
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Présentation d'un Knockout Génique Directement dans la phase Amastigote de Trypanosoma cruzi à l'aide du système CRISPR/Cas9
Dans cet article
Résumé
Ici, nous décrivons un protocole pour introduire un KO de gène dans l'amastigote extracellulaire de Trypanosoma cruzi, utilisant le système CRISPR/Cas9. Le phénotype de croissance peut être suivi soit par le comptage cellulaire de la culture amastigote axenique ou par la prolifération des amastigotes intracellulaires après l'invasion de cellules hôtes.
Résumé
Trypanosoma cruzi est un parasite protozoaire pathogène qui cause la maladie de Chagas principalement en Amérique latine. Afin d'identifier une nouvelle cible médicamentelle contre T. cruzi, il est important de valider l'essentiel du gène cible au stade des mammifères du parasite, l'amastigote. Les amastigotes de T. cruzi se reproduisent à l'intérieur de la cellule hôte; ainsi, il est difficile de mener une expérience knock-out sans passer par d'autres stades de développement. Récemment, notre groupe a signalé une condition de croissance dans laquelle l'amastigote peut se répliquer axenically pendant jusqu'à 10 jours sans perdre ses propriétés amastigote-like. En utilisant cette culture temporelle d'amastigote axenique, nous avons introduit avec succès des gARN directement dans l'amastigote cas9-exprimant pour causer des knockouts de gène et avons analysé leurs phénotypes exclusivement dans le stade d'amastigote. Dans ce rapport, nous décrivons un protocole détaillé pour produire des amastigotes extracellulaires dérivés in vitro, et pour employer la culture axenic dans une expérience de KNOCK-ball CRISPR/Cas9-négociée. Le phénotype de croissance des amastigotes knock-out peut être évalué soit par le nombre de cellules de la culture axenique, soit par la réplication de l'amastigote intracellulaire après l'invasion de cellules hôtes. Cette méthode contourne la différenciation du stade parasite normalement impliquée dans la production d'un amastigote transgénique ou kok. L'utilisation de la culture temporelle de l'amastigote axenique a le potentiel d'élargir la liberté expérimentale des études spécifiques à la scène dans T. cruzi.
Introduction
Trypanosoma cruzi est l'agent causal de la maladie de Chagas, qui est répandue principalement en Amérique latine1. T. cruzi a des stades distinctifs du cycle de vie comme il se déplace entre un insecte vecteur et un hôte de mammifères2. T. cruzi se reproduit comme épimastigote dans le milieu d'un insecte triatomine suceur de sang et se différencie en un trypomastigote métacyclique infectieux dans son intestin postérieur avant d'être déposé sur un hôte humain ou animal. Une fois que le trypomastigote pénètre dans le corps hôte par le site de morsure ou par une muqueuse, le parasite envahit une cellule hôte et se transforme en une forme ronde sans flagella appelée amastigote. L'amastigote se reproduit dans la cellule hôte et finit par se différencier en trypomastigote, qui éclate hors de la cellule hôte et pénètre dans la circulation sanguine pour infecter une autre cellule hôte.
Puisque les agents chimiothérapeutiques actuellement disponibles, le benznidazole et le nifurtimox, causent des effets secondaires défavorables et sont inefficaces dans la phase chronique de la maladie3,il est d'un grand intérêt d'identifier de nouvelles cibles de drogue contre T. cruzi. Ces dernières années, le système CRISPR/Cas9 est devenu un outil puissant pour effectuer efficacement ko en gène dans T. cruzi, soit par transfection de plasmides distincts ou simples contenant gRNA et Cas94, par expression stable de Cas9 et subséquent introduction de gRNA5,6,7 ou modèle de transcription de gRNA8, ou par électroporation du complexe pré-formé gRNA/Cas9 RNP7,9. Cette avancée technologique devrait accélérer la recherche sur les médicaments ciblés dans la maladie de Chagas.
Pour procéder à la mise au point du médicament, il est crucial de valider l'essentiel du gène cible ou l'efficacité des composés candidats médicamenteux dans l'amastigote de T. cruzi, car c'est l'étape de réplication du parasite chez l'hôte mammifère. Cependant, il s'agit d'une tâche difficile, car les amastigotes ne peuvent pas être manipulés directement en raison de la présence d'une cellule hôte obstructive. Dans Leishmania, un parasite protozoaire étroitement lié à T. cruzi, une méthode de culture amastigote axenique a été développé et a été utilisé dans les essais de dépistage de drogues10,11,12, 13. Bien qu'il existe certaines divergences de susceptibilité aux composés entre les amastigotes axeniques et les amastigotes intracellulaires14, la capacité de maintenir la culture axenique fournit néanmoins des outils expérimentaux précieux pour étudier le biologie de base de l'étape cliniquement pertinente de Leishmania15,16. Dans le cas de T. cruzi, les littératures concernant la présence d'amastigotes extracellulaires naturels (EA)17 et la production in vitro d'EA17,18,19 remontent à Décennies. En outre, EA est connu pour avoir une capacité infectieuse20, bien que inférieure à celle de trypomastigote, et le mécanisme de l'invasion hôte amastigote a été élucidé ces dernières années (passé en revue par Bonfim-Melo et al.21 ). Cependant, contrairement à Leishmania, EA n'avait pas été utilisé comme un outil expérimental dans T. cruzi, principalement parce que EA avait été considéré comme un parasite intracellulaire obligatoire, et n'avait donc pas été considéré comme «forme réplicatrice» dans une pratique bon sens.
Récemment, notre groupe a proposé d'utiliser EA de T. cruzi comme culture axenique temporelle22. Les amastigotes de la souche T. cruzi Tulahuen peuvent se répliquer sans cellules hôtes dans le milieu de LIT à 37 oC pendant 10 jours sans détérioration majeure ou perte de propriétés semblables à des amastigotes. Pendant la période de croissance hôte-libre, EA a été avec succès utilisé pour l'expression génique exogène par l'électroporation conventionnelle, essai de titration de drogue avec des composés trypanocidal, et KO CRISPR/Cas9-négocié suivi de la surveillance de phénotype de croissance. Dans ce rapport, nous décrivons le protocole détaillé pour produire l'EE dérivé in vitro et pour employer l'amastigote axenic dans des expériences de knock-out.
Access restricted. Please log in or start a trial to view this content.
Protocole
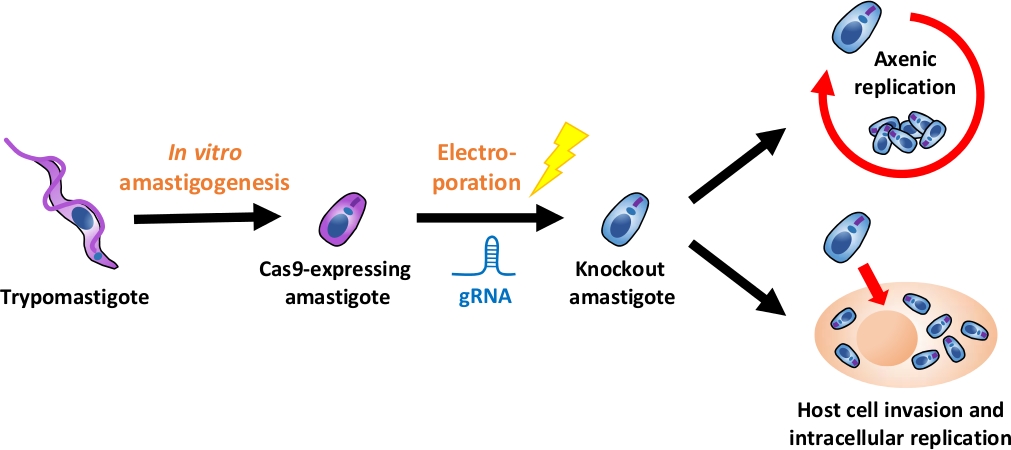
REMARQUE: Un aperçu de l'ensemble du flux expérimental est représenté dans la figure 1.

Figure 1 : Aperçu de l'expérience par KO à l'aide d'EA. Les trypomastigotes dérivés de la culture tissulaire sont récoltés et différenciés en EA. gRNA est transfecté en amastigotes exprimant Cas9 par électroporation, et le phénotype de croissance de l'amastigote knock-out est évalué soit par la réplication axenique ou par réplication intracellulaire après l'invasion des cellules hôtes. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
1. Préparations à la culture parasite
- La souche Tulahuen de Trypanosoma cruzi est utilisée tout au long de ce rapport. Maintenir les épimastigotes de T. cruzi dans le milieu LIT (10% FCS, voir Tableau supplémentaire 1). Fermez solidement le bouchon et maintenez le flacon de culture à 28 oC.
- Générer une souche transgénique de T. cruzi qui abrite l'endonucléase Cas9. Des exemples de plasmides d'expression qui contiennent laséquence de codage Cas9 et le marqueur de sélection G418 (néo r) peuvent être trouvés dans Lander et al.4 et Peng et al.5
- Transfect le plasmide ci-dessus en épimastigote par électroporation, en utilisant le kit énuméré dans le tableau des matériaux. Pour 1 cuvette, faire tourner 2 x 107 cellules, jeter le supernatant, et resuspendre avec 100 L de tampon d'électroporation contenant la solution de supplément fournie.
REMARQUE: Le tampon d'électroporation peut être remplacé par un tampon EM24 (3:1 mélange de cytomix25 et de tampon phosphate-saccharose). - Ajouter 20-40 g de plasmide et transférer le mélange dans une cuvette d'électroporation de 2 mm. Appliquer l'impulsion à l'aide d'un dispositif d'électroporation (voir Tableau des matériaux),en utilisant le programme X-1426.
- Transférer le contenu de la cuvette dans un flacon T-25 contenant 5 ml de milieu LIT (10 % de FCS). Incuber le flacon à 28 oC pendant 24 h.
- Ajouter le G418 à une concentration finale de 250 g/mL et poursuivre l'incubation à 28 oC. Il faut environ 1 semaine pour tuer les épimastigotes non transfectés. Une fois que la population résistante au G418 commence à se rétablir, passageez la culture une ou deux fois par semaine pour éviter la saturation et diluer les cellules mortes. L'établissement d'une lignée cellulaire stable prend habituellement au total 4 semaines.
REMARQUE: Si l'expression constitutive de Cas9 est un fardeau pour la cellule5, rendre l'expression spécifique à l'étape amastigote en conjuguant le 3'-UTR du gène amastin immédiatement en aval du cadre de lecture ouvert Cas927. La description détaillée du plasmide d'expression Cas9 spécifique à l'amastigote se trouve dans Takagi et al.22
- Transfect le plasmide ci-dessus en épimastigote par électroporation, en utilisant le kit énuméré dans le tableau des matériaux. Pour 1 cuvette, faire tourner 2 x 107 cellules, jeter le supernatant, et resuspendre avec 100 L de tampon d'électroporation contenant la solution de supplément fournie.
- Établir une co-culture hôte-parasite de T. cruzi exprimant Cas9 et de cellules hôtes mammifères. Dans ce rapport, utilisez la cellule fibroblaste 3T3-Swiss Albino comme hôte.
- Différencier les épimastigotes de T. cruzi en trypomastigotes métacycliques. Déterminer la densité cellulaire de la culture des épimastigotes à l'aide d'un hémocytomètre et recueillir 5 x 107 cellules par centrifugation pendant 15 min à 2 100 x g. Jeter le supernatant et resuspendre les cellules avec 10 ml de milieu RPMI. Incuber le parasite à 28 oC pendant 1 semaine28.
- Recueillir des trypomastigotes métacycliques dans le milieu RPMI. Inclinez soigneusement le flacon et déviez la solution sans déranger les parasites adhérés à la surface inférieure. Transférer le milieu sur un tube conique et une centrifugeuse pendant 15 min à 2 100 x g. Jeter le supernatant et resuspendre le parasite avec 5 ml de DMEM (10% FCS).
REMARQUE: La population de parasites est un mélange de trypomastigotes métacycliques, d'épimastigotes et de certaines formes intermédiaires. Bien que pas nécessaire, vous pouvez isoler trypomastigote par DEAE ion échange chromatographie29. Alternativement, les épimastigotes peuvent être éliminés en incuber les parasites avec le sérum actif pour les soumettre pour compléter la lyse30. - Seed 3T3-Swiss Albino fibroblast cell to 60-70% confluency, or about 1.7-2.0 x 106 cells in 5 mL of DMEM (10% FCS) in T-25 culture flask. Retirer le milieu de croissance et appliquer le mélange parasite de l'étape 1.3.2. Incuber pendant 24 h à 37 oC sous 5 % de CO2 dans un incubateur humidifié pour établir l'infection.
- Enlever les parasites restant à l'extérieur des cellules 3T3 de l'hôte en lavant la co-culture avec DMEM (10% FCS) deux fois.
- Une fois que la co-culture est saturée, passer les cellules hôtes infectées par l'amastigote par trypsinisation. Aspirer le médium et rincer la culture une fois avec PBS. Appliquer 1 ml de solution trypsine de 0,05 % pour couvrir toute la surface de la culture, et couver pendant quelques minutes à température ambiante jusqu'à ce que les cellules hôtes attachées se détachent. Détachez les cellules de la surface du flacon en rinçant 3 mL de DMEM (10 % de FCS) sur les cellules.
- Transférer les cellules hôtes détachées dans un tube conique et la centrifugeuse pendant 3 min à 300 x g. Aspirer le supernatant et resuspendre les cellules avec 3 ml de DMEM frais (10% FCS). Cette étape permet d'éliminer les épimastigotes restants. Transférer le tout dans un flacon T-75 propre contenant 9 ml de DMEM (10% FCS). Continuer à cultiver jusqu'à ce que trypomastigote est libéré dans la culture supernatant.
- Maintenir la co-culture en passant avec trypsinisation deux fois par semaine. Une fois que 70-80% de la population hôte sont infectées, ajoutez régulièrement des cellules 3T3 non infectées d'hôte au rapport de 5:1 (carryover : frais), afin d'éviter la détérioration de culture. Trypomastigote s'égresse continuellement si l'équilibre entre les cellules hôtes et T. cruzi est correctement maintenu.
2. Différenciation des trypomastigotes en EA
- La veille de cette expérience, retirez le milieu de croissance de la co-culture hôte-parasite et ajoutez du DMEM frais (10% FCS) pour laver l'EA et les trypomastigotes qui avaient déjà été libérés des cellules hôtes dans les jours précédents. Les expériences régulières nécessitent au moins deux flacons T-75 de co-culture confluente.
-
Recueillir supernatant de culture dans un tube conique pour récolter les trypomastigotes fraîchement émergés. Vérifier l'échantillon au microscope (10x ou 20x objectif objectif) pour la qualité. S'il y a des débris de cellules hôtes, centrifuge brièvement l'échantillon et transférer le supernatant dans un nouveau tube. S'il y a un nombre important d'EE, isolez les trypomastigotes par la procédure suivante de nage-out.
- Faites tourner le mélange de trypomastigotes et d'amastigotes pendant 15 min à 2 100 x g. Jeter la plupart du supernatant, en laissant 0,5-1,0 ml de milieu dans le tube.
REMARQUE : La réduction du volume est facultative, mais elle facilite l'étape suivante. - Incuber la pastille à 37 oC pendant 1 à 2 h, ce qui permet aux trypomastigotes actifs de nager hors de la pastille (Figure 2).
- Transférer le supernatant contenant des trypomastigotes dans un tube microcentrifuge de 1,5 ml.
- Faites tourner le mélange de trypomastigotes et d'amastigotes pendant 15 min à 2 100 x g. Jeter la plupart du supernatant, en laissant 0,5-1,0 ml de milieu dans le tube.
- Centrifuger le tube conique pendant 15 min à 2 100 x g pour recueillir les trypomastigotes. Si la procédure de nage-out a été effectuée ci-dessus, centrifuger le tube de 1,5 ml pendant 2 min à 4.000 x g pour granuler le trypomastigote. Jetez le supernatant.
- Resuspendre la pastille avec 5 ml de DMEM tamponné avec 20 mM MES (pH 5.0), complété par 0,4% BSA19. Transférer le parasite dans un flacon de culture T-25. Laisse le bouchon en vrac. La densité cellulaire doit être d'environ ou en dessous de 1 x 107 cellules/mL, car la sursaturation augmente le risque de mort cellulaire.
REMARQUE: La couleur du DMEM doit être jaune, pas orange. Si le DMEM d'origine avait une capacité tampon élevée, 20 mM MES (pH 5.0) ne suffit pas à abaisser le pH. Le pH du milieu doit être ajusté par ajout de HCl dans ce cas. Le milieu acide peut être maintenu à 4 oC, mais pas plus d'un mois. - Incuber le flacon de culture à 37 oC sous 5 % de CO2 dans un incubateur humidifié. Environ 95% des parasites se différencient en amastigotes après 24 h.
3. Électroporation de l'EE
- Préparer le gRNA pour l'électroporation. Cela peut être fait par transcription in vitro, ou tout simplement en achetant des oligonucléotides à ARN synthétique d'un fabricant. Dans ce rapport, crRNA et tracrRNA de Integrated DNA Technologies, Inc. sont utilisés.
- Centrifugelant la culture des EA pendant 15 min à 2 100 x g. Jetez le supernatant.
- Resuspendre le granule avec tampon d'électroporation contenant fourni une solution de supplément à la densité cellulaire finale de 1 x 108 cellules/mL.
REMARQUE : Le tampon EM cause plus de décès cellulaires que le tampon d'électroporation énuméré dans tableau des matériaux; ainsi, il n'est pas recommandé pour la transfection amastigote (Figure supplémentaire 1). - Aliquot 100 l de parasites en suspension (1 x 107 cellules) en tubes microcentrifuges de 1,5 ml. Ajouter 5 à 10 g de gRNA et mélanger délicatement par pipetting.
- Transférer le mélange dans une cuvette d'électroporation de 2 mm. Appliquer l'impulsion avec le dispositif d'électroporation, en utilisant le programme X-14.
- Transférer le contenu de la cuvette dans un flacon T-25 contenant 5 ml de milieu LIT préréchauffé (10 % de FCS). Laisser le bouchon en vrac et couver le flacon à 37 oC sous 5 % de CO2.
- Surveiller la croissance cellulaire soit en poursuivant la culture axenique (section 4) soit en tant qu'amastigotes intracellulaires après une infection des cellules hôtes (section 5).
4. Suivi de la croissance des cellules Knockout comme Amastigotes Axenic
- Les EE s'installent au bas du milieu de culture, si doucement secouer le flacon pour les resuspendre dans la solution. Le lavage de la surface de la fiole par pipetting aide, comme certaines cellules sont adhérées à la fiole.
- Mélanger 1 l l de solution d'iodure de propidium (PI) (20 g/mL) avec 20 oL de culture amastigote.
REMARQUE: Ne laissez pas le flacon de culture à l'extérieur de l'incubateur plus longtemps que nécessaire. La température est l'un des facteurs qui permet la prolifération axenique22. - Appliquer l'échantillon sur l'hémocytomètre et observer sous un microscope à fluorescence. L'IP est permeant à la membrane cellulaire endommagée mais est exclu des cellules vivantes. Comptez le nombre d'amastigotes viables qui ne sont pas tachés par PI (ex/em 570 nm/602 nm).
5. Suivi de la croissance des cellules Knockout en tant qu'amastigotes intracellulaires
- Les cellules 3T3 d'hôte de graine dans une plaque de 12 puits avec DMEM (10% FCS). Ajuster la densité cellulaire à 70-80% de confluence, ou environ 3 x 105 cellules par puits.
REMARQUE: Puisque les amastigotes ne sont pas motiles, couvrant la majeure partie de la surface de croissance par les cellules hôtes améliore l'efficacité d'infection. - Recueillir les amastigotes knock-out de l'étape 3.6 par centrifugation un jour après l'électroporation. Jeter le supernatant, et resuspendre le parasite avec 2 ml de DMEM (10% FCS).
- Retirer le milieu de la culture de la cellule hôte et appliquer les amastigotes resuspendus. La multiplicité de l'infection devrait être de 20 ou plus. Incuber la plaque à 37 oC sous 5 % de CO2 pendant 2 jours pour permettre aux amastigotes d'établir l'infection.
REMARQUE: La période d'infection peut être de 1 jour, selon le but. - Les EE de lavage sont restés à l'extérieur des cellules hôtes deux fois avec le DMEM (10% FCS).
- Ajouter du DMEM frais (10 % FCS) à la co-culture hôte-parasite et poursuivre l'incubation à 37 oC pendant 2 jours supplémentaires.
-
Pour évaluer l'efficacité de l'infection, visualisez les noyaux des cellules hôtes et de l'amastigote intracellulaire.
REMARQUE : Les noyaux ont tendance à se chevaucher dans la co-culture saturée. Re-placage des cellules à faible densité cellulaire (comme 1:5 dilution) avant la fixation et la coloration aide à compter les noyaux plus facilement.- Retirez le milieu de culture et appliquez 1 ml de solution formaline de 10% dans LE PBS pour fixer les cellules. Incuber 10 min à température ambiante.
- Remplacez la solution formaline par 1 ml de PBS contenant 1 Hoechst 33342 et 0,1 % Triton X-100. Incuber 5 min à température ambiante.
- Retirez la solution Hoechst et rincez les cellules une fois avec pbS. Ajouter 1 ml de PBS frais.
- Observez sous le microscope à fluorescence et identifiez les noyaux des cellules hôtes qui sont associés à de plus petits noyaux parasites (figure 4). Les cellules hôtes qui contiennent plus de 2 amastigotes doivent être considérées comme infectées, sans inclure l'infection initiale non productive ou les EE non lavés.
Access restricted. Please log in or start a trial to view this content.
Résultats
Isolement des trypomastigotes par la procédure de nage-out
Pour récolter les trypomastigotes frais de la contamination des anciens EE par la procédure de nage-out, les granulés de cellules doivent être incubés au moins pendant 1 h. Incubating les granulés pendant plus de 2 h n'augmente pas de manière significative le nombre de trypomastigotes nageant dans la solution ( Figure 2B). Dans cette expérience particulière, le pourcentage de try...
Access restricted. Please log in or start a trial to view this content.
Discussion
Nous avons démontré que la culture axenique des amastigotes de T. cruzi peut être utilisée dans LE knock-out de gène CRISPR/Cas9-négocié, en électroporating gRNA directement dans Cas9-exprimant EA. De cette façon, l'essentiel du gène cible spécifiquement au stade amastigote peut être évalué sans passer par d'autres stades de développement.
Un autre aspect bénéfique de la transfection amastigote est la commodité dans l'essai d'un grand nombre de gènes cibles. Une foi...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs n'ont aucun conflit d'intérêts à divulguer.
Remerciements
Ce travail a été soutenu en partie par JSPS KAKENHI Grant Number 18K15141 à Y.T.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
Références
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.