
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
DNA含有量測定法による哺乳類の複製タイミングのゲノムワイドな決意
要約
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
要約
ゲノムの複製は、DNA複製の忠実度を保証する高度に調節されたプロセスにおける細胞周期のS期の間に起こります。それぞれのゲノム領域は、複製の複数の起源の同時活性化を介して、S期の間に明確な時に複製されます。レプリケーション(ToRの)の時間は、多くのゲノムおよびエピジェネティックな機能と相関し、突然変異率やがんにリンクされています。健康と病気で、複製プログラムの完全なゲノムビューを理解することは、主要な将来の目標や課題です。
、哺乳動物細胞のゲノムワイドのToRをマップするための簡単な方法:この記事では詳細に(CNR-のToRここでいう)メソッド」レプリケーションのゲノム時間をマッピングするためのS / G1のコピー数の比率」を説明しています。この方法は、S期細胞及びG1期の細胞の間のコピー数の差異に基づいています。 CNR-のToR法は6工程で行われる:ヨウ化プロピジウム(PI)で細胞を染色の調製。 2.ソル(FACS)を蛍光活性化細胞選別を用いた定G1及びS期細胞。 3. DNA精製。 4.超音波処理; 5.ライブラリの準備および配列決定;そして6.バイオインフォマティクス解析。 CNR-のToR方法は、詳細なレプリケーション・マップになり、迅速かつ簡単なアプローチです。
概要
哺乳動物のDNA複製は、細胞周期の間に正確に一度、各染色体の正確な複製を確保するために厳密に調節されています。レプリケーションは、高度に制御順序に従って発生-他のゲノム領域は、中間または後期S期(中期および後期複製ドメイン)で、後に複製する一方、複数の大規模なゲノム領域(〜Mbが)、S期(早期複製ドメイン)の先頭に複製する1。ゲノムの50%、また、癌の形質転換5中分化3,4の間及びより少ない程度に、組織2との間ののToRを変更-ゲノムのほとんど30%が、全ての組織(構成のToRドメイン)で同時に複製します。また、特定のゲノム領域は非同期6、7、8、すなわち差が複製します2対立遺伝子間のToRインチ
Torは転写レベル、GC含量、クロマチン状態、遺伝子密度など 1,9を含む多くのゲノムおよびエピ機能と相関します。 ToRのも突然変異率とタイプ10、11、したがって、当然に関連付けられている、複製プログラムの摂動は、がん12、13にリンクされています。 ToRとクロマチン構造の間の因果関係は未だ解明されていません。開いたクロマチンが早い複製を容易にすることが可能です。しかし、代替モデルは14、クロマチンは、初期および後期複製領域1のパッケージングを差動にS期リードの開始時と終了時に複製し、異なるクロマチン制御因子存在の間に組み立てられていることを示唆しています。我々は最近のToRは異なるゲノム領域11に発生する突然変異のタイプに影響を与えることによって、GC含量を整形することが示されています。
蛍光in situハイブリダイゼーション(FISH)は、個々の遺伝子座でのToRを測定するための主要な方法です。これは、単一のFISHシグナル対与えられた対立遺伝子15、16二重の割合を示すS期の細胞の割合をカウントすることによって簡単に行われます。別の方法は、Sに沿って複数の時点にそれらのDNA含有量に応じて細胞を選別、BrdUでDNAを標識のBrdUを含むDNAを免疫沈降し、定量PCR 17で沈殿したDNAの存在量をチェックするパルスで構成されています。
ゲノムのToRマッピングは、2つの方法によって達成することができます。第一の方法は、上述したBrdU-IPに基づく方法のゲノムのバージョンであり、ここで量の定量各分画中の沈殿したDNAは、マイクロアレイへのハイブリダイゼーションを介して、全ゲノムまたはディープシークエンシングにより同時に行われます。第二の方法、CNR-Torは、G1細胞におけるDNA含有量によってS期細胞と正規化の各ゲノム領域のコピー数を測定することに基づいています。この方法では、細胞は非複製(G1期)および(S期)基( 図1)の複製にFACSによってソートされます。 G1の細胞はすべてのゲノム領域において同じコピー数を持っているので、それらのDNA含有量は同じである必要があります。一方、SにおけるDNAコピー数が遅く複製領域は、したがって、それらのDNA含量がするほとんどの細胞ではまだ複製していない一方で、初期の複製領域は、ほとんどの細胞で複製を受け、したがって、それらのDNA含有量が倍になるので、のToRに依存しますG1細胞のものと同様です。したがって、DNA含量のG1比Sは、のToRを示します。各ゲノム領域のためのDNAの量は、ハイブリダイゼーションによってのいずれかで測定されますマイクロアレイまたはディープシークエンシング2、8による。 CNR-のToRの方法の利点をさらに説明します。
図2で説明したように本論文では、ゲノムのToRマッピング用のCNR-のToR方法について説明します。紙は、結果の基本的な分析およびゲノムのToRマップの作成まで、細胞を採取からプロセス全体の細部を説明します。この論文に記載されているプロトコルが正常に培養で増殖させた種々の細胞型上で実行されています。このプロトコルの将来の改善は、in vivoでのToRのマッピングに、希少細胞タイプにつながることができます。
プロトコル
注:Torは唯一の成長、同期されていない細胞で測定することができます。通常、S期にある〜1×10 5個の細胞になり、2×10 6急成長する細胞、(律速段階) -手順は、少なくとも1で開始する必要があります。 2または3つの複製を使用して、各実験を実施することをお勧めします。 CNR-のToRの全体のプロセスは、1週間以内に完了することができます - 2日には、ライブラリの準備までのすべての段階に専念する必要があり、1〜2日を配列決定し、さらに1日に必要とされる初期データを解析するために必要です。
文化からの細胞の1コレクション
注:プロトコルは、(約2を含む- 5×10 6細胞)を10cmプレートでの培養中で増殖する細胞のために書かれているが、簡単に他のプラットフォームに調整することができます。
- 懸濁液中で増殖させた細胞については、固定(セクション2)に進みます。
- 接着細胞の場合、吸引およびPLを洗いますCa 2+およびMg 2+なしで3 mLのPBSで食べました。
- PBSを捨て、細胞が剥がれるまで1 mLの商業トリプシン-EDTAを用いて37℃で5分間、細胞をインキュベートします。
注:トリプシン処理の持続時間は、各細胞型に調整されるべきです。 - トリプシンを中和し、15 mLのコニカルチューブまたは5mLのポリスチレンチューブ中の細胞を収集するために3 mLの培地を加えます。氷の上に保管してください。
2.固定
注:この部分については、すべての手順は4℃で行われるべきです。
- 4℃で5分間、300×gで細胞を遠心。
- 1 mLの冷PBSで細胞を2回吸引し、洗います。
- 250μL(合計)冷PBSで細胞を再懸濁し。
- チューブを穏やかにボルテックスしながら、ゆっくりと-20℃の100%エタノールを滴下800μLを追加します。 80% - これは、70の最終エタノール濃度をもたらします。
注:高純度のエタノールをこの段階で推奨されています。 - foを氷上で細胞をインキュベートR 30分間。
注:この段階で細胞を4℃で数日間、-20℃で数ヶ月間保つことができます。
3. PI染色
- 4℃で10分間、500×gで細胞を遠心。
- 注意深く上清を吸引し、1mLの冷PBSで細胞を2回洗浄します。
- 以下の混合物で吸引し、再懸濁し、各サンプル:1 mLのPBS、5μLを10mg / mL RNaseAを、50μLの1mg / mLのヨウ化プロピジウム(PI;使用前にボトルを混ぜます)。 〜2×10 6細胞/ mL)までの濃度を調整します。
注:光のうちキープ - PIは、光に敏感です。 - 5 mLのポリスチレンチューブに35μmのメッシュでろ過し、パラフィルムであります。
- 30分 - 15のために、暗所で室温でインキュベートします。
注:細胞は、今、FACS分析のために準備ができています。必要であれば、染色された細胞を、暗所で4℃で少なくとも24時間保存することができます。
4.ソート
- FACS機を用いてソート細胞。米国そのPI強度に基づいて細胞を区別するために561 nmのレーザーを電子。 488ナノメートルまたは532のような535 nmの励起極大付近の他のレーザは、FACS機械構成に応じて使用することができます。
- 最適な結果を得るためには、特定のセルサイズに推奨最小のノズル(ほとんどの細胞のための85μm)を使用します。収量を超える純度のモードに優先順位を付けます。 45 PSIのシース圧で、通常300〜500イベント/秒まで、安定した遅い流れを使用してください。
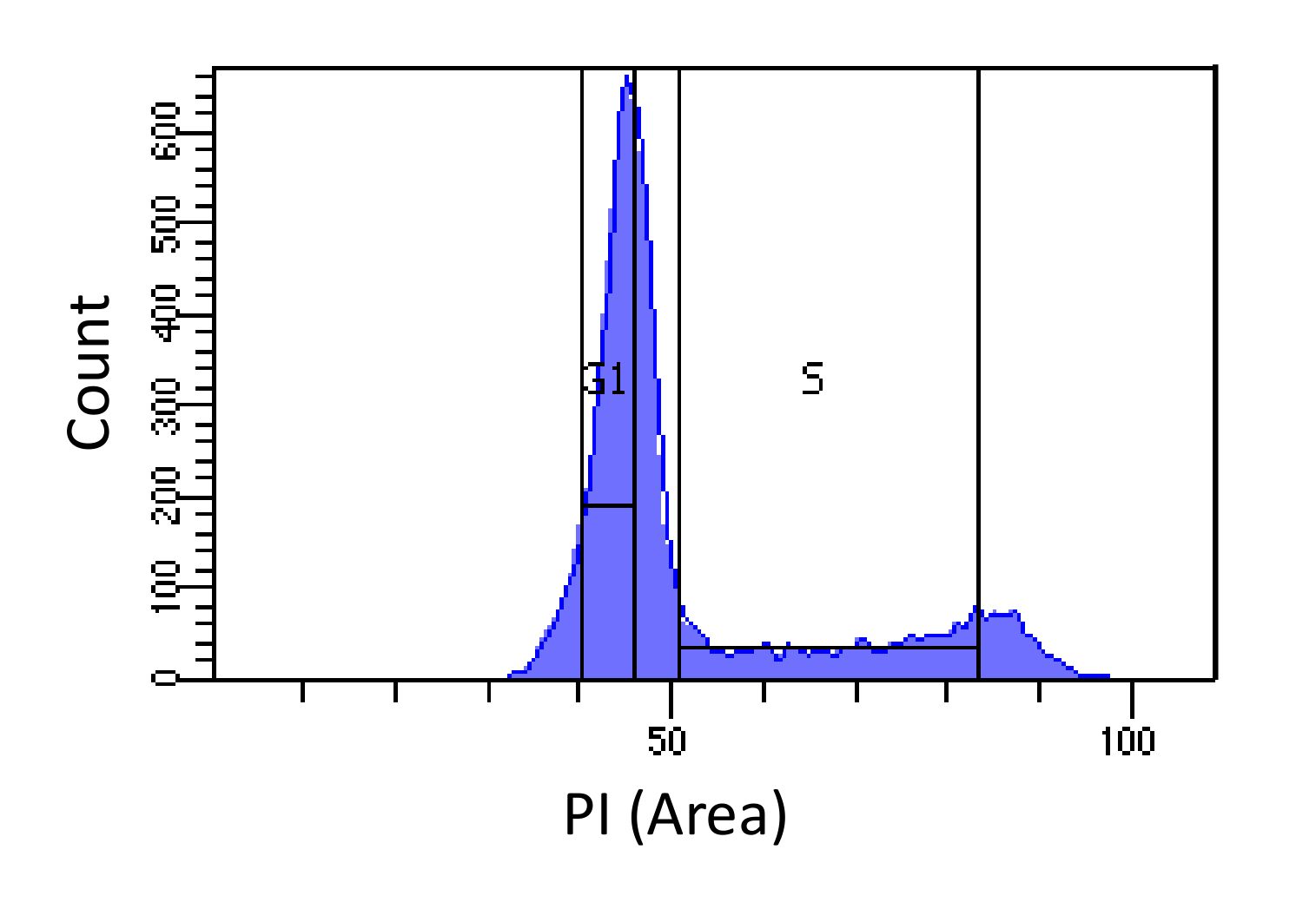
- ゲーティングを使用して、SSC 対 FCSをプロットすることにより、死細胞と細胞内残骸(低FCSおよび高SSC)を判別します。生存細胞から(ダブレットが同じH-値を持つことになりますSSC-幅(W)SSC-高さ(H)は、PI-H 対 FSC-W FSC-Hプロット対によっておよびPI-Wに続いて対をプロットすることによりダブレットを区別しかし、大きなW値)。実行可能な単一細胞では、細胞のDNA含有量を表すPI-エリア(A)強度のヒストグラムを描きます。
- 図1に示すように、G1およびS相へソート細胞、。 G1・ゲーティングは限りSから可能な限り狭くするべきでありながら、Sのためのゲーティングは、広いことと、G1とG2期に侵入する必要があります。

PI強度に基づいて、図1細胞周期の決意。マウス胚線維芽細胞(MEF)集団(PI-面積によって測定される)細胞のDNA含量の分布を示すヒストグラム。マークされた領域を使用して、4N DNA含量) - DNA量は、2つのサブ集団I)G1細胞(2NのDNA含量)及びii)S期細胞(2Nに集団をソートするために使用されます。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
注:G1細胞の収集の目的は、異なるゲノム領域間のシーケンシング効率のバイアスを考慮するためです。代替アプリローチは、同じ細胞型からG1停止細胞を使用することです。このアプローチは、クリーンな結果を与える(これはS期の汚染を最小限に抑えるため)が、停止した細胞と測定された細胞との間の遺伝的差異に起因するバイアスを導入することができます。
- 寒い条件でソートし、1.5 / 2 mLチューブにソートされた細胞を回収します。ソート以下のチューブを氷上に保管してください。
注:4°C 18で3時間- 1 - 5%BSA DNAの回収率を向上させるためには、低結合チューブを使用すること、または4で被覆された管を使用した方がよいです。
5. DNA精製
- 各サンプル(G1およびS)のためのDNA精製キットを用いてDNAを精製します。
注:市販のキットを使用する場合は、製造業者によって推奨されるように、高収率のために2 mLのチューブに400μLの溶出緩衝液で溶出します。 - 蛍光光度計を用いてDNA濃度を確認してください。
注:FACSによって収集10万、哺乳動物細胞から、1はDNAの〜1μgのを取得する必要があります。 ATは、この段階のDNAを、4℃で、または長期保存のために-20℃で数日間保存することができます。
6.超音波処理
- 使用される磁気スタンドと互換性のある1.7 mLチューブにDNAを移します。
- 磁気スタンドを用いて、製造業者の指示に従って2×SPRIビーズを用いてDNAを濃縮し、そして50μLの溶出緩衝液で溶出します。
- 250塩基対の平均目標ピークの大きさに集束超音波装置とせん断DNA。 50 W、20%のデューティ・ファクタ、バーストあたり200サイクル、20℃、120秒:50μLのDNAサンプルを次のように設定します。
- 電気泳動により剪断されたDNAのサイズを確認してください。 〜250bpのピークと700 bpの - 推奨サイズ分布は200です。
注:この段階ではDNAは4℃で、または長期保管のために-20℃で数日間保存することができます。
7.図書館準備、および配列決定
注:多くのライブラリ作成キットと異なり耳鼻咽喉科シーケンシングプラットフォームは、私たちと材料の項で述べたことにより、使用されているものと同様に動作するはずです。実際に、過去に、ToRのマップは、マイクロアレイプラットフォーム2と非常に類似した方法を用いて作製しました。
- すべての市販のライブラリー調製キットを使用してライブラリを準備します。
- 800 bpの - ライブラリの準備の終わりには、300のために磁気ビーズを使用してサイズを選択します。
- ライブラリーを調製した後、蛍光光度計を用いてDNA濃度を測定。
- 電気泳動を用いたDNAのサイズを測定します。
- 任意のプラットフォームでシーケンシングを実行します。
注:シーケンシング、少なくとも10 Mはサンプル毎に読み込みが推奨されます。この深さは読んごとに約300塩基対(3〜のためのGbサイズのゲノム)に相当し、50の分解能でのToRの測定のために十分である - 100キロバイト深さを大きくすると、ウィンドウのサイズの減少につながるため、より確実に高解像度化が可能になります。ペアエンド配列決定は、中に必要はありません唯一のカバレッジ情報が収集されているので、このプロトコル。しかしながら、反復配列を含有する読み取りの位置を解決するのに役立つことができます。
8.分析
注:データ分析A.コレンらによって使用される方法に基づいています。 19。
- bowtie2または任意の信頼性の高い、短いリード・アライナを使用して、対応するゲノムにマップする配列データ。様々なサイズを定義し、同じカバレッジ染色体窓200で覆われたセグメントは、G1画分を読み込むようにし、S期は、同じウィンドウに読み込む数えます。
- ウィンドウごとにS / G1比を計算します。これは、ゲノム( 図3)に沿ってS / G1比の大きな変動を持つマップを生成する必要があります。 ToRの測定の信頼性のための良好な制御はずっと平坦でなければならない(G1の二つの別々の測定から)G1 / G1比にこのマップを比較することです。
- 各Vから減算することにより、0平均と1 SDにデータを正規化(X染色体を除く)すべてのウィンドウの平均値をALUEし、すべてのウィンドウのS / G1の標準偏差で結果を割ます。これは、zスコアに変換し、異なる実験間の比較を可能にするために行われます。
- UCSCゲノムブラウザだけでなく、15未満のデータウィンドウを含む残りの各間のギャップフラグメントに記載されているすべてのギャップ領域を削除してください。
- 10のパラメータでMATLAB関数csaps経由立方平滑化スプラインとの残りのフラグメントを滑らかに- 16とセットポイントですべての100キロバイトを補間します。
注:平滑化及び補間のパラメータは、データの深さに基づいて調整されるべきです。他の適切な平滑化方法および機能は存在し、使用することができます。 - 視覚的に、各複製の信頼性を確認した後、すべての読み取りマージし、このデータに対して上述と同様の処理を行うことにより、より深い解像度プロファイルを計算します。
結果
典型的なToRのマップは、マウス胚性線維芽細胞(MEF)は、 図3に示されています。それは個々のウィンドウのための正規化されたS / G1比(ステップ8.3)、ならびに立方平滑化及び補間(ステップ8.5)から得られるラインであるドット、両方を示しているので、この図は、分析プロセスを示しています。
初期、?...
ディスカッション
CNR-Torは(Rhind N.およびGilbert DM 20によってレビュー)SにFACSおよびG1相により分割することができる任意の真核生物増殖細胞集団の原理で行うことができます。ここに記載される方法は、ヒト及びマウスのような約3ギガビットのゲノムサイズを有する哺乳動物細胞に調整しました。 (細胞調製および配列決定の深さ)CNR-のToRプロトコルにおける小さな変化は、他の真核生物?...
開示事項
No conflicts of interest declared.
謝辞
私たちは、数字を生成する際に支援するためのオリヤー語ヴァルディに感謝します。 ISグループでの作業は、イスラエル科学財団(助成番号10分の567)とグラント(#281306)を開始し、欧州研究評議会によってサポートされていました。
資料
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
参考文献
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved