
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
使用CRISPR/Cas9系统将基因挖空直接引入锥虫瘤的肌瘤阶段
摘要
在这里,我们描述了一个协议,使用CRISPR/Cas9系统将基因敲除引入锥虫的细胞外肌瘤。生长表型可以通过细胞计数来追踪轴酸乳腺培养物,或者通过宿主细胞入侵后细胞内乳腺增殖。
摘要
锥虫病是一种致病原生动物寄生虫,主要在拉丁美洲引起恰加斯病。为了识别针对T.cruzi的新型药物靶点,验证靶向基因在寄生虫的哺乳动物阶段(乳腺)中的重要性是很重要的。T. 克鲁西的阿马斯蒂在宿主细胞内复制;因此,如果不经过其他发展阶段,就很难进行淘汰实验。最近,我们小组报告了一种生长状况,其中乳腺可异种复制长达10天,而不会失去其类似乳腺的特性。通过使用这种时间轴质乳腺培养,我们成功地将gRNA直接引入Cas9表达的乳腺,导致基因敲除,并仅在乳腺阶段分析其表型。在本报告中,我们描述了一个详细的方案,用于产生体外衍生的细胞外乳腺,并在CRISPR/Cas9介导的敲除实验中利用轴培养。敲除的乳腺的生长表型可以通过轴培养的细胞计数或宿主细胞入侵后细胞内乳腺的复制来评估。该方法绕过寄生虫阶段分化通常涉及生产转基因或敲除的乳腺。利用时间轴突培养具有扩大T.cruzi特定阶段研究的实验自由的潜力。
引言
锥虫病是恰加斯病的致病剂,主要在拉丁美洲1流行。T. cruzi具有独特的生命周期阶段,因为它在昆虫载体和哺乳动物宿主2之间传播。T. cruzi在吸血三胺虫的中肠道中复制为表皮球菌,在被沉积在人类或动物宿主之前,在后肠中分化成传染性代谢性胰小体球菌。一旦锥虫通过叮咬部位或通过粘膜进入宿主体,寄生虫就会侵入宿主细胞,并转化为称为乳腺的无旗体圆形。乳腺在宿主细胞内复制,并最终分化成锥虫,从宿主细胞中爆发并进入血流,感染另一个宿主细胞。
由于目前可用的化疗药物,苯甲酰氨基苯甲酸和尼富蒂莫克斯,引起不良副作用,并在疾病的慢性阶段无效3,这是极大的兴趣,确定针对T.cruzi的新药物靶点。近年来,CRISPR/Cas9系统已成为一个强大的工具,有效地执行基因敲除在T.cruzi,无论是通过转染单独或单质粒含有gRNA和Cas9 4,通过稳定表达Cas9和随后的引入gRNA 5,6,7或转录模板的gRNA8,或通过电穿孔预成型的gRNA/Cas9 RNP复合物7,9。这一技术进步被高度期待加速恰加斯病的药物靶点研究。
为了继续药物开发,验证靶基因或药物候选化合物在T.cruzi的药效中的重要性至关重要,因为它是寄生虫在哺乳动物宿主中的复制阶段。然而,这是一项具有挑战性的任务,因为由于存在阻塞宿主细胞,因此不能直接操纵乳腺。在利什曼尼亚,一种与T.cruzi密切相关的原生动物寄生虫,开发了一种斧头阿马斯蒂戈特培养方法,并已用于药物筛选测定方法10,11,12, 13.虽然轴突和细胞内乳腺14在化合物的易感性上有一些差异,但维持轴培养的能力为研究利什曼病临床相关阶段的基本生物学15、16。在T.cruzi的情况下,关于自然发生的细胞外乳腺(EA)17和EA 17,18,19的体外生产文献可追溯到几十年前。此外,EA已知具有传染性能力20,虽然小于锥形雄蒂戈特,和马斯蒂戈特宿主入侵的机制已阐明近年来(审查由Bonfim-Melo等人21)。然而,与利什曼尼亚不同,EA在T.cruzi中没有被用作实验工具,主要是因为EA被认为是细胞内寄生虫,因此在实际操作中并未被视为"复制形式"感。
最近,我们小组提议利用T.克鲁兹的EA作为时间轴文化22。T. 克鲁西图拉胡恩菌株的Amastigotes可在37°C下在LIT培养基中无宿主细胞复制长达10天,而不会严重恶化或丧失类似亲子的特性。在无宿主生长期,EA通过常规电穿孔、药物滴定测定与胰蛋白酶化合物和CRISPR/Cas9介导敲除,然后进行生长表型监测,成功地用于外源性基因表达。在本报告中,我们描述了在敲除实验中产生体外衍生EA和利用轴突腺体的详细方案。
Access restricted. Please log in or start a trial to view this content.
研究方案
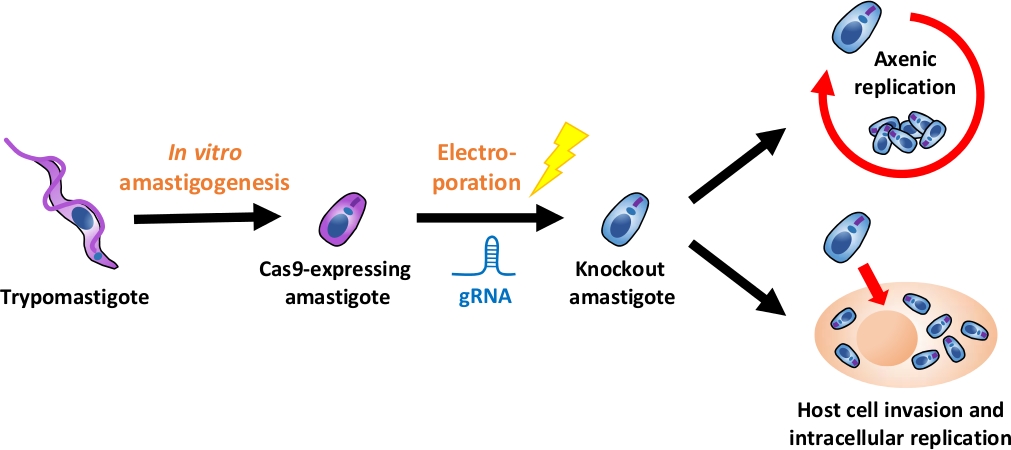
注:图 1描述了整个实验流程的概述。

图 1:使用 EA 的挖空实验概述。组织培养衍生的胰腺炎被收获并分化成EA.gRNA通过电穿孔转染成Cas9表达的乳腺,并且通过轴仿或通过宿主细胞入侵后的细胞内复制。请点击此处查看此图的较大版本。
{kind=link}
1. 寄生虫培养准备
- 图拉胡恩菌株的锥虫病在整个报告中使用。在 LIT 介质中保持T. cruzi的表位(10% FCS,参见补充表 1)。牢固地关闭盖子,并将文化瓶保持在 28°C。
- 生成含有Cas9内西的T.克鲁西转基因菌株。包含 Cas9 编码序列和 G418 (neo r)选择标记的表达质粒示例可在 Lander 等人4和彭等人5中找到。
- 使用材料表中列出的试剂盒,通过电穿孔将上述质粒转染到表皮质。对于 1 个比色皿,旋转 2 x 107个电池,丢弃上清液,然后用含有提供的补充溶液的 100 μL 电穿孔缓冲液重新悬浮。
注:电穿孔缓冲液可用EM缓冲液24(细胞混合25和磷酸-蔗糖缓冲液的3:1混合物)替代。 - 加入20-40μg质粒,将混合物转移到2毫米间隙电穿孔的彩粒中。使用X-14程序26应用电穿孔装置(见材料表)。。
- 将比色皿内容物转移到含有 5 mL 的 LIT 介质(10% FCS)的 T-25 烧瓶中。在28°C下孵育烧瓶24小时。
- 将G418添加到250μg/mL的最终浓度中,并在28°C下继续孵育。杀死非转染的表皮约1周。一旦G418耐药人群开始恢复,每周通过一次或两次培养,以避免饱和并稀释死细胞。建立一个稳定的细胞系通常需要总共4周。
注:如果Cas9的构成表达是细胞5的负担,则通过结合紧接Cas9开放阅读框架27下游的amastin基因的3'-UTR,使该表达特定于乳腺阶段。在Takagi等人22中可以找到对马斯蒂戈特特异性Cas9表达质粒的详细描述。
- 使用材料表中列出的试剂盒,通过电穿孔将上述质粒转染到表皮质。对于 1 个比色皿,旋转 2 x 107个电池,丢弃上清液,然后用含有提供的补充溶液的 100 μL 电穿孔缓冲液重新悬浮。
- 建立Cas9表达的T.克鲁西和哺乳动物宿主细胞的宿主-寄生虫共培养。在本报告中,使用3T3-瑞士白化细胞作为宿主。
- 将T.克鲁西表皮菌分化成代谢性胰体虫。使用血细胞计确定表皮球培养基的细胞密度,并通过离心收集5 x 107细胞,在2,100 x g下收集15分钟。丢弃上清液,用 10 mL 的 RPMI 培养基重新悬浮细胞。在28°C孵育寄生虫1周28。
- 在 RPMI 培养基中收集代谢性胰小板。小心地倾斜烧瓶并移出溶液,而不会干扰粘附在底面的寄生虫。将介质转移到锥形管和离心机15分钟,在2,100 x g下。丢弃上清液,用 5 mL 的 DMEM (10% FCS) 重新悬浮寄生虫。
注:寄生虫群是代谢性胰体炎、表皮质和一些中间形态的混合物。虽然没有必要,但您可以通过DEAE电联色谱29分离锥形。或者,通过用活性血清孵育寄生虫来补充利沙30,可以消灭表皮。 - 种子3T3-瑞士白化细胞到60-70%汇合,或约1.7-2.0 x 106细胞在5mL的DMEM(10%FCS)在T-25培养瓶。去除生长介质,从步骤1.3.2中涂抹寄生虫混合物。在加湿培养箱中,在37°C下在5%CO2下孵育24小时,以建立感染。
- 用DMEM(10%FCS)洗涤共培养,两次去除宿主3T3细胞外的寄生虫。
- 一旦共培养饱和,通过胰蛋白酶化传播受肌炎感染的宿主细胞。吸气介质,用PBS冲洗培养液。应用1 mL的0.05%胰蛋白酶溶液覆盖整个培养表面,并在室温下孵育几分钟,直到附着的宿主细胞松动。通过冲洗 3 mL 的 DMEM (10% FCS) 在电池上分离从烧瓶表面的细胞。
- 将分离的宿主细胞转移到锥形管中,并在300 x g下离心3分钟。吸出上清液,用3 mL的新鲜DMEM(10%FCS)重新悬浮细胞。此步骤有助于消除剩余的表皮。将所有产品转移到含有 9 mL DMEM (10% FCS) 的清洁 T-75 烧瓶中。继续培养,直到胰蛋白酶体被释放到文化上清液。
- 通过每周两次的胰蛋白酶化来保持共同文化。一旦70-80%的宿主被感染,定期以5:1的比例添加未感染的宿主3T3细胞,以避免培养恶化。如果宿主细胞和T.cruzi之间的平衡得到适当维护,胰小球会不断出退。
2. 将试探药分化为 EA
- 在实验的前一天,去除宿主寄生虫共培养物的生长培养基,加入新鲜的DMEM(10%FCS),以洗去过去几天已经从宿主细胞中释放的EA和胰小孔。常规实验至少需要两个T-75瓶的汇合培养。
-
收集文化上清液放入锥形管中,收获新鲜出现的锥形锥形锥形。在显微镜下(10 倍或 20 倍物镜)下检查样品的质量。如果存在宿主细胞碎片,则短暂地将样品离心并将上清液转移到新管中。如果存在大量 EA,请通过以下游出程序分离胰小孔。
- 在2,100 x g下旋转锥形和乳腺的混合物15分钟。丢弃大部分上清液,在管中留下 0.5-1.0 mL 的介质。
注: 减小音量是可选的,但它使以下步骤更容易。 - 在37°C孵育颗粒1-2小时,允许活性胰小球从颗粒中游出(图2)。
- 将含有胰蛋白酶的上清液转移到1.5 mL微离心管中。
- 在2,100 x g下旋转锥形和乳腺的混合物15分钟。丢弃大部分上清液,在管中留下 0.5-1.0 mL 的介质。
- 在2,100 x g下将锥形管离心15分钟,以收集锥形胶。如果在上面执行游出程序,在 4,000 x g下将 1.5 mL 管离心 2 分钟,以颗粒锥形胶。丢弃上清液。
- 用5 mL的DMEM缓冲颗粒重新悬浮20mM MES(pH 5.0),辅以0.4%BSA19。将寄生虫转移到T-25培养瓶。保持盖子松弛。细胞密度必须围绕或低于1 x 107细胞/mL,因为过度饱和会增加细胞死亡的机会。
注:DMEM 的颜色必须为黄色,而不是橙色。如果原始 DMEM 具有高缓冲容量,则 20 mM MES (pH 5.0) 不足以降低 pH。在这种情况下,必须通过添加 HCl 来调整介质的 pHH。酸性介质可保持在4°C,但不超过1个月。 - 在加湿培养箱中孵育在37°C以下的5%CO2下培养瓶。约95%的寄生虫在24小时后分化成乳腺。
3. EA的电穿孔
- 准备gRNA进行电穿孔。这可以通过体外转录,或者简单地从制造商购买合成RNA寡核苷酸。在这份报告中,使用了来自综合DNA技术公司的crRNA和tracRNA。
- 在 2,100 x g下将 EA 培养值离心 15 分钟。丢弃上清液。
- 用含有电穿孔缓冲液的悬浮颗粒,为1 x 108细胞/mL的最终细胞密度提供补充溶液。
注:与材料表中列出的电穿孔缓冲液相比,EM缓冲液导致更多的细胞死亡;因此,不建议进行乳腺转染(补充图1)。 - 将再悬浮寄生虫(1 x 107细胞)的100 μL等值液放入1.5 mL微离心管中。加入5-10μgRNA,通过移液轻轻混合。
- 将混合物转移到 2 mm 间隙电穿孔。使用 X-14 程序使用电穿孔装置应用脉冲。
- 将比色皿内容物转移到含有 5 mL 预加热 LIT 介质(10% FCS)的 T-25 烧瓶中。保持盖子松弛,在 37°C 下孵育烧瓶,温度低于 5% CO2。
- 通过继续进行斧头培养(第4节)或在宿主细胞感染后作为细胞内乳腺(第5节)监测细胞生长。
4. 监测作为斧头阿马斯蒂戈特的挖空细胞的生长情况
- EA 位于培养基底,因此轻轻摇动烧瓶以将其重新悬浮到溶液中。通过移液清洗烧瓶表面有助于,因为有些细胞粘附在烧瓶上。
- 将 1 μL 碘化钠 (PI) 溶液 (20 μg/mL) 与 20 μL 的乳腺培养物混合。
注:请勿将培养瓶留在培养箱外的时间超过必要的时间。温度是使轴增增22的因素之一。 - 将样品涂在血细胞仪上,在荧光显微镜下观察。PI 是被损坏的细胞膜,但从活细胞排除。计算未被 PI 染色的可存活的乳腺的数量(例如 570 nm/602 nm)。
5. 监测作为细胞内肌要件的挖空细胞的生长情况
- 种子宿主 3T3 细胞在 12 孔板与 DMEM (10% FCS)。将细胞密度调整到70-80%的汇合度,或每孔约3 x 105个细胞。
注:由于乳腺不是可活动性,由宿主细胞覆盖大部分生长表面可提高感染效率。 - 电穿孔后一天通过离心从步骤 3.6 收集敲除乳石。丢弃上清液,用 2 mL 的 DMEM (10% FCS) 重新悬浮寄生虫。
- 从宿主细胞培养物中取出培养基,并应用重新悬浮的乳腺。感染的倍增率应为20或更高。在37°C下5%CO2孵育板2天,使乳腺建立感染。
注:感染期可以是1天,视用途而定。 - 使用 DMEM(10% FCS)将 EA 洗掉两次留在主机单元之外。
- 向宿主寄生虫共培养中加入新鲜的DMEM(10%FCS),并在37°C下继续孵育2天。
-
为了评估感染效率,可视化宿主细胞和细胞内乳腺的细胞核。
注:核在饱和共培养中往往重叠。在固定和染色之前以较低的细胞密度(如1:5稀释)重新镀层细胞有助于更容易地计数细胞核。- 去除培养基,在PBS中应用1mL的10%形式内溶液来修复细胞。在室温下孵育10分钟。
- 用含有 1 μg/mL Hoechst 33342 和 0.1% Triton X-100 的 1 mL PBS 替换形式溶液。在室温下孵育5分钟。
- 取出 Hoechst 溶液,用 PBS 冲洗细胞一次。添加 1 mL 的新鲜 PBS。
- 在荧光显微镜下观察,识别与较小寄生虫核相关的宿主细胞核(图4)。含有2个以上乳腺的宿主细胞应视为受感染,不包括非生产性初始感染或未洗涤的EA。
Access restricted. Please log in or start a trial to view this content.
结果
通过游出程序隔离胰岛
为了通过游出程序从污染旧的EA中收获新鲜的胰小球,细胞颗粒至少需要孵育1小时。 孵育颗粒超过2小时不会显著增加在溶液中游泳的胰小球的数量(图 2B.在这个特殊的实验中,在初始混合物中,胰小体测定的百分比为38%,在任意给定时间点,游出后百分比超过98%。从两个T-75瓶的汇合培养,我们经常获得3-4 x 107细胞的?...
Access restricted. Please log in or start a trial to view this content.
讨论
我们证明,在CRISPR/Cas9介导的基因敲除中,通过将gRNA直接电化到Cas9表达的EA中,可以利用T.克鲁兹的乳腺的轴培养。这样,在乳腺发育阶段,可以评估靶基因的本质,而无需经历其他发育阶段。
乳腺转染的另一个有益方面是对大量靶基因的检测方便。一旦Cas9表达T.cruzi和宿主哺乳动物细胞的共同培养建立,它只需要几天时间将组织衍生的胰腺炎转化为EA和转染gRNA,以获得敲?...
Access restricted. Please log in or start a trial to view this content.
披露声明
提交人没有利益冲突可披露。
致谢
这项工作部分得到了JSPS KAKENHI授予编号18K15141到Y.T.的部分支持。
Access restricted. Please log in or start a trial to view this content.
材料
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
参考文献
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。