
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
CRISPR/Cas9システムを用いたトリパノソマ・クルジのアマスティゴットステージに遺伝子ノックアウトを直接導入
要約
ここでは、CRISPR/Cas9システムを用いてトリパノノーマ・クルジの細胞外アマスティゴテに遺伝子ノックアウトを導入するプロトコルについて述べた。成長表現型は、アックスアンマスティゴテ培養の細胞カウントまたは宿主細胞侵入後の細胞内アマスティゴテスの増殖のいずれかによって追跡することができる。
要約
トリパノソマ・クルジは、主にラテンアメリカでシャーガス病を引き起こす病原性原虫寄生虫である。T.クルジに対する新しい薬物標的を同定するためには、寄生虫の哺乳動物期における標的遺伝子の本質性を検証することが重要である。T.クルジのアマスティゴットは、宿主細胞内で複製する。したがって、他の発達段階を経ずにノックアウト実験を行うのは困難です。最近、我々のグループは、アマスティゴテのような特性を失うことなく、最大10日間軸を複製することができる成長条件を報告した。この側軸アミスティゴテ培養を用いることで、遺伝子ノックアウトを引き起こすCas9発現アマスティゴテに直接gRNAを導入し、その表現型をアマスティゴテ段階でのみ解析することに成功した。本報告では、インビトロ由来細胞外アマティストを生成し、CRISPR/Cas9媒介ノックアウト実験で軸培養を利用するための詳細なプロトコルについて述べる。ノックアウト・アマスティゴテスの増殖表現型は、軸培養の細胞数、または宿主細胞侵入後の細胞内アマスティゴットの複製によって評価することができる。この方法は、通常、トランスジェニックまたはノックアウト・アマスティゴテの産生に関与する寄生虫段階の分化をバイパスする。時間的軸アマスティゴテ培養の利用は、T.クルジにおけるステージ特異的研究の実験的自由を拡大する可能性を有する。
概要
トリパノソマ・クルジは、主にラテンアメリカ1で流行しているシャーガス病の原因となる薬剤である。T. クルジは、昆虫ベクターと哺乳類の宿主2の間を移動する独特のライフサイクルステージを持っています。T.クルジは、血液を吸うトリアトミンバグの中腸内のエピマスチゴテとして複製し、ヒトまたは動物の宿主に沈着する前に、その後腸内の感染性形而上皮トリポマスティゴに分化する。トリポマスティゴテが咬傷部位または粘膜を通して宿主体内に入ると、寄生虫は宿主細胞に侵入し、アマスティゴテと呼ばれるフラゲラレス丸型に変形する。amastigoteは宿主細胞内で複製し、最終的にはトリポマスティゴテに分化し、宿主細胞から破裂し、別の宿主細胞に感染するために血流に入る。
現在利用可能な化学療法剤、ベンズニダゾールおよびニフルティモックスは、有害な副作用を引き起こし、疾患3の慢性相に効果がないため、T.クルジに対する新規な薬物標的を同定することは大きな関心事である。近年、CRISPR/Cas9システムは、GRNAとCas94を含む別個または単一プラスミドのトランスフェクションによって、Cas9およびその後の安定した発現によって、T.クルジで遺伝子ノックアウトを効果的に行う強力なツールとなっています。gRNA 5、6、7またはgRNA8の転写テンプレートの導入、または予形成されたgRNA/Cas9 RNP複合体7、9のエレクトロポレーションによる。この技術の進歩は、シャーガス病の薬物標的研究を加速することが非常に期待されています。
薬剤開発を進めるためには、哺乳類宿主における寄生虫の複製段階であるT.クルジのアマスティゴテにおける標的遺伝子または薬剤候補化合物の有効性を検証することが重要である。しかし、閉塞性宿主細胞が存在するため、アマスティゴットを直接操作することはできないため、これは困難な作業です。リーシュマニアでは、T.クルジに密接に関連する原虫寄生虫、アックスアンスアミゴット培養法が開発され、薬物スクリーニングアッセイ10、11、12、および、 13.アックスアマスティゴテスと細胞内アマスチゴテス14との間の化合物に対する感受性にはいくつかの不一致があるものの、それでもアックス培養を維持する能力は、リーシュマニア15、16の臨床的に関連する段階の基礎生物学。T.クルジの場合、自然発生する細胞外アマスティゴテス(EA)17の存在に関する文献とEA 17、18、19のインビトロ産生何十年も前のことださらに、EAは、トリポスチゴテのそれよりも少ないが、感染能力20を有することが知られており、近年、アマスティゴット宿主侵入のメカニズムが解明されている(Bonfim-Melo et al.21によるレビュー)。しかし、リーシュマニアとは異なり、EAはT.クルジの実験ツールとして利用されていなかった。意味。
最近、我々のグループは、T.クルジのEAを一時的な軸文化22として利用することを提案した。T.クルシトゥラフエン株のアマスティゴテスは、アマスティゴット様特性の大きな劣化または損失なしに最大10日間、37°CでLIT培地中の宿主細胞を含まない複製が可能です。宿主フリー成長期間中、EAは、従来のエレクトロポレーション、トリパノシダル化合物による薬物滴定アッセイ、およびCRISPR/Cas9媒介ノックアウトに続いて成長表現型モニタリングを行い、外因性遺伝子発現に成功しました。本報告では、インビトロ由来EAを生成し、ノックアウト実験でアックスアンマスタゴテを利用するための詳細なプロトコルについて述べた。
Access restricted. Please log in or start a trial to view this content.
プロトコル
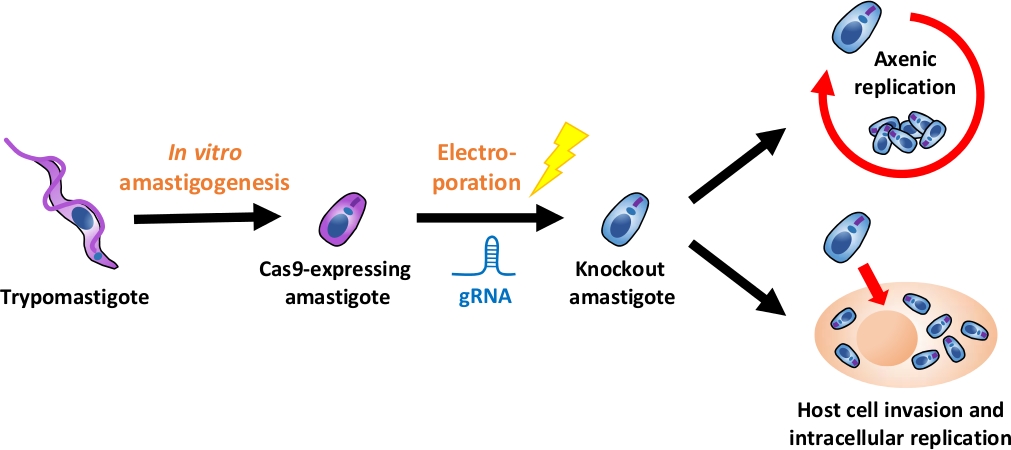
注:実験フロー全体の概要を図 1に示します。

図1:EAを用いたノックアウト実験の概要組織培養由来トリポスチゴテスは、EAに収穫され、EAに分化される. gRNAは、エレクトロポレーションによってCas9発現のアマスティゴテスにトランスフェクトされ、ノックアウトアマスティゴテスの成長表現型は、アックスレプリケーションまたはによって評価される宿主細胞侵入後の細胞内複製。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
1. 寄生虫培養製剤
- トリパノソマクルシのトゥラフエン株は、このレポート全体を通じて使用されます。LIT培地におけるT.クルジのエピマスチゴテスを維持する(10%FCS、補足表1参照)。キャップをしっかりと閉じ、培養フラスコを28°Cに保ちます。
- Cas9エンドヌクレアーゼを保有するT.クルジのトランスジェニック株を生成します。Cas9コードシーケンスとG418(ネオr)選択マーカーを含む発現プラスミドの例は、Lander et al.4およびPeng et al.5で見つけることができます。
- 上記のプラスミドをエレクトロポレーションによってエピマスチゴットにトランスフェクトし、材料表に記載のキットを使用する。1キュベットの場合は、2 x 107細胞をスピンダウンし、上清を廃棄し、提供されたサプリメント溶液を含む100μLのエレクトロポレーションバッファーで再懸濁する。
注:エレクトロポレーションバッファーは、EMバッファー24(細胞ミックス25およびリン酸スクロースバッファーの3:1混合物)で置換することができる。 - プラスミドの20-40 μgを追加し、2ミリメートルのギャップエレクトロポレーションキュベットに混合物を転送します。X-14プログラム26を使用して、エレクトロポレーション装置(材料の表を参照)でパルスを適用します。
- キュベットの内容物をLIT培地の5mL(10%FCS)を含むT-25フラスコに移します。フラスコを28°Cで24時間インキュベートします。
- G418を250 μg/mLの最終濃度に加え、28°Cでインキュベーションを続けます。非トランスフェクトされたエピマスチゴを殺すのに約1週間かかります。G418耐性集団が回復し始めたら、飽和を避け、死んだ細胞を希釈するために週に1~2回培養を行う。安定した細胞株の確立は、通常、合計4週間かかります。
注:Cas9の構成的発現が細胞5の負担である場合、Cas9オープンリーディングフレーム27の直下流のアマスティン遺伝子の3'-UTRを結合することにより、アマスティゴット期に特異的な発現を行う。アマスティゴット特異的Cas9発現プラスミドの詳細な説明は、高木ら22で見つけることができます
- 上記のプラスミドをエレクトロポレーションによってエピマスチゴットにトランスフェクトし、材料表に記載のキットを使用する。1キュベットの場合は、2 x 107細胞をスピンダウンし、上清を廃棄し、提供されたサプリメント溶液を含む100μLのエレクトロポレーションバッファーで再懸濁する。
- Cas9発現T.クルジおよび哺乳動物宿主細胞の宿主寄生虫共培養を確立する。このレポートでは、宿主として3T3-スイスアルビノ線維芽細胞を使用する。
- T. クルジエピマスチゴテスをメタ環型トリポマスティゴに区別する。血球計を用いてエピマスチゴテス培養の細胞密度を決定し、2,100 x gで15分間遠心分離により5 x 107細胞を集集める。上清を廃棄し、RPMI培地の10mLで細胞を再懸濁する。寄生虫を28°Cで1週間28°Cでインキュベートする。
- RPMI培地でメタ環型トリポスチゴテスを収集する。フラスコを慎重に傾け、底面に付着した寄生虫を邪魔することなく、溶液をピペトアウトします。2,100 x gで15分間円錐形の管と遠心分離機に媒体を移します。上清を廃棄し、DMEMの5 mL(10%FCS)で寄生虫を再懸濁させる。
注:寄生虫集団は、メタ環型トリポスチゴテス、エピマスチゴット、およびいくつかの中間形態の混合物である。必要ではないが、DEAEイオン交換クロマトグラフィー29によりトリポスチゴテを分離することができる。あるいは、エピマスチゴットは、活性血清で寄生虫をインキュベートして、それらを補体するリシス30を補和することによって排除することができる。 - 種子3T3-スイスアルビノ線維芽細胞は60〜70%合流、またはT-25培養フラスコにおけるDMEMの5 mL(10%FCS)で約1.7-2.0 x 106細胞に。成長培地を除去し、ステップ1.3.2から寄生虫混合物を適用します。加湿インキュベーターで37°C以下で24時間インキュベートし、感染を確立します。
- DMEM(10%FCS)で共培養を2回洗浄することにより、宿主3T3細胞の外側に残った寄生虫を除去する。
- 共培養が飽和したら、トリプシン化によりアマスティゴット感染宿主細胞を通過させる。培地を吸引し、PBSで一度培養をすすいでくす。0.05%トリプシン溶液の1mLを塗布し、培養面全体を覆い、付着した宿主細胞が緩むまで室温で数分間インキュベートする。セル上に DMEM (10% FCS) の 3 mL をフラッシュしてフラスコ表面からセルを取り外します。
- 離れた宿主細胞を円錐形のチューブに移し、遠心分離機を300xgで3分間遠心分離する。上清を吸引し、新鮮なDMEM(10%FCS)の3 mLで細胞を再中断します。このステップは、残りのエピマスチゴットを排除するのに役立ちます。DMEM(10%FCS)の9 mLを含むきれいなT-75フラスコにすべてを移す。トリポマスティゴテが培養上清に放出されるまで培養を続ける。
- 週に2回、トリプシン化で受け継ぐことで共培養を維持します。宿主集団の70~80%が感染すると、培養の悪化を避けるために、感染していない宿主3T3細胞を5:1(持ち越し:フレッシュ)の割合で定期的に追加します。宿主細胞とT.クルジの間のバランスが適切に維持されている場合、トリポスチゴテは連続的に出口を出る。
2. トリポマスティゴットをEAに分化
- この実験の前日に、宿主寄生虫共培養の増殖培地を取り除き、新鮮なDMEM(10%FCS)を加えて、前日に宿主細胞から既に放出されていたEAとトリポマスティゴットを洗い流す。定期的な実験は、コンフルエント共培養の少なくとも2つのT-75フラスコを必要とします。
-
培養上清を円錐管に集め、新鮮なトリポマスティゴットを収穫します。顕微鏡(10倍または20倍の対物レンズ)でサンプルを確認して品質を確認してください。宿主細胞の破片がある場合は、サンプルを簡単に遠心分離し、上清を新しいチューブに移します。EA の数が多い場合は、次のスイムアウト手順でトリポスティクスを分離します。
- 2,100 x gで15分間、トリポスチゴットとアマスティゴットの混合物をスピンダウンします。上清のほとんどを廃棄し、チューブ内に0.5〜1.0 mLの培地を残します。
注: 音量を減らすことは任意ですが、次の手順を簡単にします。 - ペレットを37°Cで1~2時間インキュベートし、活性トリポスチゴテスがペレットから泳ぎ出せるようにします(図2)。
- トリポスチゴットを含む上清を1.5mLマイクロ遠心管に移します。
- 2,100 x gで15分間、トリポスチゴットとアマスティゴットの混合物をスピンダウンします。上清のほとんどを廃棄し、チューブ内に0.5〜1.0 mLの培地を残します。
- 2,100 x gで15分間円錐管を遠心分離し、トリポスチゴットを収集します。上記のスイムアウト手順を行った場合は、1.5mLチューブを4,000 x gで2分間遠心分離し、トリポスチゴテをペレットする。上清を捨てます。
- 20 mM MES(pH 5.0)でバッファリングされたDMEMの5 mLでペレットを再中断し、0.4%BSA19を補充した。寄生虫をT-25培養フラスコに移す。キャップを緩めたままにしておきます。細胞密度は、過飽和が細胞死の可能性を増加させるので、1 x 107細胞/mLの周りまたは以下でなければなりません。
注:DMEM の色はオレンジではなく黄色でなければなりません。元の DMEM のバッファリング容量が高い場合、20 mM MES (pH 5.0) は pH を下げるには不十分です。その場合、培地のpHはHClを添加して調整しなければならない。酸性培地は4°Cに保つことができるが、1ヶ月以下である。 - 加湿インキュベーターで培養フラスコを37°C以下で5%CO2でインキュベートします。寄生虫の約95%は24時間後にアマスティゴットに分化する。
3. EAのエレクトロポレーション
- エレクトロポレーション用のgRNAを準備します。これは、インビトロ転写によって、または単にメーカーから合成RNAオリゴヌクレオチドを購入することによって行うことができます。本報告では、統合DNAテクノロジーズ社のcrRNAおよびtracrRNAを用いる。
- 2,100 x gで15分間EAの培養を遠心分離します。上清を捨てる。
- 1 x 108細胞/mLの最終細胞密度に対する補足溶液を含むエレクトロポレーションバッファーでペレットを再中断する。
注:EMバッファは、材料の表に記載されているエレクトロポレーションバッファと比較して、より多くの細胞死を引き起こします。したがって、アマスティゴットトランスフェクションにはお勧めしません(補足図1)。 - アリコット100 μLの再懸濁寄生虫(1 x 107細胞)を1.5mLマイクロ遠心管に入れる。gRNAの5-10 μgを追加し、ピペッティングによって穏やかに混合します。
- 混合物を2mmのギャップエレクトロポレーションキュベットに移します。X-14プログラムを使用して、エレクトロポレーションデバイスでパルスを適用します。
- キュベットの内容物を、5mLの予め温められたLIT培地(10%FCS)を含むT-25フラスコに移します。キャップを緩めたままにし、5%CO2以下でフラスコを37°Cでインキュベートします。
- アックス培養の継続(セクション4)または宿主細胞感染後の細胞内アマスチゴットとして細胞増殖を監視する(セクション5)。
4. アクセニック・アマスティゴテスとしてのノックアウト細胞の増殖をモニタリングする
- EAは培養培地の底に落ち着くので、フラスコをゆっくりと振って溶液中に再び振ります。一部の細胞がフラスコに付着しているので、ピペッティングによってフラスコ表面を洗浄するのに役立ちます。
- ヨウ化プロピジウム(PI)溶液(20 μg/mL)を20μLのアマスティゴテ培養と混合します。
注:培養フラスコを必要以上に長くインキュベーターの外に放置しないでください。温度は、軸増殖22を可能にする要因の一つである。 - サンプルをヘモサイトメーターに適用し、蛍光顕微鏡下で観察します。PIは損傷した細胞膜に対してパーセされているが、生細胞から除外される。PI(例/em 570 nm/602 nm)によって染色されていない生存可能なアマスティゴットの数を数えます。
5. 細胞内アマスティゴテスとしてのノックアウト細胞の増殖をモニタリングする
- シードホスト3T3細胞をDMEM(10%FCS)を用した12ウェルプレート内に入れる。細胞密度を70~80%の合流性、またはウェルあたり約3 x 105細胞に調整します。
注:アマスティゴットはモチルではないため、宿主細胞による増殖表面の大部分を覆うことで感染効率が向上する。 - エレクトロポレーションの1日後に遠心分離によってステップ3.6からノックアウトアマスティゴテスを収集します。上清を廃棄し、DMEMの2 mL(10%FCS)で寄生虫を再懸濁させる。
- 宿主細胞培養から培地を取り出し、再懸濁したアマスティゴットを適用する。感染の多重性は20以上である必要があります。37°Cでプレートを5%CO2以下で2日間インキュベートし、アマスティゴットが感染を起こした。
注:感染期間は、目的に応じて1日することができます。 - DMEM(10%FCS)で2回、宿主細胞の外に残ったEAを洗い流します。
- 宿主寄生虫共培養に新鮮なDMEM(10%FCS)を加え、さらに2日間37°Cでインキュベーションを続けます。
-
感染効率を評価するために、宿主細胞および細胞内アマスティゴテの核を可視化する。
注:核は飽和共培養で重なり合う傾向がある。固定および染色の前に、より低い細胞密度(1:5希釈など)で細胞を再めっきすることは、核をより簡単に数えるのに役立ちます。- 培地を取り出し、10%ホルマリン溶液の1mLをPBSに塗布し、細胞を固定する。室温で10分間インキュベートします。
- ホルマリン溶液を1 μg/mLのホエヒト33342と0.1%トリトンX-100を含むPBSの1 mLに置き換えます。室温で5分間インキュベートします。
- Hoechst溶液を取り出し、PBSで細胞を一度すすいで下す。新鮮なPBSの1 mLを追加します。
- 蛍光顕微鏡下で観察し、より小さな寄生虫核に関連する宿主細胞核を同定する(図4)。2以上のアマスティゴットを含む宿主細胞は、非生産的な初期感染または洗浄されていないEAを含まない、感染していると考えるべきである。
Access restricted. Please log in or start a trial to view this content.
結果
スイムアウト手順によるトリポマスティゴットの分離
スイムアウト手順によって古いEAを汚染から新鮮なトリポスチゴテスを収穫するには、細胞ペレットを少なくとも1時間インキュベートする必要があります。図2B)。この特定の実験では、最初の混合物におけるトリポマスティゴテの割合は38%であり、泳ぎ後の割合は任意の時点で98%を...
Access restricted. Please log in or start a trial to view this content.
ディスカッション
我々は、T.クルジアマスティゴテスのアックスカルバチカルが、GRNAをCas9発現EAに直接エレクトロポレートすることにより、CRISPR/Cas9媒介遺伝子ノックアウトで利用できることを実証した。このようにして、amastigote期に特異的に標的遺伝子の本質性は、他の発達段階を経ることなく評価することができる。
アマスティゴットトランスフェクションのもう一つの有益?...
Access restricted. Please log in or start a trial to view this content.
開示事項
著者は開示する利益相反を持っていない。
謝辞
この作品は、JSPS KAKENHI助成金番号18K15141からY.Tまで一部サポートされました。
Access restricted. Please log in or start a trial to view this content.
資料
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
参考文献
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved