Карта секвенирования данные соответствующему генома с использованием bowtie2 или из любого надежного чтения короткий выравнивателя. Определить изменения размера, равного охвата хромосомные окна в виде сегментов, охватываемых 200 считывает в G1 фракции и рассчитывать фазы S читает в тех же окнах.
- Рассчитать отношение S / G1 для каждого окна. Это должно создать карту с большими колебаниями в соотношении S / G1 вдоль генома (рисунок 3). Хороший контроль за достоверность измерений ToR, чтобы сравнить эту карту с соотношением G1 / G1 (от двух отдельных измерений G1), которая должна быть гораздо более пологой.
- Нормализация Данные 0 означают и 1 SD путем вычитания из каждого VALUE среднее значение всех окон (за исключением Х-хромосомы) и разделив результат на стандартное отклонение S / G1 всех окон. Это делается для того, чтобы преобразовать в г баллов и возможность сравнения между различными экспериментами.
- Удалите все щелевые участки, перечисленные в геноме браузера УСК, а также каждого оставшегося фрагмента между зазором, содержащего менее 15 окон данных.
- Гладкая оставшиеся фрагменты с кубической сглаживающего сплайна через функцию csaps Matlab с параметром 10 - 16 и интерполировать в заданных точках через каждые 100 кб.
Примечание: Параметры сглаживания и интерполяции должны быть скорректированы на основе глубины данных. Другие подходящие способы и функции сглаживающих существуют и могут быть использованы. - После того, как визуально подтверждения достоверности каждой репликации, объединить все операции чтения и вычислить более глубокий профиль разрешения, выполняя тот же процесс, описанный выше, по этим данным.
Результаты
Типичная карта ToR показана на рисунке 3 для эмбриональных фибробластов мыши (МЭФ). На этом рисунке показано, процесс анализа, так как он показывает как точки, которые являются нормализованное отношение / G1, S для отдельных окон (этап 8.3), а также линии, возникающее...
Обсуждение
CNR-TOR может быть выполнена в принципе на любой эукариотической популяции пролиферирующих клеток , которые могут быть разделены на FACS к S и G1 фазы (обзор Ринда Н. и Гилберта DM 20). Метод, описанный здесь, был скорректирован к клеткам млекопитающих с размером генома ~ 3 Gb, таких как ...
Раскрытие информации
No conflicts of interest declared.
Благодарности
Мы благодарим ория Варди за помощь в создании фигуры. Работа в группе была поддержана Израильского научного фонда (грант № 567/10) и Европейского исследовательского совета Начиная Грант (# 281306).
Материалы
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Ссылки
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены