
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Presentazione di un Gene Knockout direttamente nello stadio Amastigote di Trypanosoma cruzi Utilizzando il sistema CRISPR/Cas9
In questo articolo
Riepilogo
Qui, descriviamo un protocollo per introdurre un knockout genico nell'amastigote extracellulare di Trypanosoma cruzi, usando il sistema CRISPR/Cas9. Il fenotipo della crescita può essere seguito sia dal conteggio cellulare della coltura degli amastigote axenico, sia dalla proliferazione degli amastigoti intracellulari dopo l'invasione delle cellule ospiti.
Abstract
Trypanosoma cruzi è un parassita protozoo patogeno che causa la malattia di Chagas principalmente in America Latina. Al fine di identificare un nuovo bersaglio farmacologico contro T. cruzi, è importante convalidare l'essenzialità del gene bersaglio nello stadio dei mammiferi del parassita, l'amastigote. Gli amastigoti di T. cruzi si replicano all'interno della cellula ospite; quindi, è difficile condurre un esperimento ad eliminazione diretta senza passare attraverso altre fasi di sviluppo. Recentemente, il nostro gruppo ha segnalato una condizione di crescita in cui l'amastigote può replicare axenically per un massimo di 10 giorni senza perdere le sue proprietà amastigote-like. Utilizzando questa coltura amastigote ascia temporale, abbiamo introdotto con successo i gRNA direttamente nell'amastigote che esprime Cas9 per causare eliminazioni genetiche e analizzato i loro fenotipi esclusivamente nella fase di amastigote. In questo rapporto, descriviamo un protocollo dettagliato per produrre amastigoti extracellulari derivati in vitro e per utilizzare la coltura ascia in un esperimento di knockout mediato da CRISPR/Cas9. Il fenotipo di crescita degli amastigoti knockout può essere valutato sia dai conteggi cellulari della coltura axenica, sia dalla replicazione dell'amastigote intracellulare dopo l'invasione delle cellule ospiti. Questo metodo bypassa la differenziazione dello stadio parassita normalmente coinvolto nella produzione di un amastigote transgenico o knockout. L'utilizzo della cultura amastigote ascia temporale ha il potenziale per espandere la libertà sperimentale di studi specifici dello stadio in T. cruzi.
Introduzione
Trypanosoma cruzi è l'agente causale della malattia di Chagas, prevalente principalmente in America Latina1. T. cruzi ha fasi del ciclo di vita distintive mentre viaggia tra un vettore di insetti e un ospite di mammiferi2. T. cruzi si replica come un epimastigote nel midgut di un insetto di triatomina succhiasangue e si differenzia in una trypomastigote metaciclica infettiva nel suo intestino posteriore prima di essere depositato su un ospite umano o animale. Una volta che il trypomastigote entra nel corpo ospite attraverso il sito del morso o attraverso una membrana mucosa, il parassita invade una cellula ospite e si trasforma in una forma rotonda senza flagelli chiamata amastigote. L'amastigote si replica all'interno della cellula ospite e alla fine si differenzia in trypostigote, che esce dalla cellula ospite ed entra nel flusso sanguigno per infettare un'altra cellula ospite.
Dal momento che gli agenti chemioterapici attualmente disponibili, benznidazolo e nifurtimox, causano effetti collaterali negativi e sono inefficaci nella fase cronica della malattia3, è di grande interesse identificare nuovi bersagli farmacologici contro T. cruzi. Negli ultimi anni, il sistema CRISPR/Cas9 è diventato un potente strumento per eseguire efficacemente l'abbattimento genico in T. cruzi, sia per trasfezione di plasmide separate o singole contenenti gRNA e Cas94, con espressione stabile di Cas9 e successiva introduzione di gRNA5,6,7 o modello di trascrizione di gRNA8, o per elettroporazione del complesso gRNA/Cas9 RNP preformato7,9. Questo progresso tecnologico è molto previsto per accelerare la ricerca sul target farmacologico nella malattia di Chagas.
Per procedere con lo sviluppo del farmaco, è fondamentale convalidare l'essenzialità del gene bersaglio o l'efficacia dei composti candidati al farmaco nell'amastigote di T. cruzi, in quanto è la fase di replicazione del parassita nell'ospite dei mammiferi. Tuttavia, questo è un compito impegnativo, perché gli amastigoti non possono essere manipolati direttamente a causa della presenza di una cellula ospite ostruttiva. In Leishmania, un parassita protozoo strettamente imparentato con T. cruzi, è stato sviluppato un metodo di coltura amastigote axenico ed è stato utilizzato nei prelievi di farmaci10,11,12, 13.Sebbene vi siano alcune discrepanze nella suscettibilità ai composti tra amastigoti axenici e amastigoti intracellulari14, la capacità di mantenere comunque la cultura ascia fornisce preziosi strumenti sperimentali per studiare la biologia di base della fase clinicamente rilevante della Leishmania15,16. Nel caso di T. cruzi, le pubblicazioni riguardanti la presenza di amastigotes extracellulari naturali (EA)17 e la produzione in vitro di EA17,18,19 risalgono a decenni fa. Inoltre, EA è noto per avere una capacità infettiva20, anche se inferiore a quella di trypomastigote, e il meccanismo di invasione dell'ospite amastigote è stato chiarito negli ultimi anni (rivisto da Bonfim-Melo et al.21). Tuttavia, a differenza della Leishmania, EA non era stato utilizzato come strumento sperimentale in T. cruzi, soprattutto perché EA era stato considerato come un parassita intracellulare obbligato, e quindi non era stato considerato come "forma replicativa" in un pratico senso.
Recentemente, il nostro gruppo ha proposto di utilizzare EA di T. cruzi come cultura axenica temporale22. Gli amastigotes del ceppo T. cruzi Tulahuen possono replicarsi senza cellule ospiti nel mezzo LIT a 37 gradi centigradi per un massimo di 10 giorni senza un grave deterioramento o perdita di proprietà simili ad amastigote. Durante il periodo di crescita senza ospiti, EA è stata utilizzata con successo per l'espressione genica esogena mediante elettroporazione convenzionale, l'analisi della titolazione dei farmaci con composti tributo e il knockout mediato da CRISPR/Cas9 seguito dal monitoraggio del fenotipo della crescita. In questo rapporto, descriviamo il protocollo dettagliato per produrre EA derivato in vitro e per utilizzare l'amastigote ascia negli esperimenti con knockout.
Access restricted. Please log in or start a trial to view this content.
Protocollo
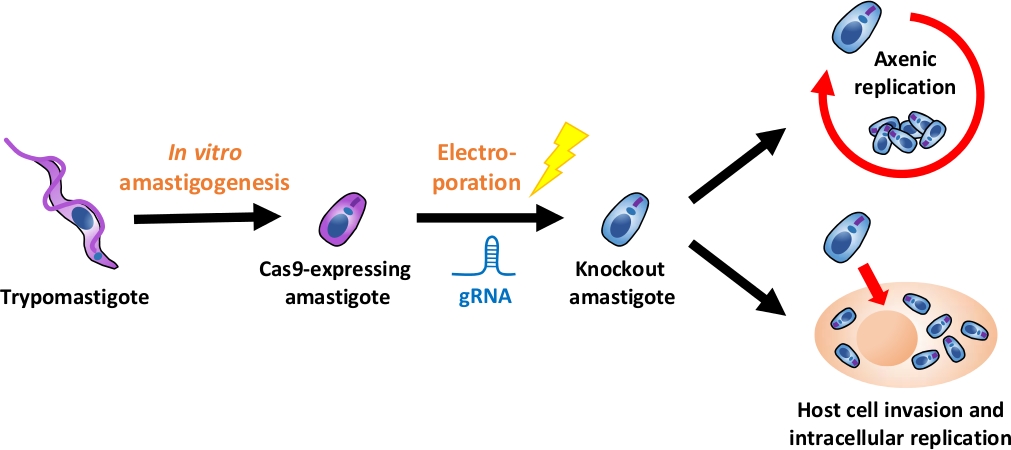
NOT: Una panoramica dell'intero flusso sperimentale è illustrata nella Figura 1.

Figura 1: Panoramica dell'esperimento a eliminazione diretta tramite EA. I trypomastigoti derivati dalla coltura dei tessuti vengono raccolti e differenziati in EA. gRNA viene trafitto in amastigoti che esprimono Cas9 per elettroporazione, e il fenotipo di crescita dell'amastigote knockout viene valutato mediante la replica axenica o mediante la replica axenica replica intracellulare dopo l'invasione delle cellule ospiti. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
1. Preparati per la coltura dei parassiti
- Il ceppo Tulahuen di Trypanosoma cruzi viene utilizzato in tutto questo rapporto. Mantenere gli epimastigoti di T. cruzi in Media LIT (10% FCS, vedi tabella supplementare 1). Chiudere in modo sicuro il tappo e mantenere la fiaschetta di coltura a 28 gradi centigradi.
- Generare un ceppo transgenico di T. cruzi che ospita l'endonucleasi Cas9. Esempi di espressione plasmidi che contengono sequenza di codifica Cas9 e G418 (neor) marcatore di selezione possono essere trovati in Lander et al.4 e Peng et al.5
- Trasfecare il plasmide di cui sopra in epimastigote per elettroporazione, utilizzando il kit elencato nella Tabella dei Materiali. Per 1 cuvette, girare verso il basso 2 x 107 celle, scartare il supernatante e risospendere con 100 l di buffer di elettroporazione contenente la soluzione di supplemento fornita.
NOT: Il buffer di elettroporazione può essere sostituito con il buffer EM24 (3:1 miscela di cytomix25 e buffer fosfato-sucrosi). - Aggiungete 20-40 g di plasmide e trasferite la miscela in una cuvette di elettroporazione gap di 2 mm. Applicare l'impulso con un dispositivo di elettroporazione (vedere Tabella dei materiali), utilizzando il programma X-1426.
- Trasferire il contenuto della cuvette in un pallone T-25 contenente 5 mL di supporto LIT (10% FCS). Incubare il pallone a 28 gradi centigradi per 24 h.
- Aggiungete il G418 ad una concentrazione finale di 250 g/mL e continuate l'incubazione a 28 gradi centigradi. Ci vuole circa 1 settimana per uccidere gli epimastigoti non trasfettati. Una volta che la popolazione resistente al G418 inizia a riprendersi, passare la coltura una o due volte alla settimana per evitare la saturazione e diluire le cellule morte. La creazione di una linea cellulare stabile di solito richiede un totale di 4 settimane.
NOT: Se l'espressione costituiscitiva di Cas9 è un onere per la cellula5, rendere l'espressione specifica per lo stadio amastigote coniugando il 3'-UTR del gene amastin immediatamente a valle del telaio di lettura aperto Cas927. La descrizione dettagliata del plasmide dell'espressione Cas9 specifica dell'amastigote si trova in Takagi et al.
- Trasfecare il plasmide di cui sopra in epimastigote per elettroporazione, utilizzando il kit elencato nella Tabella dei Materiali. Per 1 cuvette, girare verso il basso 2 x 107 celle, scartare il supernatante e risospendere con 100 l di buffer di elettroporazione contenente la soluzione di supplemento fornita.
- Stabilire una co-coltura ospite-parassita di Cellule ospiti T. cruzi e mammiferi che esprimono Cas9. In questo rapporto, utilizzare la cellula fibroblasta 3T3-Svizzera di Albino come ospite.
- Differenziare T. cruzi epimastigotes in metaciclici trypostigotes. Determinare la densità cellulare della coltura dell'epimastigote utilizzando un emocitometro e raccogliere 5 x 107 cellule mediante centrifugazione per 15 min a 2.100 x g. Eliminare il supernatante e risospendere nuovamente le cellule con 10 mL di mezzo RPMI. Incubare il parassita a 28 gradi centigradi per 1 settimana28.
- Raccogli le metamastigote metacicliche nel mezzo RPMI. Inclinare con attenzione il pallone e pipet fuori la soluzione senza disturbare i parassiti aderitati alla superficie inferiore. Trasferire il mezzo in un tubo conico e centrifugare per 15 min a 2.100 x g. Eliminare il supernatante e risospendere nuovamente il parassita con 5 mL di DMEM (10% FCS).
NOT: La popolazione di parassiti è una miscela di trypomastigotes metacicliche, epimastigotes e alcune forme intermedie. Anche se non è necessario, è possibile isolare trypostigote da DEAE ion exchange cromatografia29. In alternativa, gli epimastigoti possono essere eliminati incubando i parassiti con siero attivo per sottoporli a completare la lisi30. - Seme 3T3-Svizzera fibroblasto Albino a 60-70% di confluenza, o circa 1.7-2.0 x 106 cellule in 5 mL di DMEM (10% FCS) in flacone di coltura T-25. Rimuovere il mezzo di crescita e applicare la miscela di parassiti dal passo 1.3.2. Incubare per 24 h a 37 gradi sotto il 5% di CO2 in un'incubatrice umidificata per stabilire l'infezione.
- Rimuovere i parassiti che rimangono al di fuori delle cellule 3T3 dell'ospite lavando la co-coltura con DMEM (10% FCS) due volte.
- Una volta che la co-cultura è satura, passaggio cellule ospiti infettati da amastigote da trypsinization. Aspirare il mezzo e risciacquare la cultura una volta con PBS. Applicare 1 mL di 0,05% soluzione di prova per coprire l'intera superficie di coltura e incubare per alcuni minuti a temperatura ambiente fino a quando le cellule ospiti collegate non si allentano. Scollegare le celle dalla superficie del pallone conungere 3 mL di DMEM (10% FCS) sulle celle.
- Trasferire le cellule ospiti staccate in un tubo conico e centrifugare per 3 min a 300 x g. Aspirare il supernatante e risospendere le cellule con 3 mL di DMEM fresco (10% FCS). Questo passaggio aiuta ad eliminare gli epimastigoti rimanenti. Trasferire tutto in un flacone T-75 pulito contenente 9 mL di DMEM (10% FCS). Continuare a coltivare fino a quando trypomastigote viene rilasciato in cultura supernatant.
- Mantenere la co-cultura passando con la prova due volte a settimana. Una volta che il 70-80% della popolazione ospite si infetta, aggiungere regolarmente cellule host 3T3 non infette al rapporto di 5:1 (carryover:fresh), al fine di evitare il deterioramento della coltura. Trypostigote scappa continuamente se l'equilibrio tra le cellule ospiti e T. cruzi è correttamente mantenuto.
2. Differenziazione dei Trypomastigotes in EA
- Il giorno prima di questo esperimento, rimuovi il mezzo di crescita della cocoltura ospite-parassita e aggiungi dMEM fresco (10% FCS) per lavare via EA e trypomastigotes che erano già stati rilasciati dalle cellule ospiti nei giorni precedenti. Esperimenti regolari richiedono almeno due flaconi T-75 di co-cultura confluente.
-
Raccogliere la coltura supernatant in un tubo conico per raccogliere trypostigotes appena emersi. Controllare il campione al microscopio (10x o 20x obiettivo obiettivo) per la qualità. Se ci sono detriti cellulari ospiti, centrifugare brevemente il campione e trasferire il supernatante in un nuovo tubo. Se c'è un numero significativo di EA, isolare trypomastigotes con la seguente procedura di swim-out.
- Girare verso il basso la miscela di trypostigotes e amastigotes per 15 min a 2.100 x g. Eliminare la maggior parte del supernatante, lasciando 0,5-1,0 mL di mezzo nel tubo.
NOTA: la riduzione del volume è facoltativa, ma semplifica il passaggio seguente. - Incubare il pellet a 37 gradi centigradi per 1-2 h, permettendo ai trypostigoti attivi di nuotare fuori dal pellet (Figura 2).
- Trasferire il supernatante contenente trypomastigotes in un tubo di microcentrifuga da 1,5 mL.
- Girare verso il basso la miscela di trypostigotes e amastigotes per 15 min a 2.100 x g. Eliminare la maggior parte del supernatante, lasciando 0,5-1,0 mL di mezzo nel tubo.
- Centrifugare il tubo conico per 15 min a 2.100 x g per raccogliere trypomastigotes. Se la procedura di nuoto è stata eseguita sopra, centrifugare il tubo da 1,5 mL per 2 min a 4.000 x g per pellet il trypomastigote. Scartare il super-attardato.
- Risospendere il pellet con 5 mL di DMEM memorizzato nel buffer con 20 mM MES (pH 5.0), integrato con lo 0,4% BSA19. Trasferire il parassita in una fiaschetta di coltura T-25. Lascia il tappo allentato. La densità cellulare deve essere pari o inferiore a 1 x 107 cellule/mL, poiché la sovrasaturazione aumenta la probabilità di morte cellulare.
NOT: Il colore del DMEM deve essere giallo, non arancione. Se il DMEM originale aveva un'elevata capacità di buffering, 20 mM MES (pH 5.0) non sono sufficienti per abbassare il pH. Il pH del mezzo deve essere regolato mediante l'aggiunta di HCl in questo caso. Il mezzo acido può essere mantenuto a 4 gradi centigradi, ma non più di 1 mese. - Incubare il flacone di coltura a 37 gradi centigradi sotto il 5% di CO2 in un'incubatrice umidificata. Circa il 95% dei parassiti si differenzia in amastigoti dopo 24 ore.
3. Elettroporazione di EA
- Preparare il gRNA per l'elettroporazione. Questo può essere fatto con la trascrizione in vitro, o semplicemente acquistando oligonucleotidi RNA sintetici da un produttore. In questo rapporto, vengono utilizzati crRNA e tracrRNA da Integrated DNA Technologies, Inc.
- Centrifugare la cultura di EAs per 15 min a 2.100 x g. Scartare supernatante.
- Risospendere il pellet con buffer di elettroporazione contenente la soluzione di supplemento fornita alla densità cellulare finale di 1 x 108 celle/mL.
NOTA: il buffer EM causa più decessi cellulari rispetto al buffer di elettroporazione elencato nella tabella dei materiali; pertanto, non è raccomandato per la trasfezione amastigote ( Figura supplementare1). - Aliquota 100 - L di parassiti risospesi (1 x 107 cellule) in tubi di microcentrifuga da 1,5 ml. Aggiungere 5-10 g di gRNA e mescolare delicatamente pipettando.
- Trasferire la miscela in una cuvette di elettroporazione gap di 2 mm. Applicare l'impulso con il dispositivo di elettroporazione, utilizzando il programma X-14.
- Trasferire il contenuto della cuvette in un flacone T-25 contenente 5 mL di mezzo LIT preriscaldato (10% FCS). Lasciare il tappo allentato e incubare il pallone a 37 gradi sotto il 5% di CO2.
- Monitorare la crescita cellulare mediante la continuazione della coltura axenica (sezione 4) o come amastigotes intracellulari dopo l'infezione delle cellule ospiti (sezione 5).
4. Monitoraggio della crescita delle cellule Knockout come Amastigotes axenic
- EAs stabilirsi nella parte inferiore del mezzo di coltura, in modo da scuotere delicatamente il pallone per rinvigorire nella soluzione. Lavare la superficie del pallone con la pipimernatura aiuta, poiché alcune cellule vengono aderite al pallone.
- Mescolare 1 - L di propidio iodide (PI) soluzione (20 g/mL) con 20 l di coltura amastigote.
NOT: Non lasciare il flacone di coltura al di fuori dell'incubatrice più a lungo del necessario. La temperatura è uno dei fattori che consente la proliferazione ascia22. - Applicare il campione sull'emocitometro e osservarlo al microscopio a fluorescenza. PI è permeant e la membrana cellulare danneggiata, ma è escluso dalle cellule vive. Contare il numero di amastigoti vitali che non sono macchiati da PI (ex/em 570 nm/602 nm).
5. Monitoraggio della crescita delle cellule knockout come Amastigotes intracellulari
- I semi ospitano celle 3T3 in una piastra da 12 pozzetti con DMEM (10% FCS). Regolare la densità cellulare al 70-80% di confluenza, o circa 3 x 105 celle per bene.
NOT: Poiché gli amastigoti non sono motili, coprire la maggior parte della superficie di crescita da parte delle cellule ospiti migliora l'efficienza dell'infezione. - Raccogliere gli amastigoti knockout dal passo 3.6 per centrifugazione un giorno dopo l'elettroporazione. Eliminare il supernatante e risospendere il parassita con 2 mL di DMEM (10% FCS).
- Rimuovere il supporto dalla coltura della cellula ospite e applicare gli amastigoti risospesi. La molteplicità dell'infezione deve essere pari o superiore a 20. Incubare la piastra a 37 gradi centigradi sotto il 5% di CO2 per 2 giorni per consentire agli amastigoti di stabilire l'infezione.
NOT: Il periodo di infezione può essere di 1 giorno, a seconda dello scopo. - Lavare via EA è rimasto al di fuori delle celle ospiti due volte con DMEM (10% FCS).
- Aggiungere dMEM fresco (10% FCS) alla co-coltura ospite-parassita e continuare l'incubazione a 37 gradi centigradi per ulteriori 2 giorni.
-
Per valutare l'efficienza dell'infezione, visualizzare i nuclei delle cellule ospiti e l'amastigote intracellulare.
NOTA: i nuclei tendono ad essere sovrapposti alla cocultura satura. Ri-placcatura delle cellule a densità cellulare inferiore (come diluizione 1:5) prima della fissazione e colorazione aiuta a contare i nuclei più facilmente.- Rimuovere il mezzo di coltura e applicare 1 mL di 10% soluzione formalina in PBS per fissare le cellule. Incubare per 10 min a temperatura ambiente.
- Sostituire la soluzione di formalina con 1 mL di PBS contenente 1 G/mL Hoechst 33342 e 0,1% Triton X-100. Incubare per 5 min a temperatura ambiente.
- Rimuovere la soluzione Hoechst e risciacquare le cellule una volta con PBS. Aggiungere 1 mL di PBS fresco.
- Osservare al microscopio a fluorescenza e identificare i nuclei delle cellule ospiti associati a nuclei parassiti più piccoli (Figura 4). Le cellule ospiti che contengono più di 2 amastigotes devono essere considerate infette, per non includere infezioni iniziali non produttive o EA non lavate.
Access restricted. Please log in or start a trial to view this content.
Risultati
Isolamento dei trypostigotes con la procedura di swim-out
Per raccogliere nuove prove da contaminanti vecchi EA con procedura di nuoto fuori, pellet cellulari devono essere incubati almeno per 1 h. Incubare i pellet per più di 2 h non aumenta significativamente il numero di trypostigotes nuotare nella soluzione ( Figura 2B). In questo particolare esperimento, la percentuale di trypomastigote nella miscela iniziale era del 38% e la percentuale dop...
Access restricted. Please log in or start a trial to view this content.
Discussione
Abbiamo dimostrato che la cultura ascia di T. cruzi amastigotes può essere utilizzata nel knockout genico mediato da CRISPR/Cas9, elettropolando gRNA direttamente nell'EA che esprime Cas9. In questo modo, l'essenzialità del gene bersaglio specificamente nella fase amastigote può essere valutata senza passare attraverso altre fasi di sviluppo.
Un altro aspetto benefico della trasfezione dell'amastigote è la convenienza nel test di un gran numero di geni bersaglio. Una volta stabili...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori non hanno alcun conflitto di interessi da divulgare.
Riconoscimenti
Questo lavoro è stato supportato in parte da JSPS KAKENHI Grant numero 18K15141 a Y.T.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
Riferimenti
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati