
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Einführung eines Gene Knockout direkt in die Amastigote-Phase von Trypanosoma cruzi mit dem CRISPR/Cas9-System
In diesem Artikel
Zusammenfassung
Hier beschreiben wir ein Protokoll zur Einführung eines Genknockouts in den extrazellulären Amastigot von Trypanosoma cruzi, mit dem CRISPR/Cas9-System. Dem Wachstumsphänotyp kann entweder durch Zellzählung der axtischen Amastigotenkultur oder durch Proliferation intrazellulärer Amastigoten nach der Invasion der Wirtszelle gefolgt werden.
Zusammenfassung
Trypanosoma cruzi ist ein pathogener Protozoenparasit, der die Chagas-Krankheit hauptsächlich in Lateinamerika verursacht. Um ein neuartiges Wirkstoffziel gegen T. cruzizu identifizieren, ist es wichtig, die Wesentlichkeit des Zielgens im Säugetierstadium des Parasiten, des Amastigots, zu validieren. Amastigotes von T. cruzi replizieren sich in der Wirtszelle; daher ist es schwierig, ein Knockout-Experiment durchzuführen, ohne andere Entwicklungsstadien zu durchlaufen. Kürzlich berichtete unsere Gruppe von einem Wachstumszustand, in dem sich der Amastigot für bis zu 10 Tage axenisch replizieren kann, ohne seine amastigotähnlichen Eigenschaften zu verlieren. Durch die Verwendung dieser zeitlichen axikonischen Amastigotenkultur führten wir erfolgreich gRNAs direkt in die Cas9-extierende Amastigote ein, um Genknockouts zu verursachen, und analysierten ihre Phänotypen ausschließlich im Amastigot-Stadium. In diesem Bericht beschreiben wir ein detailliertes Protokoll zur Herstellung von in vitro abgeleiteten extrazellulären Amastigoten und zur Nutzung der axtischen Kultur in einem CRISPR/Cas9-vermittelten Knockout-Experiment. Der Wachstumsphänotyp von Knockout-Amastigoten kann entweder anhand der Zellzahl der Axtkultur oder durch Replikation des intrazellulären Amastigots nach der Invasion der Wirtszelle bewertet werden. Diese Methode umgeht die Parasitenstadiumdifferenzierung, die normalerweise bei der Herstellung eines transgenen oder knockout amastigote beteiligt ist. Die Nutzung der zeitlichen aatonischen Amastigotenkultur hat das Potenzial, die experimentelle Freiheit bühnenspezifischer Studien in T. cruzi zu erweitern.
Einleitung
Trypanosoma cruzi ist der Erreger der Chagas-Krankheit, die vor allem in Lateinamerika vorherrscht1. T. cruzi hat auf der Reise zwischen einem Insektenvektor und einem Säugetierwirt2unterschiedliche Lebenszyklusstadien. T. cruzi repliziert als Epimastigot im Mittelgut eines blutsaugenden Triatomin-Bugs und differenziert sich in seinem Hintergut zu einem infektiösen metazyklischen Trypomastigot, bevor er auf einem menschlichen oder tierischen Wirt abgelagert wird. Sobald der Trypomastigot durch die Bissstelle oder durch eine Schleimhaut in den Wirtskörper gelangt, dringt der Parasit in eine Wirtszelle ein und verwandelt sich in eine flagellalose runde Form, die als Amastigot bezeichnet wird. Der Amastigot repliziert sich innerhalb der Wirtszelle und differenziert sich schließlich in Trypomastigote, das aus der Wirtszelle platzt und in den Blutkreislauf eintritt, um eine andere Wirtszelle zu infizieren.
Da derzeit verfügbare Chemotherapeutika, Benznidazol und Nifurtimox Nebenwirkungen verursachen und in der chronischen Phase der Krankheit3unwirksam sind, ist es von großem Interesse, neuartige Wirkstoffziele gegen T. cruzi zu identifizieren. In den letzten Jahren hat sich das CRISPR/Cas9-System zu einem leistungsfähigen Werkzeug entwickelt, um Gen Knockout in T. cruzieffektiv durchzuführen, entweder durch Transfektion von separaten oder einzelnen Plasmiden, die gRNA und Cas94enthalten, durch stabile Expression von Cas9 und Einführung von gRNA5,6,7 oder Transkriptionsschablon von gRNA8, oder durch Elektroporation des vorgeformten gRNA/Cas9 RNP-Komplexes7,9. Dieser technologische Fortschritt wird mit Spannung erwartet, um die Drogenzielforschung bei der Chagas-Krankheit zu beschleunigen.
Um mit der Arzneimittelentwicklung fortzufahren, ist es entscheidend, die Wesentlichkeit des Zielgens oder die Wirksamkeit von Wirkstoff-Kandidatenverbindungen im Amastigot von T. cruzizu validieren, da es sich um die Replikationsstufe des Parasiten im Säugetierwirt handelt. Dies ist jedoch eine schwierige Aufgabe, da Amastigoten aufgrund des Vorhandenseins einer obstruktiven Wirtszelle nicht direkt manipuliert werden können. In Leishmaniawurde ein eng verwandter protozoischer Parasit zu T. cruzientwickelt, eine axenische Amastigote-Kultivierungsmethode entwickelt und in den Arzneimittelscreening-Assays10,11,12 , 13. Obwohl es einige Unterschiede in der Anfälligkeit für Verbindungen zwischen axtischen Amastigoten und intrazellulären Amastigoten14gibt, bietet die Fähigkeit, die axenische Kultur aufrechtzuerhalten, dennoch wertvolle experimentelle Werkzeuge, um die Grundbiologie des klinisch relevanten Stadiums von Leishmania15,16. Im Fall von T. cruzistammen Literaturen über das Vorhandensein natürlich vorkommender extrazellulärer Amastigoten (EA)17 und die In-vitro-Produktion von EA17,18,19 Jahrzehnten. Darüber hinaus ist EA bekannt, eine infektiöse Fähigkeit20haben , wenn auch weniger als die von Trypomastigote, und der Mechanismus der amastigote Host Invasion wurde in den letzten Jahren aufgeklärt (rezensiert von Bonfim-Melo et al.21). Im Gegensatz zu Leishmaniawar EA jedoch nicht als Versuchswerkzeug in T. cruziverwendet worden, vor allem weil EA als obligate intrazellulärer Parasit angesehen worden war und daher in einer praktischen Form nicht als "replikative Form" betrachtet worden war. sinn.
Kürzlich schlug unsere Gruppe vor, EA von T. cruzi als zeitliche axenische Kultur zu nutzen22. Amastigotes des T. cruzi Tulahuen Stammes können sich bis zu 10 Tage lang frei von Wirtszellen in LIT-Medium bei 37 °C replizieren, ohne dass eine größere Verschlechterung oder Verlust amastigotähnlicher Eigenschaften entsteht. Während der wirtfreien Wachstumsphase wurde EA erfolgreich für die exogene Genexpression durch konventionelle Elektroporation, Arzneimitteltitrationstest mit trypanocidalen Verbindungen und CRISPR/Cas9-vermittelten Knockout gefolgt von einer Wachstumsphänotypüberwachung eingesetzt. In diesem Bericht beschreiben wir das detaillierte Protokoll zur Herstellung von in vitro abgeleiteten EA und zur Nutzung des axtischen Amastigots in Knockout-Experimenten.
Access restricted. Please log in or start a trial to view this content.
Protokoll
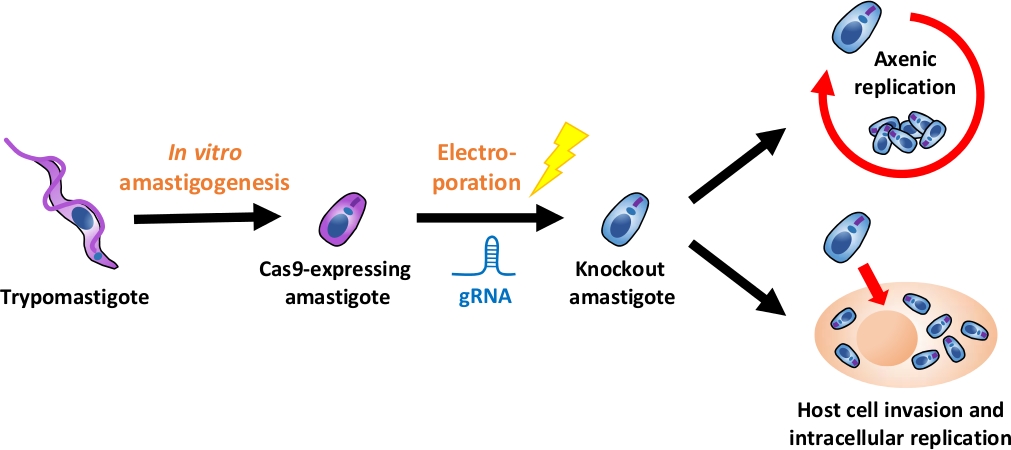
HINWEIS: Eine Übersicht über den gesamten experimentellen Fluss ist in Abbildung 1dargestellt.

Abbildung 1: Übersicht über das Knockout-Experiment mit EA. Gewebekultur-abgeleitete Trypomastigoten werden geerntet und in EA differenziert. gRNA wird durch Elektroporation in Cas9-exektierende Amastigoten transfiziert, und der Wachstumsphänotyp des Knockout-Amastigots wird entweder durch axtische Replikation oder durch intrazelluläre Replikation nach der Invasion der Wirtszelle. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
1. Parasitenkultur präparate
- Tulahuen Stamm von Trypanosoma cruzi wird während dieses Berichts verwendet. Epimastigoten von T. cruzi in LIT-Medium pflegen (10% FCS, siehe Zusatztabelle 1). Schließen Sie die Kappe sicher und halten Sie den Kulturkolben bei 28 °C.
- Generieren Sie einen transgenen Stamm von T. cruzi, der die Cas9 Endonuklease beherbergt. Beispiele für die Expressionplasmide, die Cas9-Kodierungssequenz und G418 (neor) Selektionsmarker enthalten, finden Sie in Lander et al.4 und Peng et al.5
- Transfect das obige Plasmid in Epimastigot durch Elektroporation, mit dem Kit in der Tabelle der Materialienaufgeführt . Für 1 Küvette 2 x 107 Zellen nach unten drehen, den Überstand entsorgen und mit 100 l Elektroporationspuffer, der die mitgelieferte Ergänzungslösung enthält, wieder aufsetzen.
HINWEIS: Der Elektroporationspuffer kann durch EM-Puffer24 (3:1-Mischung aus Cytomix25 und Phosphat-Sucrose-Puffer) ersetzt werden. - Fügen Sie 20-40 g Plasmid hinzu und übertragen Sie das Gemisch in eine 2 mm Spalt-Elektroporationsküvette. Tragen Sie den Puls mit einem Elektroporationsgerät auf (siehe Materialtabelle),mit dem X-14-Programm26.
- Übertragen Sie den Küvetteninhalt in einen T-25-Kolben mit 5 ml LIT-Medium (10% FCS). Den Kolben bei 28 °C für 24 h inkubieren.
- G418 auf eine Endkonzentration von 250 g/ml geben und die Inkubation bei 28 °C fortsetzen. Es dauert etwa 1 Woche, um die nicht transfizierten Epimastigoten abzutöten. Sobald die G418-resistente Population beginnt, sich zu erholen, durchdiestobdiele die Kultur ein- bis zweimal pro Woche, um Sättigung zu vermeiden und die abgestorbenen Zellen zu verdünnen. Die Einrichtung einer stabilen Zelllinie dauert in der Regel insgesamt 4 Wochen.
HINWEIS: Wenn die konstitutive Expression von Cas9 eine Belastung für die Zelle5ist, machen Sie den Ausdruck spezifisch für das Amastigot-Stadium, indem Sie die 3'-UTR des Amastin-Gens unmittelbar unterhalb des offenen Lesrahmens Cas9 konjugieren27. Die detaillierte Beschreibung des amastigotspezifischen Cas9-Expressionsplasmids findet sich in Takagi et al.22
- Transfect das obige Plasmid in Epimastigot durch Elektroporation, mit dem Kit in der Tabelle der Materialienaufgeführt . Für 1 Küvette 2 x 107 Zellen nach unten drehen, den Überstand entsorgen und mit 100 l Elektroporationspuffer, der die mitgelieferte Ergänzungslösung enthält, wieder aufsetzen.
- Etablieren Sie eine Wirtsparasiten-Kokultur von Cas9-exezierenden T. cruzi und Säugetier-Wirtszellen. Verwenden Sie in diesem Bericht 3T3-Schweizer Albino-Fibroblastenzellen als Wirt.
- Exzessen T. cruzi epimastigotes in metazyklische Trypomastigoten. Bestimmen Sie die Zelldichte der Epimastigotenkultur mit einem Hämozytometer und sammeln Sie 5 x 107 Zellen durch Zentrifugation für 15 min bei 2.100 x g. Entsorgen Sie den Überstand und setzen Sie die Zellen mit 10 ml RPMI-Medium wieder auf. Inkubieren Sie den Parasiten bei 28 °C für 1 Woche28.
- Sammeln Sie metazyklische Trypomastigoten in RPMI Medium. Neigen Sie den Kolben vorsichtig und pipet die Lösung heraus, ohne die an der Bodenoberfläche haftenden Parasiten zu stören. Das Medium für 15 min bei 2.100 x g in ein konisches Rohr und eine Zentrifuge übertragen. Entsorgen Sie den Überstand und setzen Sie den Parasiten mit 5 ml DMEM (10% FCS) wieder aus.
HINWEIS: Die Parasitenpopulation ist eine Mischung aus metazyklischen Trypomastigoten, Epimastigoten und einigen Zwischenformen. Obwohl nicht notwendig, können Sie Trypomastigote durch DEAE Ionenaustauschchromatographie29isolieren. Alternativ können Epimastigoten eliminiert werden, indem die Parasiten mit aktivem Serum inkubiert werden, um sie der Lyse30zu ergänzen. - Samen 3T3-Schweizer Albino-Fibroblastenzelle auf 60-70% Konfluenz oder etwa 1,7-2,0 x 106 Zellen in 5 ml DMEM (10% FCS) in T-25-Kulturkolben. Entfernen Sie das Wachstumsmedium und wenden Sie die Parasitenmischung aus Schritt 1.3.2 an. 24 h bei 37 °C unter 5%CO2 in einem befeuchteten Inkubator inkubieren, um eine Infektion zu etablieren.
- Entfernen Sie die Parasiten, die außerhalb der Host-3T3-Zellen verbleiben, indem Sie die Co-Kultur zweimal mit DMEM (10% FCS) waschen.
- Sobald die Kokultur gesättigt ist, Durchgang amastigot-infizierten Wirtszellen durch Trypsinisierung. Aspirieren Sie das Medium und spülen Sie die Kultur einmal mit PBS. Tragen Sie 1 ml 0,05% Trypsin-Lösung auf, um die gesamte Kulturoberfläche abzudecken, und bebrüten Sie einige Minuten bei Raumtemperatur, bis sich die angeschlossenen Wirtszellen lösen. Lösen Sie die Zellen von der Kolbenoberfläche, indem Sie 3 ml DMEM (10% FCS) über die Zellen spülen.
- Abgetrennte Wirtszellen in ein konisches Rohr und Zentrifuge für 3 min bei 300 x g übertragen. Aspirieren Sie den Überstand und setzen Sie die Zellen mit 3 ml frischem DMEM (10% FCS) wieder aus. Dieser Schritt hilft, verbleibende Epimastigoten zu beseitigen. Alles in einen sauberen T-75-Kolben mit 9 ml DMEM (10% FCS) übertragen. Fahren Sie mit der Kultivierung fort, bis Trypomastigote in den Kulturüberstand entlassen wird.
- Bewahren Sie die Co-Kultur, indem Sie zweimal pro Woche mit Trypsinisierung übergeben. Sobald 70-80% der Wirtspopulation infiziert sind, fügen Sie regelmäßig nicht infizierte Wirts-3T3-Zellen im Verhältnis 5:1 (Übertrag:frisch) hinzu, um eine Verschlechterung der Kultur zu vermeiden. Trypomastigote geht kontinuierlich aus, wenn das Gleichgewicht zwischen Wirtszellen und T. cruzi ordnungsgemäß aufrechterhalten wird.
2. Differenzierung von Trypomastigoten in EA
- Entfernen Sie am Tag vor diesem Experiment das Wachstumsmedium der Wirtsparasiten-Co-Kultur und fügen Sie frisches DMEM (10% FCS) hinzu, um EA und Trypomastigotes, die bereits in den vorherigen Tagen aus den Wirtszellen freigesetzt worden waren, wegzuwaschen. Regelmäßige Experimente erfordern mindestens zwei T-75-Flaschen konfluenter Kokultur.
-
Sammeln Sie Kultur überstand in eine konische Röhre, um frisch entstandene Trypomastigoten zu ernten. Überprüfen Sie die Probe unter dem Mikroskop (10x oder 20x Objektivlinse) auf die Qualität. Wenn es Wirtzellablagerungen gibt, zentrieren Sie die Probe kurz und übertragen Sie den Überstand in ein neues Rohr. Wenn es eine signifikante Anzahl von EAs gibt, isolieren Trypomastigotes durch das folgende Ausschwimmverfahren.
- Die Mischung aus Trypomastigoten und Amastigoten 15 min bei 2.100 x g abdrehen. Entsorgen Sie den größten Teil des Überstandes und lassen Sie 0,5-1,0 ml Medium in der Röhre zurück.
HINWEIS: Das Reduzieren des Volumes ist optional, erleichtert jedoch den folgenden Schritt. - Inkubieren Sie das Pellet bei 37 °C für 1-2 h, so dass aktive Trypomastigoten aus dem Pellet schwimmen können (Abbildung 2).
- Übertragen Sie den Überstand, der Trypomastigoten enthält, in ein 1,5 ml Mikrozentrifugenrohr.
- Die Mischung aus Trypomastigoten und Amastigoten 15 min bei 2.100 x g abdrehen. Entsorgen Sie den größten Teil des Überstandes und lassen Sie 0,5-1,0 ml Medium in der Röhre zurück.
- Zentrifugieren Sie das konische Rohr für 15 min bei 2.100 x g, um Trypomastigoten zu sammeln. Wenn das Schwimmen durchgeführt wurde oben durchgeführt wurde, zentrifugieren Sie das 1,5 ml Rohr für 2 min bei 4.000 x g, um das Trypomastigot ekeln. Entsorgen Sie den Überstand.
- Das Pellet mit 5 ml DMEM gepuffert mit 20 mM MES (pH 5.0), ergänzt mit 0,4% BSA19,wieder aufsetzen. Übertragen Sie den Parasiten in einen T-25-Kulturkolben. Lassen Sie die Kappe locker. Die Zelldichte muss um oder unter 1 x 107 Zellen/ml liegen, da eine Übersättigung die Wahrscheinlichkeit eines Zelltodes erhöht.
HINWEIS: Die Farbe des DMEM muss gelb und nicht orange sein. Wenn das ursprüngliche DMEM eine hohe Pufferkapazität hatte, reichen 20 mM MES (pH 5.0) nicht aus, um den pH-Wert zu senken. Der pH-Wert des Mediums muss in diesem Fall durch Zugabe von HCl angepasst werden. Das saure Medium kann bei 4 °C gehalten werden, jedoch nicht länger als 1 Monat. - Inkubieren Sie den Kulturkolben bei 37 °C unter 5% CO2 in einem befeuchteten Inkubator. Rund 95% der Parasiten unterscheiden sich nach 24 h in Amastigoten.
3. Elektroporation von EA
- Bereiten Sie gRNA für die Elektroporation vor. Dies kann durch In-vitro-Transkription oder einfach durch den Kauf synthetischer RNA-Oligonukleotide von einem Hersteller erfolgen. In diesem Bericht werden crRNA und tracrRNA von Integrated DNA Technologies, Inc. verwendet.
- Zentrifugieren Sie die Kultur der EAs für 15 min bei 2.100 x g. Entsorgen Sie Überstand.
- Setzen Sie das Pellet mit Elektroporationspuffer mit bereitgestellter Ergänzungslösung für die endgültige Zelldichte von 1 x 108 Zellen/ml wieder auf.
ANMERKUNG: EM-Puffer verursacht mehr Zellsterblichkeit im Vergleich zu dem in der Materialtabelleaufgeführten Elektroporationspuffer; daher wird es nicht für amastigote Transfektion empfohlen (Ergänzungsfigur 1). - Aliquot 100 l resuspendierte Parasiten (1 x 107 Zellen) in 1,5 ml Mikrozentrifugenröhren. Fügen Sie 5-10 gRNA hinzu und mischen Sie vorsichtig durch Pipettieren.
- Übertragen Sie das Gemisch auf eine 2 mm Spalt-Elektroporationsküvette. Tragen Sie den Puls mit dem Elektroporationsgerät auf, indem Sie das X-14-Programm verwenden.
- Übertragen Sie den Küvetteninhalt in einen T-25-Kolben mit 5 ml vorgewärmten LIT-Medium (10% FCS). Lassen Sie die Kappe locker und brüten Sie den Kolben bei 37 °C unter 5%CO2.
- Überwachen Sie das Zellwachstum entweder durch Fortsetzung der axtischen Kultivierung (Abschnitt 4) oder als intrazelluläre Amastigoten nach einer Wirtszellinfektion (Abschnitt 5).
4. Überwachung des Wachstums von Knockout-Zellen als Axenic Amastigotes
- EAs setzen sich am unteren Ende des Kulturmediums an, schütteln Sie den Kolben also sanft, um sie in die Lösung zurückzusetzen. Das Waschen der Kolbenoberfläche durch Pipetten hilft, da einige Zellen an den Kolben kleben.
- Mischen Sie die Propidiumjodid-Lösung (PI) mit 20 l Amastigotenkultur.
HINWEIS: Lassen Sie den Kulturkolben nicht länger als nötig außerhalb des Inkubators. Die Temperatur ist einer der Faktoren, die axenische Proliferationermöglicht 22. - Tragen Sie die Probe auf das Hämozytometer auf und beobachten Sie sie unter einem Fluoreszenzmikroskop. PI ist auf beschädigte Zellmembran, aber von lebenden Zellen ausgeschlossen. Zählen Sie die Anzahl der lebensfähigen Amastigoten, die nicht von PI gefärbt sind (ex/em 570 nm/602 nm).
5. Überwachung des Wachstums von Knockout-Zellen als intrazelluläre Amastigoten
- Seed Host 3T3 Zellen in einer 12-Well-Platte mit DMEM (10% FCS). Passen Sie die Zelldichte auf 70-80% Konfluenz oder etwa 3 x 105 Zellen pro Brunnen an.
HINWEIS: Da Amastigoten nicht motil sind, verbessert die Abdeckung des größten Teils der Wachstumsfläche durch die Wirtszellen die Infektionseffizienz. - Sammeln Sie Knockout-Amastigoten aus Schritt 3.6 durch Zentrifugation einen Tag nach der Elektroporation. Entsorgen Sie den Überstand und setzen Sie den Parasiten mit 2 ml DMEM (10% FCS) wieder aus.
- Entfernen Sie medium aus der Hostzellenkultur, und wenden Sie resuspendierte Amastigoten an. Die Vielzahl der Infektionen sollte 20 oder höher sein. Inkubieren Sie die Platte bei 37 °C unter 5% CO2 für 2 Tage, damit Amastigoten eine Infektion feststellen können.
HINWEIS: Die Infektionsdauer kann je nach Zweck 1 Tag betragen. - Wash away EAs blieb mit DMEM (10% FCS) zweimal außerhalb der Hostzellen.
- Fügen Sie der Wirts-Parasiten-Kokultur frisches DMEM (10% FCS) hinzu und setzen Sie die Inkubation bei 37 °C für weitere 2 Tage fort.
-
Um die Infektionseffizienz zu bewerten, visualisieren Sie Diekerne der Wirtszellen und intrazellulären Amastigoten.
HINWEIS: Kerne neigen dazu, sich in gesättigter Kokultur zu überlappen. Die Neubeschichtung der Zellen bei geringerer Zelldichte (z. B. 1:5 Verdünnung) vor fixierender und färbung hilft, Die Kerne leichter zu zählen.- Entfernen Sie das Kulturmedium und wenden Sie 1 ml 10% Formalinlösung in PBS an, um die Zellen zu reparieren. 10 min bei Raumtemperatur inkubieren.
- Ersetzen Sie die Formalinlösung durch 1 ml PBS mit 1 g/ml Hoechst 33342 und 0,1% Triton X-100. 5 min bei Raumtemperatur inkubieren.
- Entfernen Sie die Hoechst-Lösung und spülen Sie die Zellen einmal mit PBS. Fügen Sie 1 ml frische PBS hinzu.
- Beobachten Sie unter Fluoreszenzmikroskop und identifizieren Wirtszellkerne, die mit kleineren Parasitenkernen verbunden sind (Abbildung 4). Wirtszellen, die mehr als 2 Amastigoten enthalten, sollten als infiziert betrachtet werden, nicht um eine unproduktive Erstinfektion oder ungewaschene EAs einzubeziehen.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Isolierung von Trypomastigoten durch das Ausschwimmverfahren
Um frische Trypomastigoten aus der Kontamination alter EAs durch Swim-out-Verfahren zu ernten, müssen Zellpellets mindestens für 1 h inkubiert werden. Abbildung 2B). In diesem speziellen Experiment betrug der Prozentsatz der Trypomastigoten in der ursprünglichen Mischung 38 %, und der Prozentsatz nach dem Ausschwimmen lag zu einem bestimmten Zeitpunkt über 98 %. Aus zwei T-75-Flasche...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Wir haben gezeigt, dass die axtische Kultur von T. cruzi amastigotes in CRISPR/Cas9-vermitteltem Gen Knockout genutzt werden kann, indem gRNA direkt in Cas9-exemittiert wird. Auf diese Weise kann die Wesentlichkeit des Zielgens speziell im Amastigot-Stadium bewertet werden, ohne andere Entwicklungsstadien durchlaufen zu müssen.
Ein weiterer vorteilhafter Aspekt der Amastigot-Transfektion ist die Bequemlichkeit bei der Prüfung auf eine große Anzahl von Zielgenen. Sobald die Kokultur...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben keinen Interessenkonflikt zu offenbaren.
Danksagungen
Diese Arbeit wurde teilweise von JSPS KAKENHI Grant Number 18K15141 an Y.T. unterstützt.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
Referenzen
- World Health Organization. (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , [updated 2017 Mar; cited 2017 Aug], at http://www.who.int/mediacentre/factsheets/fs340/en/ (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012(2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388(2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547(1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341(2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088(2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370(2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960(2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten