
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Determinazione genoma a livello di timing di replicazione del DNA dei mammiferi da contenuti di misura
In questo articolo
Riepilogo
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Abstract
La replicazione del genoma si verifica durante la fase S del ciclo cellulare in un processo altamente regolato che assicura la fedeltà di duplicazione del DNA. Ogni regione genomica si replica in un momento distinto durante la fase S attraverso l'attivazione simultanea di più origini di replicazione. Tempo di replica (RPT) correla con molte caratteristiche genomiche ed epigenetici ed è collegato a tassi di mutazione e il cancro. Comprendere la visualizzazione completa del genoma del programma di replica, in salute e malattia è un importante obiettivo futuro e una sfida.
Questo articolo descrive in dettaglio il "Numero rapporto di riproduzione di S / G1 per la mappatura genomica tempo di replica" il metodo (di seguito denominato: CNR-Tor), un approccio semplice per mappare il genoma ToR di cellule di mammifero. Il metodo si basa sulle differenze del numero di copie tra le cellule in fase S e le cellule in fase G1. Il metodo CNR-TOR viene eseguita in 6 fasi: 1. Preparazione delle cellule e colorazione con ioduro di propidio (PI); 2. Sorting G1 cellule nella fase S utilizzando cellule di fluorescenza-attivato (FACS); 3. purificazione del DNA; 4. Sonicazione; 5. preparazione biblioteca e sequenziamento; e 6. L'analisi bioinformatica. Il metodo CNR-TOR è un approccio facile e veloce che si traduce in mappe dettagliate di replica.
Introduzione
replicazione del DNA Mammalian è strettamente regolata per assicurare la replica esatta di ciascun cromosoma esattamente una volta durante il ciclo cellulare. La replica avviene secondo un ordine altamente regolamentato - più grandi regioni genomiche (~ Mb) replicano all'inizio della fase S (inizio replicare i domini), mentre altre regioni genomiche replicano dopo a (domini media e tarda replicanti) medio o tarda fase S 1. La maggior parte del genoma replicati contemporaneamente in tutti i tessuti (domini ToR costitutive), mentre il 30% - 50% del genoma, cambia ToR tra tessuti 2, durante la differenziazione 3, 4 e, in misura minore, anche durante trasformazione di cancro 5 . Inoltre, alcune regioni genomiche replicano asincrono 6, 7, 8, cioè vi è una differenzanel capitolato tra i due alleli.
ToR correla con molte caratteristiche genomiche e epigenomiche compresi i livelli di trascrizione, contenuto GC, lo stato della cromatina, la densità genica, ecc 1, 9. TR è anche associato con tassi di mutazione e tipi 10, 11 e quindi non sorprende, perturbazioni del programma di replica sono collegati al cancro 12, 13. La relazione causale tra Tor e la struttura della cromatina non è ancora capito. E 'possibile che aperto cromatina facilita replica presto. Tuttavia, un modello alternativo suggerisce che la cromatina è assemblato durante la replica e le diverse autorità di regolamentazione cromatina presenti all'inizio e alla fine di piombo fase S al differenziale confezionamento delle regioni replicanti precoce e tardiva 1, 14 . Abbiamo recentemente dimostrato che i TR modella il contenuto di GC influenzando il tipo di mutazioni che si verificano in diverse regioni genomiche 11.
Ibridazione in situ fluorescente (FISH) è il metodo principale per la misurazione ToR a loci individuale. Si esegue semplicemente contando la percentuale di cellule in fase S che presentano segnali FISH singoli contro la percentuale di doppietti per un dato allele 15, 16. Un metodo alternativo, costituito da impulsi etichettatura del DNA con BrdU, l'ordinamento delle cellule in base al loro contenuto di DNA a più punti di tempo lungo S, immunoprecipitating DNA contenente BrdU, e controllando l'abbondanza di DNA precipitato con qPCR 17.
mappatura ToR genomico può essere raggiunto in due modi. Il primo metodo è una versione genomico del metodo basato BrdU-IP sopra descritta, in cui la quantificazione della quantitàdi DNA precipitato in ogni frazione viene effettuata contemporaneamente per l'intero genoma mediante ibridazione di microarray o sequenziamento. Il secondo metodo, CNR-TOR, si basa sulla misurazione il numero di copie di ciascuna regione genomica delle cellule in fase S e normalizzazione dal contenuto di DNA nelle cellule G1. In questo metodo, le cellule sono ordinati per FACS in non-replicante (fase G1) e replicare (fase S) gruppi (Figura 1). Le cellule in G1 hanno lo stesso numero di copie in tutte le regioni genomiche e quindi il loro contenuto di DNA dovrebbero essere gli stessi. D'altra parte, il numero di copie di DNA in S dipende TR, poiché le regioni replicanti primi sottoposti replicazione in maggior parte delle cellule e quindi il loro contenuto di DNA è raddoppiata, considerando che le regioni tardive replicanti non hanno ancora replicato in maggior parte delle cellule e quindi il loro contenuto di DNA sarà essere simile a quello delle cellule G1. Quindi il rapporto S G1 del contenuto di DNA è indicativa dei TR. La quantità di DNA per ciascuna regione genomica è misurata mediante ibridazionemicroarrays o dal sequenziamento profondo 2, 8. I vantaggi del metodo CNR-TR saranno ulteriormente discussi.
Questo documento descrive il metodo CNR-TOR per la mappatura genomica TR come descritto nella Figura 2. L'articolo discute i piccoli dettagli di tutto il processo dalla raccolta delle cellule fino a quando l'analisi di base dei risultati e la creazione di mappe genomiche Tor. Il protocollo descritto in questo documento è stata eseguita con successo su vari tipi di cellule in coltura. Futuri miglioramenti di questo protocollo può portare alla mappatura del capitolato in vivo e in tipi di cellule rare.
Protocollo
Nota: Tor può essere misurata solo sulla crescita, le cellule non sincronizzati. La procedura dovrebbe iniziare con almeno 1 - 2 x 10 6 cellule in rapida crescita, che di solito provoca ~ 1 x 10 5 cellule in fase S (il passo rate limiting). Si raccomanda di condurre ogni esperimento utilizzando due o tre repliche. L'intero processo di CNR-Tor può essere completato entro una settimana - due giorni dovrebbero essere dedicati a tutte le fasi fino alla preparazione biblioteca, uno o due giorni sono necessari per il sequenziamento e un giorno in più è necessario per l'analisi dei dati iniziali.
1. Raccolta delle cellule dalla cultura
NOTA: Il protocollo è scritto per cellule in crescita in coltura in piastre di 10 cm (contenente circa 2 - 5 x 10 6 cellule), ma può essere facilmente regolata per altre piattaforme.
- Per le celle che sono state coltivate in sospensione, procedere alla fissazione (sezione 2).
- Per cellule aderenti, aspirare e lavare il plmangiato con 3 ml di PBS senza Ca 2+ e Mg 2+.
- Eliminare il PBS e incubare le cellule per 5 minuti a 37 ° C con 1 ml commerciale tripsina-EDTA fino cellule si staccano.
NOTA: La durata del trattamento tripsina deve essere regolata per ciascun tipo di cellula. - Aggiungere 3 ml terreni di coltura per neutralizzare la tripsina e raccogliere le cellule in un tubo da 15 ml o una provetta 5 polistirolo. Tenere in ghiaccio.
2. Fissazione
NOTA: Per questa parte, tutti i passaggi dovrebbe essere fatto a 4 ° C.
- Centrifugare le cellule a 300 xg per 5 minuti a 4 ° C.
- Aspirare e lavare le cellule due volte con 1 ml di PBS freddo.
- Risospendere le cellule in 250 microlitri (totale) PBS freddo.
- Mentre vortex delicatamente il tubo, aggiungere lentamente goccia a goccia 800 ml di -20 ° C al 100% di etanolo. Questo porta ad una concentrazione di etanolo finale del 70 - 80%.
NOTA: Alta etanolo purezza è consigliabile in questa fase. - Incubare le cellule in ghiaccio FOr 30 min.
NOTA: A questo stadio le cellule possono essere conservate per un paio di giorni a 4 ° C o per un paio di mesi a -20 ° C.
3. PI colorazione
- Centrifugare le cellule a 500 xg per 10 min a 4 ° C.
- Aspirare il surnatante accuratamente e lavare le cellule due volte con 1 ml di PBS freddo.
- Aspirare e risospendere ogni campione con la seguente miscela: 1 ml di PBS, 5 microlitri 10 mg / mL RNaseA, 50 microlitri di 1 mg / ml di ioduro di propidio (PI; miscela flacone prima dell'uso). Regolare la concentrazione fino a ~ 2 x 10 6 cellule / ml).
NOTA: Tenere fuori dalla luce - PI è sensibile alla luce. - Filtrare 35 micron mesh una provetta 5 polistirolo, e chiudere con Parafilm.
- Incubare a RT al buio, per il 15 - 30 min.
NOTA: Le cellule sono ora pronti per l'analisi FACS. Se necessario, le cellule colorate possono essere conservate per almeno 24 ore a 4 ° C al buio.
4. Sort
- cellule ordinare utilizzando una macchina FACS. Noivia e il laser 561 nm a differenziare le cellule in base alla loro intensità PI. Altri laser vicino al 535 nm di eccitazione massima come 488 nm o 532 possono essere utilizzati a seconda della configurazione della macchina FACS.
- Per ottenere risultati ottimali, utilizzare il più piccolo ugello raccomandata per la dimensione specifica cella (per la maggior parte delle cellule di 85 micron). Dare priorità modalità di purezza oltre rendimento. Utilizzare un costante flusso lento, di solito fino a 300-500 eventi / s, con una pressione guaina 45 psi.
- Utilizzando gating, discriminare le cellule morte e detriti subcellulare (basso FCS e alta SSC) tracciando FCS vs SSC. Dalle cellule vitali discriminare doppietti tracciando SSC-Larghezza (W) vs SSC-altezza (H) seguito da un FSC-W vs plot FSC-H e PI- W PI-H vs (doppietti avrà lo stesso valore H ma più grande W-valore). Per le singole cellule vitali disegnare un istogramma dell'intensità PI-Area (A), che rappresenta il contenuto di DNA delle cellule.
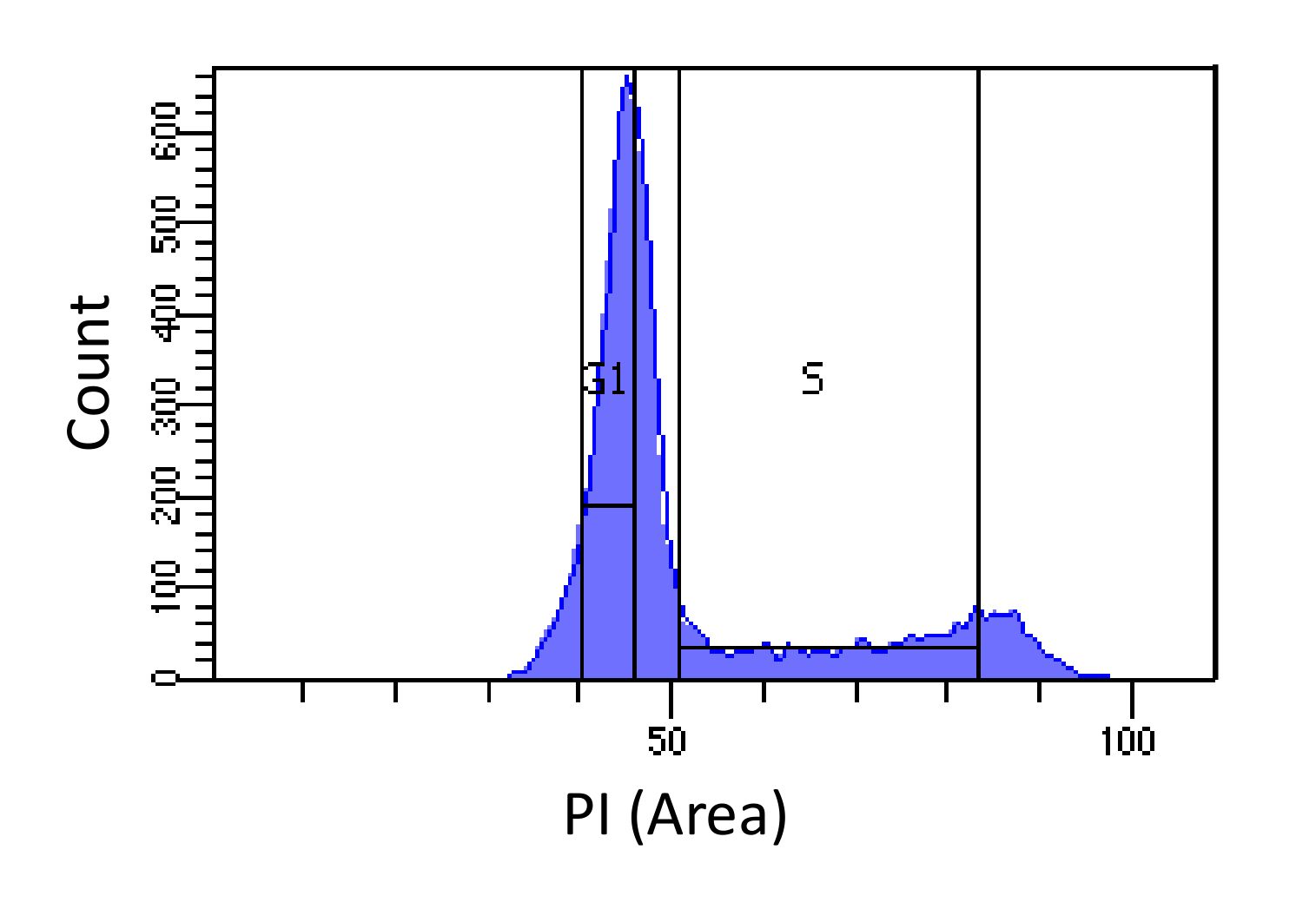
- Cellule ordinarli in fasi G1 e S, come mostrato in Figura 1. Gating per S dovrebbe essere ampia e introdursi nelle fasi G1 e G2, mentre G1 gating deve essere stretto e più lontano possibile da S possibile.

Figura determinazione fase del ciclo 1. cellulare basato sulla intensità PI. Istogramma che mostra la distribuzione del contenuto di DNA cellulare (misurata dal PI-Area) embrionali di topo fibroblasti (MEF) popolazione. Il contenuto di DNA viene utilizzato per ordinare la popolazione in due sotto-popolazioni i) cellule G1 (contenuto 2N DNA) e ii) le cellule in fase S (2N - contenuto di DNA 4N), utilizzando le regioni marcate. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
NOTA: Lo scopo della raccolta di cellule G1 è rendere conto distorsioni nel sequenziamento efficienza tra le diverse regioni genomiche. Un'applicazione alternativascarafaggio è usare cellule arrestate G1 dello stesso tipo cellulare. Questo approccio dà risultati più puliti (in quanto minimizza la contaminazione fase S), ma può introdurre distorsioni derivanti dalle differenze genetiche tra le cellule arrestate e delle celle di misura.
- Ordina in condizioni di freddo e raccogliere le cellule ordinati in 1,5 / 2 mL. Mantenere i tubi in ghiaccio dopo l'ordinamento.
NOTA: Al fine di migliorare il recupero del DNA, è meglio usare basse tubi vincolanti o usare tubi rivestiti di 4 - 5% BSA per 1 - 3 ore a 4 ° C 18.
5. DNA Purificazione
- Per ogni campione (G1 e S) purificare DNA utilizzando un kit di purificazione del DNA.
NOTA: Quando si utilizza il kit commerciale, eluire con 400 microlitri di tampone di eluizione in una provetta da 2 ml per alta resa, come raccomandato dal produttore. - Controllare la concentrazione di DNA mediante fluorimetro.
NOTA: da 100.000 cellule di mammifero raccolti dal FACS, si dovrebbe ottenere ~ 1 mg di DNA. UNt questo DNA fase può essere conservato per alcuni giorni a 4 ° C oa -20 ° C per una lunga conservazione.
6. Sonicazione
- Trasferire DNA in una provetta 1,7 ml che è compatibile con il supporto magnetico utilizzato.
- Concentrato DNA utilizzando perline 2x SPRI in base alle istruzioni del produttore, utilizzando un supporto magnetico, ed eluire in 50 microlitri di tampone di eluizione.
- DNA Shear con un ultrasonicatore mirato per una dimensione media obiettivo di picco pari a 250 bp. Utilizzare le seguenti impostazioni per un campione di DNA 50 ml: 50 W, 20% Fattore di utilizzo, 200 cicli per raffica, 20 ° C, 120 s.
- Verificare la dimensione del DNA tranciata mediante elettroforesi. La granulometria consigliata è 200 - 700 bp con un picco a ~ 250 bp.
NOTA: In questa fase il DNA può essere conservato per alcuni giorni a 4 ° C oa -20 ° C per una lunga conservazione.
7. Biblioteca Preparazione, e Sequencing
NOTA: molti kit di preparazione biblioteca e si differenzianopiattaforme di sequenziamento ent dovrebbero funzionare in modo simile a quelli usati da noi e menzionati nella sezione materiali. In realtà nel passato, mappe ToR sono stati generati utilizzando un metodo molto simile con piattaforme microarray 2.
- Preparare le librerie utilizzando qualsiasi kit di preparazione libreria commerciale.
- Alla fine della preparazione libreria, selezionare dimensioni utilizzando biglie magnetiche per 300 - 800 bp.
- Dopo aver preparato le biblioteche, misurare la concentrazione di DNA utilizzando fluorimetro.
- Misurare le dimensioni del DNA mediante elettroforesi.
- Eseguire il sequenziamento su qualsiasi piattaforma.
NOTA: Sequencing almeno 10 M legge per campione è raccomandato. Questa profondità è equivalente ad una lettura circa ogni 300 bp (per ~ 3 dimensioni genomi Gb), ed è sufficiente per misurazioni ToR ad una risoluzione di 50 - 100 kb. Aumentando la profondità si tradurrà in una diminuzione della dimensione delle finestre e quindi permetterà aumentando la risoluzione con maggiore certezza. Accoppiato sequenza finale non è necessariaquesto protocollo poiché le informazioni solo una copertura viene raccolto. E ', tuttavia, può aiutare a risolvere la posizione di legge che contengono sequenze ripetitive.
8. Analisi
NOTA: L'analisi dei dati è basata sul metodo usato da A. Koren et al. 19.
- Dati Mappa di sequenziamento ai corrispondenti genoma utilizzando bowtie2 o qualsiasi affidabile breve allineatore lettura. Definire varia grandezza, pari-copertura Windows cromosomiche come segmenti coperti da 200 legge nella frazione G1 e contano fase S legge nelle stesse finestre.
- Calcolare il rapporto S / G1 per ogni finestra. Questo dovrebbe generare una mappa con ampie variazioni del rapporto S / G1 lungo il genoma (Figura 3). Un buon controllo per l'affidabilità delle misurazioni Tor è confrontare questa mappa al rapporto G1 / G1 (da due misurazioni separate di G1) che dovrebbe essere molto più piatta.
- Normalizzare i dati a 0 media e 1 SD sottraendo dal ciascun valore il valore medio di tutte le finestre (escluso il cromosoma X) e dividendo il risultato per la deviazione standard della S / G1 di tutte le finestre. Questo viene fatto al fine di convertire in z punteggi e consentire una comparazione tra i diversi esperimenti.
- Rimuovere tutte le regioni gap elencati dal browser genoma UCSC così come ogni rimanente frammento tra divario che contiene meno di 15 finestre di dati.
- Smooth i frammenti rimanenti con una spline cubica smoothing tramite la funzione csaps Matlab con un parametro di 10 - 16 e interpolare al set point ogni 100 kb.
NOTA: Parametri di levigatura e interpolazione devono essere regolati in base alla profondità dei dati. Esistono altri metodi di smoothing adatti e funzioni e possono essere utilizzati. - Dopo la conferma visivamente l'affidabilità di ogni replicano, unire tutte le letture e calcolare un profilo più profondo risoluzione eseguendo la stessa procedura descritta in precedenza su questi dati.
Risultati
Una tipica mappa ToR è mostrato in Figura 3 per fibroblasti embrionali di topo (MEF). Questa figura illustra il processo di analisi in quanto mostra entrambi i punti, che sono normalizzate rapporto S / G1 per singole finestre (passo 8.3), così come la linea che risulta dalla levigatura cubica e interpolazione (passo 8.5).
Tali mappe catturano l'organizzazione del programma di replica, che è un mosaico d...
Discussione
CNR-tor può essere eseguita in linea di principio su una popolazione di cellule proliferanti eucariotiche che può essere diviso per FACS a S e le fasi G1 (recensito da Rhind N. e Gilbert 20 DM). Il metodo descritto qui è stato adattato per le cellule di mammifero con una dimensione del genoma di ~ 3 Gb come uomo e topo. Sono necessari Piccoli cambiamenti nel protocollo CNR-TR (in preparazione cellulare e profondità sequenziamento), al fine di adeguare ad altri eucarioti. L'attenzione deve...
Divulgazioni
No conflicts of interest declared.
Riconoscimenti
Ringraziamo Oriya Vardi per l'assistenza nella generazione di figure. Il lavoro del gruppo è stato sostenuto dalla Israel Science Foundation (Grant No. 567/10) e il Consiglio europeo della ricerca Starting Grant (# 281306).
Materiali
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Riferimenti
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati