
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Представляя ген нокаут непосредственно в Amastigote Этап trypanosoma cruzi Использование CRISPR / Cas9 системы
В этой статье
Резюме
Здесь мы описываем протокол о введении генного нокаута во внеклеточном аматиготе трипаносома крузи, с использованием системы CRISPR/Cas9. За фенотипом роста может последовать либо подсчет клеток агостической культуры аматигота, либо распространение внутриклеточного амастигота после вторжения клеток-хозяев.
Аннотация
Трипаносома крузи является патогенным протозоанским паразитом, который вызывает болезнь Шагаса в основном в Латинской Америке. Для того, чтобы определить новый целевой препарат против T. cruzi, важно проверить существенность гена-митариона в стадии млекопитающих паразита, амастигот. Аматиготы T. cruzi реплицируются внутри клетки-хозяина; таким образом, трудно провести нокаут эксперимент, не проходя через другие этапы развития. Недавно наша группа сообщила о состоянии роста, в котором амастигот может реплицировать аксенически до 10 дней, не теряя своих амастигот-подобных свойств. Используя эту временную апорную культуру амасигота, мы успешно внедрили GРНС непосредственно в Cas9-expressing amastigote, чтобы вызвать генные нокауты и проанализировали их фенотипы исключительно в стадии амасигота. В этом отчете мы описываем подробный протокол для производства in vitro, полученных внеклеточными амазиготами, и для использования опорной культуры в эксперименте по нокауту, опосредованным CRISPR/Cas9. Фенотип роста нокаут амастиготов может быть оценен либо по клеточным подсчетам аксеников, либо путем репликации внутриклеточного аматигота после вторжения клеток-хозяев. Этот метод обходит дифференциацию стадии паразита, обычно участвующую в производстве трансгенного или нокаутирующего аматигота. Использование временной апорической культуры амастигот имеет потенциал для расширения экспериментальной свободы сценических исследований в T. cruzi.
Введение
Трипаносома крузи является возбудимым возбудите болезнь Шагаса, которая распространена в основном в Латинской Америке1. T. cruzi имеет отличительные этапы жизненного цикла, как он путешествует между насекомым вектор и млекопитающих хозяин2. T. cruzi реплицирует сярлица в качестве эпиматигота в середине г. кровососущего триатомина и дифференцируется в инфекционный метациклический трипомэтигот в его заднухе перед тем, как оставить на человека или животного хозяина. Как только trypomastigote попадает в организм хозяина через место укуса или через слизистую оболочку, паразит вторгается в клетку-хозяина и превращается в флагелла менее круглая форма называется амасигот. Амасигот реплицируется внутри клетки-хозяина и в конечном итоге дифференцируется в трипоматигот, который вырывается из клетки-хозяина и входит в кровоток, чтобы заразить другую клетку-хозяина.
Поскольку в настоящее время доступны химиотерапевтические агенты, бензнидазол и нифуртимокс, вызывают неблагоприятные побочные эффекты и неэффективны в хронической фазе заболевания3, это представляет большой интерес для выявления новых целей наркотиков против T. cruzi. В последние годы, система CRISPR/Cas9 стала мощным инструментом для эффективного выполнения генного нокаута в T. cruzi, либо путем трансфекции отдельных или одной плазмиды (ы), содержащей gRNA и Cas94, путем стабильного выражения Cas9 и последующего введение gRNA5,6,7 или транскрипции шаблона gRNA8, или путем электропорации предварительно сформированного комплекса gRNA/Cas9 RNP7,9. Этот технологический прогресс, как ожидается, ускорить целевое исследование наркотиков в области болезни Шагаса.
Чтобы продолжить разработку препарата, очень важно проверить существенность гена-мишени или эффективность соединений кандидата препарата в amastigote T. cruzi, так как это этап репликации паразита в млекопитающих хозяина. Однако, это трудная задача, потому что amastigotes не может быть непосредственно манипулировать из-за присутствия обструктивной ячейки хозяина. В Лейшмании, тесно связанных простейшие паразита T. cruzi, апорный метод культивирования amastigote был разработан и был использован в анализе скрининга наркотиков10,11,12, 13. Хотя Есть некоторые расхождения в восприимчивости к соединениям между апорическими амастиготами и внутриклеточными амастиготами14, способность поддерживать топорную культуру, тем не менее, предоставляет ценные экспериментальные инструменты для изучения базовая биология клинически релевантной стадии Лейшмания15,16. В случае T. cruzi, литературы о наличии естественных внеклеточных амастиготов (EA)17 и в пробирке производства EA17,18,19 датируются десятилетия назад. Кроме того, Е.А., как известно, инфекционные возможности20, хотя и меньше, чем у trypomastigote, и механизм вторжения амасигота принимающей была выяснена в последние годы (обзор Bonfim-Мело и др.21). Однако, в отличие от Leishmania,EA не был использован в качестве экспериментального инструмента в T. cruzi, в первую очередь потому, что EA считался обязанным внутриклеточным паразитом, и, таким образом, не рассматривался как "репликативная форма" в практической Смысле.
Недавно наша группа предложила использовать EA T. cruzi в качестве временной апорической культуры22. Amastigotes штамма T. cruzi Tulahuen может размножаться без клеток-хозяев в среде LIT при 37 градусах По Цельсия в течение 10 дней без серьезного ухудшения или потери амасиготных свойств. В период роста без хозяина, EA был успешно использован для экспрессии экзогенного гена обычным электропорированием, проверкой титрации препарата с трипаноцидальными соединениями и нокаутом CRISPR/Cas9 с последующим мониторингом фенотипа роста. В этом отчете мы описываем подробный протокол для производства in vitro, полученного EA, и использования аксена в экспериментах нокаута.
Access restricted. Please log in or start a trial to view this content.
протокол
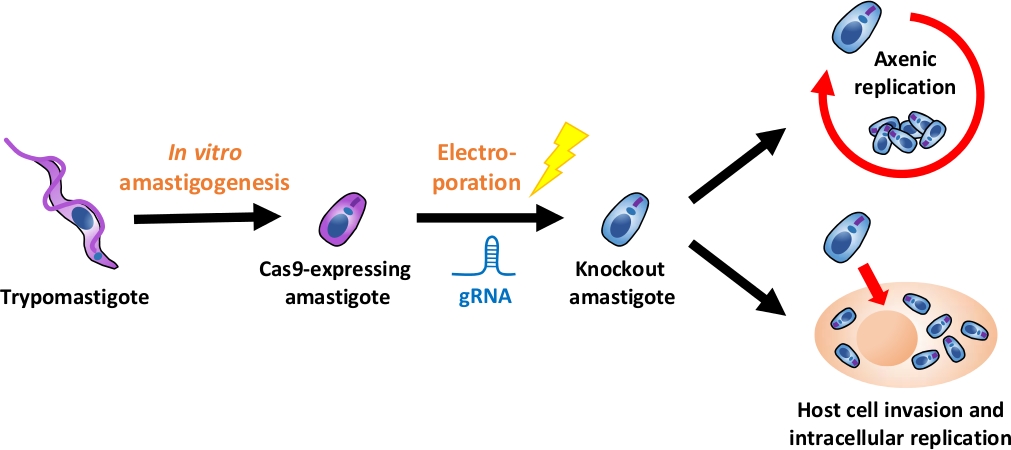
ПРИМЕЧАНИЕ: Обзор всего экспериментального потока изображен на рисунке 1.

Рисунок 1: Обзор эксперимента нокаута с использованием EA. Ткань культуры полученных trypomastigotes собираюти и дифференцируются в EA. gRNA трансфицируется в Cas9-выражения амастиготы путем электропорации, и рост фенотипа нокаут амастигот оценивается либо по осяжливой репликации или по внутриклеточной репликации после вторжения клеток-хозяев. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
1. Подготовка культуры паразитов
- Тулахуэн штамм трипаносома крузи используется во всем этом докладе. Поддержание эпимастиготов T. cruzi в lit среде (10% FCS, см. Дополнительную таблицу 1). Надежно закройте крышку и держите культурную колбу при 28 градусах Цельсия.
- Создайте трансгенный штамм T. cruzi, который улавливает эндонуклеазы Cas9. Примеры плазмидов выражения, которые содержат последовательность кодирования Cas9и маркер выбора G418 (neo r) можно найти в Ландер и др.4 и Пэн и др.5
- Трансфект выше плазмиды в эпимастигот путем электропорации, используя комплект, перечисленные в таблице материалов. Для 1 cuvette, спина вниз 2 х 107 клеток, отказаться от супернатанта, и resuspend с 100 зликатбуфер, содержащий предусмотренные дополнения решения.
ПРИМЕЧАНИЕ: Электропорный буфер можно заменить буфером EM24 (3:1 смесь цитомикса25 и фосфатно-сахарозного буфера). - Добавьте 20-40 мкг плазмиды и перенесите смесь в 2 мм зазор электропорации cuvette. Нанесите пульс с электропорационным устройством (см. Таблицаматериалов), используя программу X-1426.
- Передача содержимого кветки в колбу Т-25, содержащую 5 мл среды LIT (10% FCS). Инкубировать колбу при 28 градусах по Цельсию в течение 24 ч.
- Добавьте G418 к конечной концентрации 250 мкг/мл и продолжайте инкубацию при 28 градусах Цельсия. Это займет около 1 недели, чтобы убить не-транс-инфицированных эпимастиготов. Как только популяция, устойчивая к G418, начинает восстанавливаться, проходите культуру один или два раза в неделю, чтобы избежать насыщения и разбавить мертвые клетки. Создание стабильной клеточной линии обычно занимает в общей сложности 4 недели.
ПРИМЕЧАНИЕ: Если составное выражение Cas9 является бременем для клетки5, сделать выражение специфичным для стадии amastigote путем сопоставления 3'-UTR гена амастин сразу вниз по течению Кас9 открытый кадр чтения27. Подробное описание плазмида выражения Cas9, специфичная для амастигота, можно найти в такаги и др.
- Трансфект выше плазмиды в эпимастигот путем электропорации, используя комплект, перечисленные в таблице материалов. Для 1 cuvette, спина вниз 2 х 107 клеток, отказаться от супернатанта, и resuspend с 100 зликатбуфер, содержащий предусмотренные дополнения решения.
- Создайте ко-культуру хоста-паразита Cas9-выражения T. cruzi и клетки-хозяева млекопитающих. В этом отчете используйте 3T3-швейцарскую ячейку фибробластов Альбино в качестве хозяина.
- Дифференцируйте эпимастиготы T. cruzi в метациклические трипомастиготы. Определите плотность клеток эпимастиготной культуры с помощью гемоцитометра и соберите 5 х 107 клеток центрифугированием в течение 15 мин при 2100 х г. Откажитесь от супернатанта и отрепримите клетки с помощью 10 мл среды RPMI. Инкубировать паразита при 28 градусах По Цельсию в течение 1 недели28.
- Сбор метациклических трипомочиготов в среде RPMI. Тщательно наклоните колбу и пипетку из раствора, не нарушая паразитов придерживаться нижней поверхности. Перенесите среду в коническую трубку и центрифугу в течение 15 мин при 2100 х г. Откажитесь от супернатанта и отразите паразита с помощью 5 мл DMEM (10% FCS).
ПРИМЕЧАНИЕ: Популяция паразитов представляет собой смесь метациклических трипоматиготов, эпимастиготов и некоторых промежуточных форм. Хотя это и не нужно, вы можете изолировать trypomastigote DEAE ионный обмен хроматографии29. Кроме того, эпимастиготы могут быть устранены путем инкубации паразитов с активной сывороткой подвергать их в дополнение к лисис30. - Семена 3T3-Швейцарский Альбино фибробластной ячейки до 60-70% confluency, или около 1,7-2,0 х 106 ячеек в 5 мл DMEM (10% FCS) в Т-25 культуры колбы. Удалить среды роста и применять смесь паразита от шага 1.3.2. Инкубировать для 24 ч при 37 градусах Цельсия под 5% CO2 в увлажненный инкубатор для установления инфекции.
- Удалите паразитов, оставшихся за пределами клетки хозяина 3T3, промывая со-культуру с DMEM (10% FCS) дважды.
- После того, как со-культура насыщена, прохождение аматигот-инфицированных клеток-хозяев путем трипсинизации. Аспирировать среды и промыть культуру один раз с PBS. Нанесите 1 мл 0,05% трипсина раствор, чтобы покрыть всю поверхность культуры, и инкубировать в течение нескольких минут при комнатной температуре, пока прилагается клетки хозяина становятся свободными. Отсоедините клетки от поверхности колбы, промыв 3 мл DMEM (10% FCS) над клетками.
- Перенесите отдельные клетки-хозяина в коническую трубку и центрифугу в течение 3 мин при 300 х г. Аспирируй супернатант и resuspend клетки с 3 мл свежего DMEM (10% FCS). Этот шаг помогает устранить оставшиеся эпимастиготы. Перенесите все в чистую колбу Т-75, содержащую 9 мл DMEM (10% FCS). Продолжайте культивировать до тех пор, пока trypomastigote не будет выпущен в культуру супернатанта.
- Поддерживайте сокультуру, проходя с трипсинизацией два раза в неделю. После того, как 70-80% принимающей популяции заражаются, регулярно добавляйте неинфицированные клетки хозяина 3Т3 в соотношении 5:1 (перенос:свежий), чтобы избежать ухудшения культуры. Трипомастигот выгравирует непрерывно, если баланс между клетками-хозяинами и T. cruzi поддерживается должным образом.
2. Дифференциация трипоматиготов в Е.А.
- За день до этого эксперимента, удалить среду роста принимающей паразита совместной культуры и добавить свежие DMEM (10% FCS), чтобы смыть Е.А. и trypomastigotes, которые уже были освобождены из клеток хозяина в предыдущие дни. Регулярные эксперименты требуют, по крайней мере, двух фляг Т-75 сопливой сокультуры.
-
Соберите культуры супернатанта в коническую трубку для сбора свежевытевшиеся trypomastigotes. Проверьте образец под микроскопом (10x или 20x объектив) на качество. Если есть мусор клетки-хозяина, кратко центрифугировать образец и перенести супернатант в новую трубку. Если есть значительное количество eAs, изолировать трипомастиготы по следующей процедуре плавания.
- Спин вниз смесь trypomastigotes и amastigotes в течение 15 мин на 2100 х г. Отбросьте большую часть супернатанта, оставив в трубке 0,5-1,0 мл среды.
ПРИМЕЧАНИЕ: Уменьшение громкости не является обязательным, но это делает следующий шаг проще. - Инкубировать гранулы при 37 градусах по Цельсию в течение 1-2 ч, что позволяет активным трипомастиготам выплыть из гранул(рисунок2).
- Перенесите супернатант, содержащий трипомастиготы, в микроцентрифугную трубку 1,5 мл.
- Спин вниз смесь trypomastigotes и amastigotes в течение 15 мин на 2100 х г. Отбросьте большую часть супернатанта, оставив в трубке 0,5-1,0 мл среды.
- Центрифуги коническая трубка в течение 15 минут на 2100 х г для сбора trypomastigotes. Если процедура плавания была выполнена выше, центрифуга 1,5 мл трубки в течение 2 минут на 4000 х г, чтобы гранулы trypomastigote. Отбросьте супернатант.
- Приостановить гранулы с 5 мл DMEM буфером с 20 мМ MES (pH 5.0), дополненный 0,4% BSA19. Перенесите паразита в культурную колбу Т-25. Оставьте крышку свободной. Плотность клеток должна быть вокруг или ниже 1 х 107 клеток /мл, так как перенасыщение увеличивает вероятность гибели клеток.
ПРИМЕЧАНИЕ: Цвет DMEM должен быть желтым, а не оранжевым. Если первоначальный DMEM имел высокую буферную емкость, 20 мМ MES (pH 5.0) недостаточно для снижения рН. РН среды должен быть скорректирован путем добавления HCl в этом случае. Кислотная среда может храниться при 4 градусах Цельсия, но не более 1 месяца. - Инкубировать культурную колбу при 37 градусах Цельсия под 5% CO2 в увлажненный инкубатор. Около 95% паразитов дифференцируются в амастиготы после 24 ч.
3. Электропорация Е.А.
- Подготовка gRNA для электропорации. Это может быть сделано путем экстракорпорации, или просто путем покупки синтетических олигонуклеотидов РНК от производителя. В этом отчете используются crRNA и tracrRNA от Integrated DNA Technologies, Inc.
- Центрифуги культуры eAs в течение 15 мин на 2100 х г. Откажитесь от супернатанта.
- Resuspend гранулы с электропорации буфера, содержащего предусмотрено дополнение решение окончательной плотности клеток 1 х 108 клеток / мл.
ПРИМЕЧАНИЕ: БУФЕР EM вызывает больше смертей клеток по сравнению с буфером электропорации, перечисленным в таблицематериалов; таким образом, не рекомендуется для трансфекции амастигота(Дополнительная рисунок 1). - Aliquot 100 л повторнотуло паразитов (1 х 107 клеток) в 1,5 мл микроцентрифуговых труб. Добавьте 5-10 мкг гРНК и аккуратно перемешайте путем пипетки.
- Перенесите смесь в 2 мм зазор электропорации cuvette. Нанесите пульс с помощью электропорационного устройства, используя программу X-14.
- Перенесите содержимое кювета в колбу Т-25, содержащую 5 мл предварительно разогретого средства LIT (10% FCS). Оставьте крышку свободной и инкубировать колбу на 37 градусов по Цельсию под 5% CO2.
- Мониторинг роста клеток либо путем продолжения апорического культивирования (раздел 4), либо как внутриклеточные амасиготы после инфекции клетки-хозяина (раздел 5).
4. Мониторинг роста нокаутов клеток, как Аксеник Amastigotes
- EAs поселиться в нижней части среды культуры, так что осторожно встряхнуть колбу, чтобы повторно приостановить их в раствор. Мытье поверхности колбы путем пайпеттингпомогает помогает, так как некоторые клетки прилипают к колбе.
- Смешайте 1 зл иодийного раствора (PI) (20 мкг/мл) с 20 зл культуры амастигота.
ПРИМЕЧАНИЕ: Не оставляйте культурную колбу за пределами инкубатора дольше, чем это необходимо. Температура является одним из факторов, который позволяет топором пролиферации22. - Нанесите образец на гемоцитометр и наблюдайте под флуоресцентным микроскопом. PI является permeant к поврежденной мембране клетки, но исключается из живых клеток. Подсчитайте количество жизнеспособных амастиготов, которые не окрашены PI (ex/em 570 nm/602 нм).
5. Мониторинг роста клеток нокаута как внутриклеточных амастиготов
- Семена хост 3T3 клетки в 12-хорошо пластины с DMEM (10% FCS). Отрегулируйте плотность клеток до 70-80% стоек, или около 3 х 105 ячеек на скважину.
ПРИМЕЧАНИЕ: Так как амастиготы не являются motile, покрывая большую часть поверхности роста клетками хозяина повышает эффективность инфекции. - Соберите нокаут амастиготы от шага 3,6 на центрифугацию на следующий день после электропорации. Откажитесь от супернатанта и приостановите работу паразита с 2 мл DMEM (10% FCS).
- Удалить среду из культуры ячейки хозяина и применить повторно amastigotes. Множественность инфекции должна быть 20 или выше. Инкубировать пластину при 37 градусах Цельсия под 5% CO2 в течение 2 дней, чтобы амастиготы установить инфекцию.
ПРИМЕЧАНИЕ: Инфекционный период может быть 1 день, в зависимости от цели. - Смыть eAs остались за пределами клетки хозяина дважды с DMEM (10% FCS).
- Добавьте свежий DMEM (10% FCS) в ко-культуру хоста-паразита и продолжайте инкубацию при 37 градусах По Цельсию в течение дополнительных 2 дней.
-
Чтобы оценить эффективность инфекции, визуализировать ядра клеток-хозяев и внутриклеточного арастигота.
ПРИМЕЧАНИЕ: Ядра, как правило, перекрываются в насыщенной совместной культуре. Повторное покрытие клеток при более низкой плотности клеток (например, 1:5 разбавления) до фиксации и окрашивания помогает легче подсчитать ядра.- Удалить среду культуры и применить 1 мл 10% формина раствор в PBS, чтобы исправить клетки. Инкубировать в течение 10 минут при комнатной температуре.
- Замените раствор формалина 1 мл PBS, содержащий 1 мкг/мл Hoechst 33342 и 0,1% Triton X-100. Инкубировать в течение 5 минут при комнатной температуре.
- Удалите раствор Hoechst и промыть клетки один раз с PBS. Добавьте 1 мл свежего PBS.
- Наблюдайте под флуоресценцией и определите ядра клеток-хозяев, которые связаны с меньшими ядрами паразита(рисунок4). Клетки-хозяина, содержащие более 2 амасиготов, следует рассматривать как инфицированные, не включая непродуктивные первоначальные инфекции или немытые советники.
Access restricted. Please log in or start a trial to view this content.
Результаты
Изоляция трипомастиготов процедурой плавания
Для сбора свежих трипомастиготов от загрязнения старых овеных аув путем плавания- искупаться клетки нужно инкубировать не менее чем на 1 ч. Инкубирование гранул более чем на 2 ч не значительно увеличивает количество...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Мы продемонстрировали, что топорная культура T. cruzi amastigotes может быть использована в CRISPR/Cas9-опосредованного гена нокаутом, путем электропорабивания gRNA непосредственно в Cas9-выражения EA. Таким образом, существенность гена-мишени конкретно в стадии amastigote может быть оценена, не проходя...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторы не имеют конфликта интересов раскрывать.
Благодарности
Эта работа была частично поддержана JSPS KAKENHI Грант номер 18K15141 к Y.T.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
Ссылки
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены