
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Genomweite Bestimmung der Mammalian Replication-Timing von Inhaltsmessung DNA
In diesem Artikel
Zusammenfassung
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Zusammenfassung
Replikation des Genoms erfolgt während der S-Phase des Zellzyklus in einem stark regulierten Prozess, der die Genauigkeit der DNA-Vervielfältigung gewährleistet. Jede genomischen Region wird an einer bestimmten Zeit während der S-Phase durch die gleichzeitige Aktivierung mehrerer Replikationsursprünge repliziert. Zeit-Replikation (ToR) korreliert mit vielen genomischen und epigenetische Merkmale und ist mit Mutationsraten und Krebs in Verbindung gebracht. Begreifen die vollständige genomische Ansicht des Replikationsprogramm, in Gesundheit und Krankheit ist ein wichtiges Ziel für die Zukunft und Herausforderung.
Dieser Artikel beschreibt im Detail die "Copy Number Verhältnis von S / G1 zum Abbilden genomischen Zeit-Replikation" Methode (hier genannt: CNR-ToR), einen einfachen Ansatz der genomweiten ToR von Säugetierzellen abzubilden. Das Verfahren basiert auf der Kopienzahlunterschiede zwischen der S-Phase-Zellen und G1-Phasen-Zellen. 1. Herstellung von Zellen und Färbung mit Propidiumiodid (PI);: Der CNR-ToR Verfahren wird in 6 Schritten 2. Sorting G1 und S-Phasen-Zellen fluoreszenz-aktivierte Zellsortierung (FACS); 3. DNA-Reinigung; 4. Ultraschall-Behandlung; 5. Bibliothek Vorbereitung und Sequenzierung; und 6. Die bioinformatische Analyse. Die CNR-ToR-Methode ist eine schnelle und einfache Ansatz, der in der detaillierten Replikation Karten führt.
Einleitung
Mammalian DNA-Replikation ist streng reguliert die exakte Replikation jedes Chromosoms während des Zellzyklus genau einmal zu gewährleisten. Die Replikation erfolgt gemäß einem stark regulierten Ordnung - mehrere große genomischen Regionen (~ Mb) zu Beginn der S - Phase repliziert (frühe Domänen replizieren) , während andere Genomregionen später bei mittleren oder späten S - Phase (Mitte und Ende der replizierenden Domänen) replizieren 1. Der größte Teil des Genoms repliziert zur gleichen Zeit in allen Geweben (konstitutive ToR Domänen), während die 30% - 50% des Genoms, ändert seine ToR zwischen Geweben 2, während der Differenzierung 3, 4 und in geringerem Maße auch während der Krebstransformation 5 . Darüber hinaus sind bestimmte replizieren genomischen Regionen asynchron 6, 7, 8, nämlich ein Unterschied bestehtin den ToR zwischen den beiden Allelen.
ToR korreliert mit vielen genomischen und epigenomischen Funktionen , einschließlich der Transkription Ebenen, GC - Gehalt, Chromatinzustand, Gendichte, usw. 1, 9. ToR ist auch mit Mutationsraten und Typen zugeordnet sind 10, 11 und daher wenig überraschend, Störungen des Replikationsprogramms an Krebs 12, verbunden 13. Der kausale Zusammenhang zwischen ToR und Chromatinstruktur ist noch nicht verstanden. Es ist möglich, dass offene Chromatin frühen Replikation ermöglicht. Jedoch schlägt ein alternatives Modell , dass das Chromatin während der Replikation und die verschiedenen Chromatin - Regulatoren am Anfang und Ende der S Phasenvoreilung Verpackung Differentials der frühen und späten replizierende Regionen 1, 14 zusammengesetzt ist . Wir haben kürzlich gezeigt , dass die ToR formt den GC - Gehalt durch die Art der Mutationen betreffen , die 11 in verschiedenen genomischen Regionen auftreten.
Fluoreszenz - in - situ - Hybridisierung (FISH) ist die wichtigste Methode für ToR an einzelnen Loci zu messen. Es wird durchgeführt , indem einfach der Prozentsatz von S - Phasen - Zellen zu zählen , die einzelne FISH - Signale gegen den Prozentsatz der Dubletten für ein bestimmtes Allel 15, 16 aufweisen. Ein alternatives Verfahren besteht aus Impuls mit BrdU die DNA - Markierung, 17 Zellen entsprechend ihrem DNA - Gehalt zu mehreren Zeitpunkten längs S, Immunpräzipitation DNA enthält BrdU und Überprüfung der Fülle von gefällter DNA mit qPCR Sortierung.
Genomic ToR Zuordnung kann durch zwei Methoden erreicht werden. Die erste Methode ist eine genomische Version des BrdU-IP basierten oben beschriebenen Verfahren, bei dem die Quantifizierung der Mengevon gefällte DNA in jeder Fraktion wird gleichzeitig für das gesamte Genom durch Hybridisierung mit Microarrays oder durch tiefe Sequenzierung durchgeführt. Die zweite Methode, CNR-TOR, basiert die Kopienzahl jeder genomischen Region von S-Phasen-Zellen und Normalisierung durch den DNA-Gehalt in Zellen G1 zu messen. Bei diesem Verfahren werden die Zellen durch FACS in nicht-replizierenden (G1 - Phase) und Replizieren (S - Phase) Gruppen (1) sortiert. Zellen in G1 haben die gleiche Kopienzahl in allen genomischen Regionen und somit sollten ihren DNA-Gehalt gleich sein. Auf der anderen Seite hängt die DNA-Kopienzahl in S auf der ToR, seit Anfang replizierende Regionen unterzog Replikation in den meisten Zellen und wird daher ihre DNA-Gehalt verdoppelt, während spät replizierende Regionen noch nicht in den meisten Zellen repliziert wurden und damit deren DNA-Gehalt werden dem von G1-Zellen ähnlich. Daraus ergibt sich die S G1 Verhältnis von DNA-Gehalt der ToR indikativ. Die Menge an DNA für jeden genomischen Region wird entweder durch Hybridisierung gemessenMicroarrays oder durch tiefe Sequenzierung 2, 8. Die Vorteile des CNR-TOR Verfahren wird weiter diskutiert.
Dieses Papier beschreibt die CNR-ToR Verfahren zur genomischen ToR - Mapping , wie in Abbildung 2 beschrieben. Das Papier beschreibt die feinen Details des gesamten Prozesses von Zellen, bis die grundlegenden Analyse der Ergebnisse und der Erstellung von genomischen ToR Karten zu sammeln. Das Protokoll in diesem Papier beschrieben wurde auf verschiedenen Zelltypen in Kultur gezüchtet erfolgreich durchgeführt. Zukünftige Verbesserungen dieses Protokolls können in vivo zu der Abbildung des ToR führen und in seltenen Zelltypen.
Protokoll
Hinweis: ToR kann nur auf das Wachstum, unsynchronisierten Zellen gemessen werden. 2 x 10 6 schnell wachsende Zellen, die in der Regel in ~ 1 x 10 5 Zellen in der S - Phase (die Rate limitierende Schritt) führt - Das Verfahren sollte mit mindestens 1 beginnen. Es wird empfohlen, jedes Experiment unter Verwendung von zwei oder drei Wiederholungen durchzuführen. Der gesamte Prozess der CNR-TOR kann innerhalb einer Woche abgeschlossen werden - 2 Tage sollten für die anfängliche Datenanalyse notwendig, bis zu Bibliotheks Vorbereitung, ein bis zwei Tage benötigt werden für die Sequenzierung und einen zusätzlichen Tag, um alle Schritte gewidmet.
1. Sammlung von Zellen, die aus Kultur
HINWEIS: Das Protokoll für die Zellen geschrieben wird in Kultur in 10 cm - Platten (die etwa 2 bis 5 x 10 6 Zellen) wachsen, kann aber leicht auf andere Plattformen angepasst werden.
- Für Zellen, die in Suspension gezüchtet wurden, gehen Sie zu einer Fixierung (Abschnitt 2).
- Für anhaftenden Zellen, absaugen und waschen die plaß mit 3 ml PBS ohne Ca 2+ und Mg 2+.
- Entsorgen Sie die PBS und Inkubation der Zellen für 5 min bei 37 ° C mit 1 ml kommerzielle Trypsin-EDTA, bis die Zellen zu lösen.
HINWEIS: Die Dauer der Trypsin-Behandlung sollte auf jeden Zelltyp eingestellt werden. - 3 ml Kulturmedium um das Trypsin zu neutralisieren und zu sammeln Zellen in einem 15 ml konischen Röhrchen oder eine 5 ml Polystyrolröhrchen. Halten Sie sich auf dem Eis.
2. Fixation
HINWEIS: Für diesen Teil sollten alle Schritte bei 4 ° C durchgeführt werden.
- Zentrifugieren der Zellen bei 300 xg für 5 min bei 4 ° C.
- Absaugen und waschen zweimal Zellen mit 1 ml kaltem PBS.
- Die Zellen in 250 & mgr; l (gesamt) kaltem PBS.
- Während sanft das Röhrchen verwirbelt, langsam tropfenweise 800 & mgr; l von -20 ° C 100% Ethanol hinzu. Dies führt zu einer endgültigen Ethanolkonzentration von 70-80%.
HINWEIS: Hohe Reinheit Ethanol wird bei diesem Schritt empfohlen. - Inkubieren Zellen auf Eis for 30 min.
HINWEIS: In dieser Phase-Zellen können für ein paar Tage bei 4 ° C oder für ein paar Monate bei -20 ° C aufbewahrt werden.
3. PI-Färbung
- Zentrifugieren der Zellen bei 500 xg für 10 min bei 4 ° C.
- Saugen Sie den Überstand vorsichtig und waschen Sie die Zellen zweimal mit 1 ml kaltem PBS.
- Aspirieren und resuspendieren jede Probe mit folgender Mischung: 1 ml PBS, 5 ul 10 mg / ml RNase A, 50 & mgr; l 1 mg / ml Propidiumjodid (PI; mischen Flasche vor Gebrauch). Einstellen Konzentration von bis zu ~ 2 x 10 6 Zellen / ml).
HINWEIS: Halten Sie aus Licht - PI Licht empfindlich ist. - Filter durch 35 & mgr; m-Mesh zu einem 5 ml Polystyrolröhrchen, und in der Nähe mit Parafilm.
- Inkubieren bei RT im Dunkeln für 15 - 30 min.
HINWEIS: Die Zellen sind nun bereit für die FACS-Analyse. Falls erforderlich, können die gefärbten Zellen für mindestens 24 h bei 4 ° C im Dunkeln gelagert werden.
4. Sortieren
- Sortieren Zellen eine FACS-Maschine. Unse die 561-nm-Laser-Zellen auf ihre PI Intensität zu unterscheiden basiert. Andere Laser in der Nähe des 535 nm Anregungsmaximum wie 488 nm oder 532 kann in Abhängigkeit von der FACS Maschinenkonfiguration verwendet werden.
- Für optimale Ergebnisse verwenden Sie die kleinste Düse für die spezifische Zellgröße empfohlen (für die meisten Zellen 85 & mgr; m). Priorisieren Reinheit Modi gegenüber der Ausbeute. Verwenden Sie einen stetigen langsamen Fluss, in der Regel bis zu 300 bis 500 Veranstaltungen / s, mit einem Mantel Druck von 45 psi.
- Mit Gating diskriminieren toten Zellen und subzellulärer Schutt (niedrige FCS und hohe SSC) durch Plotten FCS vs SSC. Von den lebenden Zellen unterscheiden Dubletten durch Auftragen SSC-Breite (W) vs SSC-Höhe (H) , gefolgt von einem FSC-W vs FSC-H Grundstück und durch PI- W vs PI-H (Dubletten die gleiche H-Wert haben aber größer W-Wert). Für die tragfähige einzelnen Zellen zeichnen ein Histogramm des PI-Bereich (A) Intensität, die den DNA-Gehalt der Zellen darstellt.
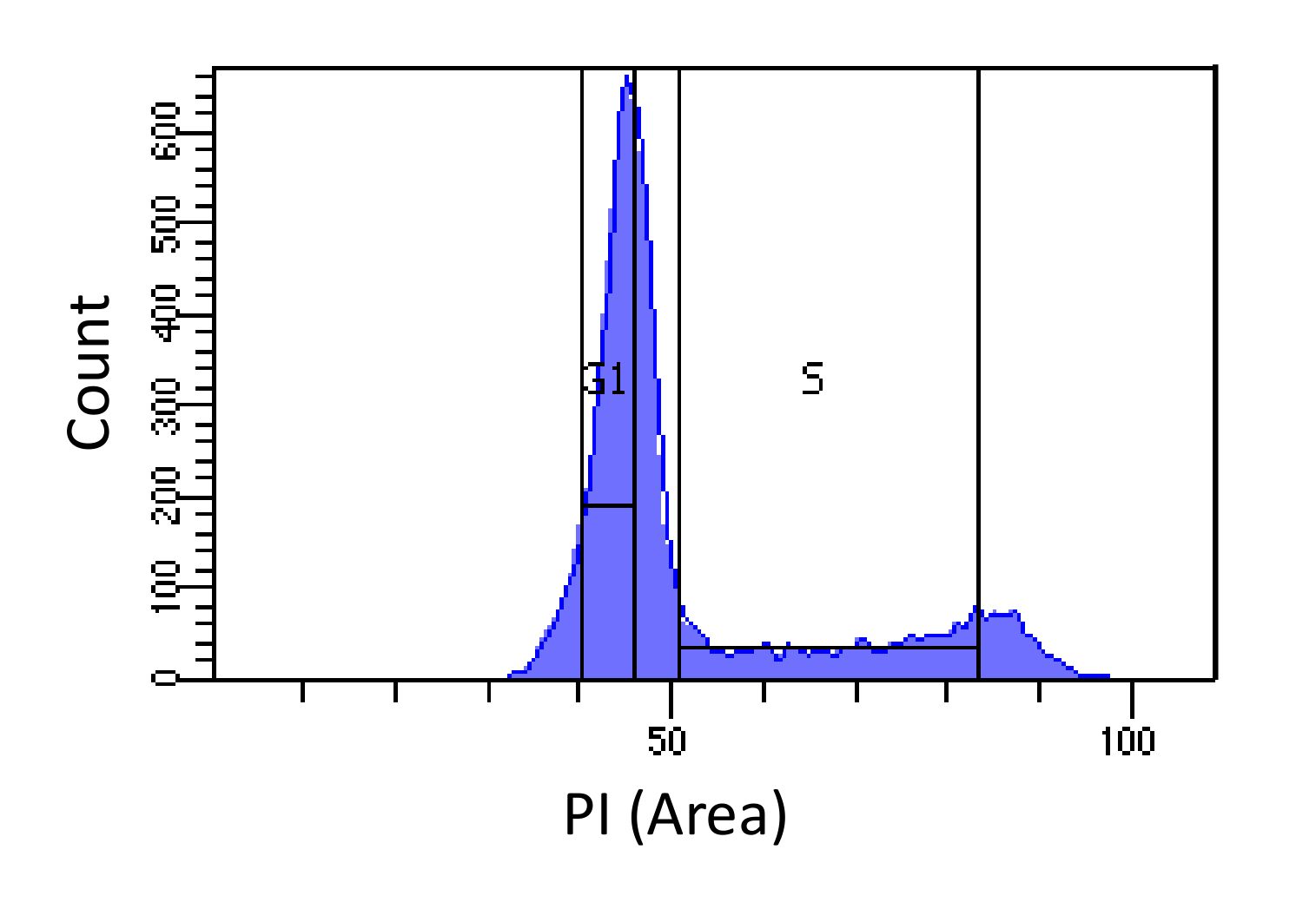
- Sortier Zellen in G1 und S - Phasen, wie in Figur 1 gezeigt. Gating für S sollte breit sein und dringen in die G1 und G2-Phase, während G1 Gating schmal sein sollte und so weit von S wie möglich.

Abbildung 1. Die Zellzyklusphasenbestimmung auf Basis von PI Intensität. Histogramm zeigt die Verteilung der zellulären DNA-Gehalt (gemessen durch PI-Area) von embryonalen Maus-Fibroblasten (MEF) Bevölkerung zeigt. Der DNA - Gehalt wird verwendet , um die Bevölkerung in zwei Subpopulationen i) G1 - Zellen (2N DNA - Gehalt) und ii) S - Phasen - Zellen (2N zu sortieren - 4N DNA - Gehalt), die markierten Regionen mit. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
HINWEIS: Der Zweck der Sammlung von G1-Zellen ist für Verzerrungen in der Sequenzierungseffizienz zwischen verschiedenen genomischen Regionen zu berücksichtigen. Eine alternative AppRotauge ist auf G1 arretierten Zellen aus dem gleichen Zelltyp verwenden. Dieser Ansatz gibt sauberere Ergebnisse (da es die S-Phase Kontamination minimiert), aber es kann Verzerrungen einführen von genetischen Unterschieden zwischen den arretierten Zellen und den gemessenen Zellen stammen.
- Sortieren in der Kälte und sortierten Zellen in 1,5 / 2 ml-Röhrchen zu sammeln. Halten Sie Röhrchen auf Eis nach Art.
ANMERKUNG: Um DNA - Gewinnung zu verbessern, ist es besser , niedrige Bindungsrohre zu verwenden oder Röhren mit 4 beschichtet zu verwenden - 5% BSA für 1 - 3 Stunden bei 4 ° C 18.
5. DNA Purification
- Für jede Probe (G1 und S) reinigen DNA, die eine DNA-Reinigung-Kit.
HINWEIS: Bei der kommerziellen Kits, eluiere mit 400 & mgr; L Elutionspuffer in eine 2 ml-Röhrchen für hohe Ausbeute, wie vom Hersteller empfohlen. - Überprüfen DNA-Konzentration Fluorometer verwendet wird.
HINWEIS: Von 100.000 Säugetieren durch die FACS gesammelten Zellen, sollte man ~ 1 ug DNA erhalten. EINt Diese Stufe DNA kann für ein paar Tage bei 4 ° C oder bei -20 ° C für lange Lagerung gehalten werden.
6. Beschallen
- Transfer-DNA in ein 1,7 ml-Röhrchen, die mit dem magnetischen Ständer kompatibel verwendet.
- Konzentrieren Sie DNA mit 2x SPRI Perlen gemäß den Anweisungen des Herstellers, einen magnetischen Ständer benutzt wird, und Eluieren in 50 & mgr; l Elutionspuffer.
- Shear-DNA mit einem fokussierten Ultraschallgerät auf eine durchschnittliche Soll-Spitzengröße von 250 bp. Verwenden Sie die folgenden Einstellungen für eine 50 & mgr; l DNA-Probe: 50 W, 20% Einschaltdauer, 200 Zyklen pro Burst, 20 ° C, 120 s.
- Überprüfen Sie die Größe der gescherten DNA durch Elektrophorese. Die empfohlene Größenverteilung von 200 bis 700 bp mit einem Peak bei ~ 250 bp.
HINWEIS: In diesem Stadium DNA kann für ein paar Tage bei 4 ° C oder bei -20 ° C für lange Lagerung gehalten werden.
7. Bibliothek Vorbereitung, und Sequenzierung
HINWEIS: Viele Bibliothek Vorbereitungs-Kits und unterscheidenent-Sequenzierungsplattformen sollten ähnlich den Abschnitt in den Materialien, die von uns und erwähnt Gebrauchten arbeiten. Tatsächlich in der Vergangenheit wurden ToR Karten 2 erzeugt ein sehr ähnliches Verfahren mit Mikroarray - Plattformen.
- Bereiten Sie Bibliotheken mit jeder kommerziellen Bibliothek Vorbereitung Kit.
- Am Ende der Bibliothek Vorbereitung, wählen Sie Größe unter Verwendung von magnetischen Kügelchen für 300 bis 800 bp.
- Nach Bibliotheken Vorbereitung messen Fluorometer DNA-Konzentration verwendet wird.
- Messen Sie DNA-Größen Elektrophorese.
- Führen Sie die Sequenzierung auf jeder Plattform.
HINWEIS: Sequencing mindestens 10 M liest pro Probe empfohlen. Diese Tiefe entspricht lesen eine etwa alle 300 bp (für ~ 3 Gb Größe Genome), und ist mit einer Auflösung von 50 für ToR Messungen ausreichend - 100 kb. die Tiefe Erhöhung in einer Verringerung der Größe der Fenster führen wird und somit ermöglicht Auflösung mit höherer Sicherheit zu erhöhen. Gepaart Ende Sequenzierung ist nicht notwendig, indieses Protokoll, da nur Abdeckung Informationen gesammelt. Es kann jedoch helfen, die Position bei der Lösung der liest, die repetitive Sequenzen enthalten.
8. Analyse
HINWEIS: Die Datenanalyse wird auf dem von A. Koren et al - Verfahren. 19.
- Karte Sequenzierungsdaten zu dem entsprechenden Genom bowtie2 oder eine zuverlässige kurze Lese Ausrichter mit. Definieren unterschiedlicher Größe, gleicher Abdeckung chromosomalen Fenster als 200 liest in der G1-Fraktion bedeckt Segmente und zählen liest S-Phase in den gleichen Fenster.
- Berechnen Sie den S / G1-Verhältnis für jedes Fenster. Dies sollte eine Karte mit großen Schwankungen in der S / G1 - Verhältnis entlang des Genoms (3) erzeugen. Eine gute Kontrolle für die Zuverlässigkeit der ToR Messungen ist diese Karte das G1 / G1-Verhältnis zu vergleichen (aus zwei separaten Messungen von G1), die wesentlich flacher sein sollte.
- Normalisieren Daten auf 0 bedeuten und 1 SD durch von jedem v Subtrahierenert den Durchschnittswert aller Fenster (mit Ausnahme des X-Chromosoms) und die Ergebnisse durch die Standardabweichung des S / G1 aller Fenster unterteilt wird. Dies wird getan, um zu Z-Werte konvertieren und den Vergleich zwischen verschiedenen Experimenten ermöglichen.
- Entfernen Sie alle Spaltbereiche durch den Browser UCSC Genom aufgelistet sowie jede verbleibende Zwischen Lücke Fragment weniger als 15 Datenfenster enthält.
- Glätten Sie die verbleibenden Fragmente mit einer kubischen Spline Glättungs über die Matlab - Funktion csaps mit einem Parameter von 10 bis 16 und interpolieren zu festgelegten Punkten alle 100 kb.
HINWEIS: Parameter der Glättung und Interpolation auf Basis der Tiefe der Daten eingestellt werden. Andere geeignete Glättungsverfahren und Funktionen vorhanden sind und verwendet werden können. - Nach visuell die Zuverlässigkeit jedes replizieren Bestätigung verschmelzen alle liest und eine tiefere Auflösung Profil berechnen, indem die gleichen Verfahren auf diesen Daten oben beschrieben durchgeführt wird.
Ergebnisse
Eine typische ToR Karte ist in Abbildung 3 für embryonalen Maus - Fibroblasten (MEF) gezeigt. Diese Figur zeigt den Analyseprozess, da es zeigt sowohl die Punkte, die die normierte S / G1-Verhältnis für einzelne Fenster (Schritt 8.3), sowie die Linie, die sich von der kubischen Glättung ergibt und Interpolation (Schritt 8.5).
Solche Karten, um die Organisation des Replikationsprogramms zu erfassen, die ein...
Diskussion
CNR-TOR kann im Prinzip auf jede eukaryotische proliferierenden Zellpopulation durchgeführt werden , die durch FACS zu S und G1 - Phasen (rezensiert von Rhind N. und Gilbert DM 20) unterteilt werden kann. Das hier beschriebene Verfahren hat sich an Säugerzellen mit einer Genomgröße von ~ 3 Gb wie Mensch und Maus angepasst. Kleine Änderungen in der CNR-TOR-Protokoll (in Zellpräparation und Sequenzierung Tiefe) werden benötigt, um es an andere Eukaryoten einzustellen. Es ist auf die Sammlung...
Offenlegungen
No conflicts of interest declared.
Danksagungen
Wir danken Orija Vardi für die Unterstützung in Zahlen zu erzeugen. Die Arbeit in der IS-Gruppe wurde von der Israel Science Foundation (Zuschuss Nr 567/10) und des European Research Council Starting Grant (# 281306) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Referenzen
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten