
Aby wyświetlić tę treść, wymagana jest subskrypcja JoVE. Zaloguj się lub rozpocznij bezpłatny okres próbny.
Method Article
Introducing a Gene Knockout Directly Into the Amastigote Stage of Trypanosoma cruzi Using the CRISPR/Cas9 System
W tym Artykule
Podsumowanie
Here, we describe a protocol to introduce a gene knockout into the extracellular amastigote of Trypanosoma cruzi, using the CRISPR/Cas9 system. The growth phenotype can be followed up either by cell counting of axenic amastigote culture or by proliferation of intracellular amastigotes after host cell invasion.
Streszczenie
Trypanosoma cruzi is a pathogenic protozoan parasite that causes Chagas’ disease mainly in Latin America. In order to identify a novel drug target against T. cruzi, it is important to validate the essentiality of the target gene in the mammalian stage of the parasite, the amastigote. Amastigotes of T. cruzi replicate inside the host cell; thus, it is difficult to conduct a knockout experiment without going through other developmental stages. Recently, our group reported a growth condition in which the amastigote can replicate axenically for up to 10 days without losing its amastigote-like properties. By using this temporal axenic amastigote culture, we successfully introduced gRNAs directly into the Cas9-expressing amastigote to cause gene knockouts and analyzed their phenotypes exclusively in the amastigote stage. In this report, we describe a detailed protocol to produce in vitro derived extracellular amastigotes, and to utilize the axenic culture in a CRISPR/Cas9-mediated knockout experiment. The growth phenotype of knockout amastigotes can be evaluated either by cell counts of the axenic culture, or by replication of intracellular amastigote after host cell invasion. This method bypasses the parasite stage differentiation normally involved in producing a transgenic or a knockout amastigote. Utilization of the temporal axenic amastigote culture has the potential to expand the experimental freedom of stage-specific studies in T. cruzi.
Wprowadzenie
Trypanosoma cruzi is the causative agent of Chagas’ disease, which is prevalent mainly in Latin America1. T. cruzi has distinctive life cycle stages as it travels between an insect vector and a mammalian host2. T. cruzi replicates as an epimastigote in the midgut of a blood-sucking triatomine bug and differentiates into an infectious metacyclic trypomastigote in its hindgut before being deposited on a human or animal host. Once the trypomastigote gets into the host body through the bite site or through a mucous membrane, the parasite invades a host cell and transforms into a flagella-less round form called an amastigote. The amastigote replicates within the host cell and eventually differentiates into trypomastigote, which bursts out of the host cell and enters the blood stream to infect another host cell.
Since currently available chemotherapeutic agents, benznidazole and nifurtimox, cause adverse side effects and are ineffective in the chronic phase of the disease3, it is of a great interest to identify novel drug targets against T. cruzi. In recent years, the CRISPR/Cas9 system has become a powerful tool to effectively perform gene knockout in T. cruzi, either by transfection of separate or single plasmid(s) containing gRNA and Cas94, by stable expression of Cas9 and subsequent introduction of gRNA5,6,7 or transcription template of gRNA8, or by electroporation of the pre-formed gRNA/Cas9 RNP complex7,9. This technological advancement is highly anticipated to accelerate the drug target research in Chagas’ disease.
To proceed with the drug development, it is crucial to validate the essentiality of the target gene or efficacy of drug candidate compounds in the amastigote of T. cruzi, as it is the replication stage of the parasite in the mammalian host. However, this is a challenging task, because amastigotes cannot be directly manipulated due to the presence of an obstructive host cell. In Leishmania, a closely related protozoan parasite to T. cruzi, an axenic amastigote culturing method was developed and has been utilized in drug screening assays10,11,12,13. Although there are some discrepancies in susceptibility to compounds between axenic amastigotes and intracellular amastigotes14, the ability to maintain the axenic culture nonetheless provides valuable experimental tools to study the basic biology of the clinically relevant stage of Leishmania15,16. In the case of T. cruzi, literatures regarding the presence of naturally occurring extracellular amastigotes (EA)17 and in vitro production of EA17,18,19 date back to decades ago. In addition, EA is known to have an infectious capability20, albeit less than that of trypomastigote, and the mechanism of amastigote host invasion has been elucidated in recent years (reviewed by Bonfim-Melo et al.21). However, unlike Leishmania, EA had not been utilized as an experimental tool in T. cruzi, primarily because EA had been regarded as an obligate intracellular parasite, and thus had not been considered as “replicative form” in a practical sense.
Recently, our group proposed to utilize EA of T. cruzi as a temporal axenic culture22. Amastigotes of T. cruzi Tulahuen strain can replicate free of host cells in LIT medium at 37 °C for up to 10 days without major deterioration or loss of amastigote-like properties. During the host-free growth period, EA was successfully utilized for exogenous gene expression by conventional electroporation, drug titration assay with trypanocidal compounds, and CRISPR/Cas9-mediated knockout followed by growth phenotype monitoring. In this report, we describe the detailed protocol to produce in vitro derived EA and to utilize the axenic amastigote in knockout experiments.
Protokół
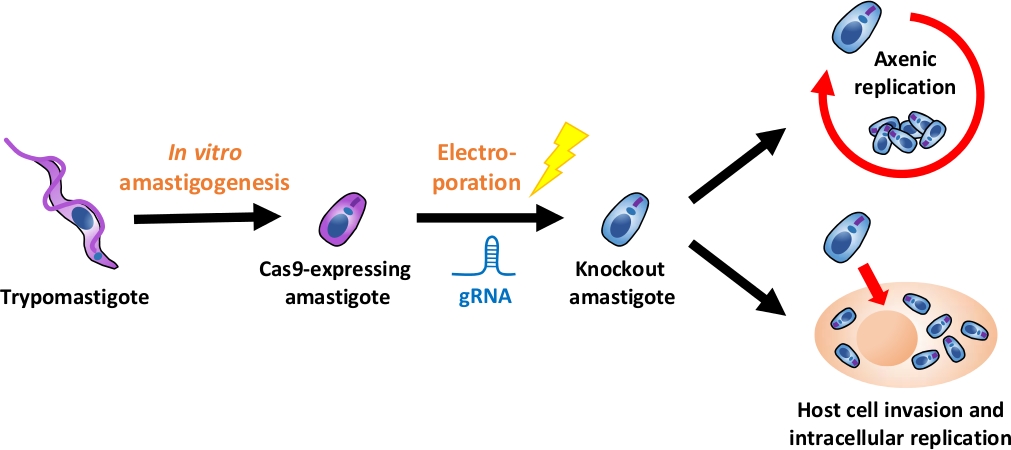
NOTE: An overview of the entire experimental flow is depicted in Figure 1.

Figure 1: Overview of the knockout experiment using EA. Tissue culture-derived trypomastigotes are harvested and differentiated into EA. gRNA is transfected into Cas9-expressing amastigotes by electroporation, and growth phenotype of the knockout amastigote is evaluated either by axenic replication or by intracellular replication after host cell invasion. Please click here to view a larger version of this figure.
{kind=link}
1. Parasite Culture Preparations
- Tulahuen strain of Trypanosoma cruzi is used throughout this report. Maintain epimastigotes of T. cruzi in LIT medium (10% FCS, see Supplemental Table 1). Securely close the cap and keep the culture flask at 28 °C.
- Generate a transgenic strain of T. cruzi that harbors the Cas9 endonuclease. Examples of the expression plasmids that contain Cas9 coding sequence and G418 (neor) selection marker can be found in Lander et al.4 and Peng et al.5
- Transfect the above plasmid into epimastigote by electroporation, using the kit listed in the Table of Materials. For 1 cuvette, spin down 2 x 107 cells, discard the supernatant, and resuspend with 100 µL of electroporation buffer containing the provided supplement solution.
NOTE: The electroporation buffer can be substituted with EM buffer24 (3:1 mixture of cytomix25 and phosphate-sucrose buffer). - Add 20-40 µg of plasmid and transfer the mixture into a 2 mm gap electroporation cuvette. Apply the pulse with an electroporation device (see Table of Materials), using the X-14 program26.
- Transfer the cuvette contents into a T-25 flask containing 5 mL of LIT medium (10% FCS). Incubate the flask at 28 °C for 24 h.
- Add G418 to a final concentration of 250 µg/mL and continue incubation at 28 ˚C. It takes about 1 week to kill off the non-transfected epimastigotes. Once the G418-resistant population starts to recover, passage the culture once or twice a week to avoid saturation and to dilute out the dead cells. Establishment of a stable cell line usually takes total of 4 weeks.
NOTE: If constitutive expression of Cas9 is a burden for the cell5, make the expression specific to the amastigote stage by conjugating the 3’-UTR of amastin gene immediately downstream of the Cas9 open reading frame27. The detailed description of the amastigote-specific Cas9 expression plasmid can be found in Takagi et al.22
- Transfect the above plasmid into epimastigote by electroporation, using the kit listed in the Table of Materials. For 1 cuvette, spin down 2 x 107 cells, discard the supernatant, and resuspend with 100 µL of electroporation buffer containing the provided supplement solution.
- Establish a host-parasite co-culture of Cas9-expressing T. cruzi and mammalian host cells. In this report, use 3T3-Swiss Albino fibroblast cell as a host.
- Differentiate T. cruzi epimastigotes into metacyclic trypomastigotes. Determine the cell density of the epimastigote culture using a hemocytometer and collect 5 x 107 cells by centrifugation for 15 min at 2,100 x g. Discard the supernatant and resuspend the cells with 10 mL of RPMI medium. Incubate the parasite at 28 °C for 1 week28.
- Collect metacyclic trypomastigotes in RPMI medium. Carefully tilt the flask and pipet out the solution without disturbing the parasites adhered to the bottom surface. Transfer the medium to a conical tube and centrifuge for 15 min at 2,100 x g. Discard the supernatant and resuspend the parasite with 5 mL of DMEM (10% FCS).
NOTE: The parasite population is a mixture of metacyclic trypomastigotes, epimastigotes, and some intermediate forms. Although not necessary, you can isolate trypomastigote by DEAE ion exchange chromatography29. Alternatively, epimastigotes can be eliminated by incubating the parasites with active serum to subject them to complement lysis30. - Seed 3T3-Swiss Albino fibroblast cell to 60-70% confluency, or about 1.7-2.0 x 106 cells in 5 mL of DMEM (10% FCS) in T-25 culture flask. Remove growth medium and apply the parasite mixture from step 1.3.2. Incubate for 24 h at 37 ˚C under 5% CO2 in a humidified incubator to establish infection.
- Remove the parasites remaining outside of the host 3T3 cells by washing the co-culture with DMEM (10% FCS) twice.
- Once the co-culture is saturated, passage amastigote-infected host cells by trypsinization. Aspirate the medium and rinse the culture once with PBS. Apply 1 mL of 0.05% trypsin solution to cover the entire culture surface, and incubate for few minutes at room temperature until the attached host cells become loose. Detach the cells from the flask surface by flushing 3 mL of DMEM (10% FCS) over the cells.
- Transfer detached host cells into a conical tube, and centrifuge for 3 min at 300 x g. Aspirate the supernatant and resuspend the cells with 3 mL of fresh DMEM (10% FCS). This step helps to eliminate remaining epimastigotes. Transfer all into a clean T-75 flask containing 9 mL of DMEM (10% FCS). Continue culturing until trypomastigote is released into culture supernatant.
- Maintain the co-culture by passaging with trypsinization twice a week. Once 70-80% of the host population become infected, regularly add uninfected host 3T3 cells at the ratio of 5:1 (carryover:fresh), in order to avoid culture deterioration. Trypomastigote egresses continuously if the balance between host cells and T. cruzi is properly maintained.
2. Differentiation of Trypomastigotes into EA
- On the day before this experiment, remove the growth medium of host-parasite co-culture and add fresh DMEM (10% FCS) to wash away EA and trypomastigotes that had already been released from the host cells in previous days. Regular experiments require at least two T-75 flasks of confluent co-culture.
- Collect culture supernatant into a conical tube to harvest freshly emerged trypomastigotes. Check the sample under a microscope (10x or 20x objective lens) for the quality. If there is host cell debris, briefly centrifuge the sample and transfer the supernatant into a new tube. If there are significant number of EAs, isolate trypomastigotes by the following swim-out procedure.
- Spin down the mixture of trypomastigotes and amastigotes for 15 min at 2,100 x g. Discard most of the supernatant, leaving 0.5-1.0 mL of medium in the tube.
NOTE: Reducing the volume is optional, but it makes the following step easier. - Incubate the pellet at 37 ˚C for 1-2 h, allowing active trypomastigotes to swim out of the pellet (Figure 2).
- Transfer the supernatant containing trypomastigotes to a 1.5 mL microcentrifuge tube.
- Spin down the mixture of trypomastigotes and amastigotes for 15 min at 2,100 x g. Discard most of the supernatant, leaving 0.5-1.0 mL of medium in the tube.
- Centrifuge the conical tube for 15 min at 2,100 x g to collect trypomastigotes. If the swim-out procedure was performed above, centrifuge the 1.5 mL tube for 2 min at 4,000 x g to pellet the trypomastigote. Discard the supernatant.
- Resuspend the pellet with 5 mL of DMEM buffered with 20 mM MES (pH 5.0), supplemented with 0.4% BSA19. Transfer the parasite to a T-25 culture flask. Leave the cap loose. The cell density must be around or below 1 x 107 cells/mL, since oversaturation increases the chance of cell death.
NOTE: The color of the DMEM must be yellow, not orange. If the original DMEM had high buffering capacity, 20 mM MES (pH 5.0) is not enough to lower the pH. The pH of the medium must be adjusted by addition of HCl in that case. The acidic medium can be kept at 4 ˚C, but no longer than 1 month. - Incubate the culture flask at 37 °C under 5% CO2 in a humidified incubator. Around 95% of parasites differentiate into amastigotes after 24 h.
3. Electroporation of EA
- Prepare gRNA for electroporation. This can be done by in vitro transcription, or simply by purchasing synthetic RNA oligonucleotides from a manufacturer. In this report, crRNA and tracrRNA from Integrated DNA Technologies, Inc. are used.
- Centrifuge the culture of EAs for 15 min at 2,100 x g. Discard supernatant.
- Resuspend the pellet with electroporation buffer containing provided supplement solution to the final cell density of 1 x 108 cells/mL.
NOTE: EM buffer causes more cell deaths compared to the electroporation buffer listed in Table of Materials; thus, it is not recommended for amastigote transfection (Supplemental Figure 1). - Aliquot 100 µL of resuspended parasites (1 x 107 cells) into 1.5 mL microcentrifuge tubes. Add 5-10 µg of gRNA and gently mix by pipetting.
- Transfer the mixture to a 2 mm gap electroporation cuvette. Apply the pulse with the electroporation device, using X-14 program.
- Transfer the cuvette contents into a T-25 flask containing 5 mL of pre-warmed LIT medium (10% FCS). Leave the cap loose and incubate the flask at 37 °C under 5% CO2.
- Monitor the cell growth either by continuation of axenic culturing (section 4) or as intracellular amastigotes after host cell infection (section 5).
4. Monitoring the Growth of Knockout Cells as Axenic Amastigotes
- EAs settle at the bottom of the culture medium, so gently shake the flask to resuspend them into the solution. Washing the flask surface by pipetting helps, as some cells are adhered to the flask.
- Mix 1 µL of propidium iodide (PI) solution (20 µg/mL) with 20 µL of amastigote culture.
NOTE: Do not leave the culture flask outside of incubator for longer than necessary. Temperature is one of factors that enables axenic proliferation22. - Apply the sample onto hemocytometer and observe under a fluorescence microscope. PI is permeant to damaged cell membrane but is excluded from live cells. Count the number of viable amastigotes that are not stained by PI (ex/em 570 nm/602 nm).
5. Monitoring the Growth of Knockout Cells as Intracellular Amastigotes
- Seed host 3T3 cells in a 12-well plate with DMEM (10% FCS). Adjust the cell density to 70-80% confluency, or about 3 x 105 cells per well.
NOTE: Since amastigotes are not motile, covering most of the growth surface by the host cells improves infection efficiency. - Collect knockout amastigotes from step 3.6 by centrifugation one day after electroporation. Discard the supernatant, and resuspend the parasite with 2 mL of DMEM (10% FCS).
- Remove medium from the host cell culture and apply resuspended amastigotes. Multiplicity of infection should be 20 or higher. Incubate the plate at 37 °C under 5% CO2 for 2 days to allow amastigotes to establish infection.
NOTE: Infection period can be 1 day, depending on the purpose. - Wash away EAs remained outside of the host cells twice with DMEM (10% FCS).
- Add fresh DMEM (10% FCS) to the host-parasite co-culture and continue the incubation at 37 °C for additional 2 days.
- To evaluate infection efficiency, visualize nuclei of the host cells and intracellular amastigote.
NOTE: Nuclei tend to be overlapped in saturated co-culture. Re-plating the cells at lower cell density (such as 1:5 dilution) prior to fixation and staining helps to count nuclei more easily.- Remove culture medium and apply 1 mL of 10% formalin solution in PBS to fix the cells. Incubate for 10 min at room temperature.
- Replace the formalin solution with 1 mL of PBS containing 1 µg/mL Hoechst 33342 and 0.1% Triton X-100. Incubate for 5 min at room temperature.
- Remove the Hoechst solution and rinse the cells once with PBS. Add 1 mL of fresh PBS.
- Observe under fluorescence microscope and identify host cell nuclei that are associated with smaller parasite nuclei (Figure 4). Host cells that contain more than 2 amastigotes should be considered as infected, not to include nonproductive initial infection or unwashed EAs.
Wyniki
Isolation of trypomastigotes by the swim-out procedure
To harvest fresh trypomastigotes from contaminating old EAs by swim-out procedure, cell pellets need to be incubated at least for 1 h. Incubating the pellets for more than 2 h does not significantly increase the number of trypomastigotes swimming in the solution (Figure 2B). In this particular experiment, the percentage of trypomastigote in the initial mixture was 38%, and the percentage after...
Dyskusje
We demonstrated that the axenic culture of T. cruzi amastigotes can be utilized in CRISPR/Cas9-mediated gene knockout, by electroporating gRNA directly into Cas9-expressing EA. This way, the essentiality of the target gene specifically in amastigote stage can be evaluated without going through other developmental stages.
Another beneficial aspect of amastigote transfection is the convenience in testing for a large number of target genes. Once the co-culture of Cas9-expressing T. c...
Ujawnienia
The authors have no conflict of interest to disclose.
Podziękowania
This work was supported in part by JSPS KAKENHI Grant Number 18K15141 to Y.T.
Materiały
| Name | Company | Catalog Number | Comments |
| 20% formalin solution | FUJIFILM Wako Pure Chemical | 068-03863 | fixing cells |
| 25 cm2 double seal cap culture flask | AGC Techno Glass | 3100-025 | |
| 75 cm2 double seal cap culture flask | AGC Techno Glass | 3110-075 | |
| All-in One Fluorescence Microscope | Keyence | BZ-X710 | |
| Alt-R CRISPR-Cas9 crRNA (for Control) | IDT | custom made | target sequence = GGACGGCACCTTCATCTACAAGG |
| Alt-R CRISPR-Cas9 crRNA (for TcCGM1) | IDT | custom made | target sequence = TAGCCGCGATGGAGAGTTTATGG |
| Alt-R CRISPR-Cas9 crRNA (for TcPAR1) | IDT | custom made | target sequence = CGTGGAGAACGCCATTGCCACGG |
| Alt-R CRISPR-Cas9 tracrRNA | IDT | 1072532 | to anneal with crRNA |
| Amaxa Nucleofector device | LONZA | AAN-1001 | electroporation |
| Basic Parasite Nucleofector Kit 2 | LONZA | VMI-1021 | electroporation |
| BSA | Sigma-Aldrich | A3294 | component of the medium for in vitro amastigogenesis |
| Burker-Turk disposable hemocytometer | Watson | 177-212C | cell counting |
| Coster 12-well Clear TC-Treated Multiple Well Plates | Corning | 3513 | |
| DMEM | FUJIFILM Wako Pure Chemical | 044-29765 | culture medium |
| Fetal bovine serum, Defined | Hyclone | SH30070.03 | heat-inactivate before use |
| G-418 Sulfate Solution | FUJIFILM Wako Pure Chemical | 077-06433 | selection of transformant |
| Hemin chloride | Sigma-Aldrich | H-5533 | component of LIT medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | staining of nuclei |
| Liver infusion broth, Difco | Becton Dickinson | 226920 | component of LIT medium |
| MES | FUJIFILM Wako Pure Chemical | 349-01623 | component of the medium for in vitro amastigogenesis |
| PBS (–) | FUJIFILM Wako Pure Chemical | 166-23555 | |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | staining of dead cells |
| RPMI 1646 | Sigma-Aldrich | R8758 | medium for metacyclogenesis |
Odniesienia
- World Health Organization. . (WHO) Fact sheet: Chagas disease (American trypanosomiasis). , (2017).
- Clayton, J. Chagas disease 101. Nature. 465 (n7301_supp), S4-S5 (2010).
- Apt, W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Design, Development and Therapy. 4, 243-253 (2010).
- Lander, N., Li, Z. H. H., Niyogi, S., Docampo, R. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio. 6 (4), e01012 (2015).
- Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., Tarleton, R. L. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio. 6 (1), e02097-e02114 (2015).
- Romagnoli, B. A. A., Picchi, G. F. A., Hiraiwa, P. M., Borges, B. S., Alves, L. R., Goldenberg, S. Improvements in the CRISPR/Cas9 system for high efficiency gene disruption in Trypanosoma cruzi. Acta Tropica. 178, 190-195 (2018).
- Burle-Caldas, G. A., Soares-Simões, M., Lemos-Pechnicki, L., DaRocha, W. D., Teixeira, S. M. R. Assessment of two CRISPR-Cas9 genome editing protocols for rapid generation of Trypanosoma cruzi gene knockout mutants. International Journal for Parasitology. 48 (8), 591-596 (2018).
- Costa, F. C. Expanding the toolbox for Trypanosoma cruzi: A parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Neglected Tropical Diseases. 12 (4), e0006388 (2018).
- Soares Medeiros, L. C. High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio. 8 (6), (2017).
- Callahan, H. L., Portal, A. C., Devereaux, R., Grogl, M. An axenic amastigote system for drug screening. Antimicrobial Agents and Chemotherapy. 41 (4), 818-822 (1997).
- Bates, P. A. Axenic culture of Leishmania amastigotes. Parasitology Today. 9 (4), 143-146 (1993).
- Ravinder, R., Bhaskar, B., Gangwar, S., Goyal, N. Development of luciferase expressing Leishmania donovani axenic amastigotes as primary model for in vitro screening of antileishmanial compounds. Current Microbiology. 65 (6), 696-700 (2012).
- Nühs, A. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Tropical Diseases. 9 (9), 1-17 (2015).
- De Rycker, M. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrobial Agents and Chemotherapy. 57 (7), 2913-2922 (2013).
- Rochette, A., Raymond, F., Corbeil, J., Ouellette, M., Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Molecular and Biochemical Parasitology. 165 (1), 32-47 (2009).
- Pescher, P., Blisnick, T., Bastin, P., Späth, G. F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cellular Microbiology. 13 (7), 978-991 (2011).
- Andrews, N. W., Hong, K. S. U., Robbins, E. S., Nussenzweig, V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Experimental Parasitology. 64 (3), 474-484 (1987).
- Pan, S. C. Trypanosoma cruzi: intracellular stages grown in a cell-free medium at 37 C. Experimental Parasitology. 45 (2), 215-224 (1978).
- Tomlinson, S., Vandekerckhove, F., Frevert, U., Nussenzweig, V. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology. 110 (05), 547 (1995).
- Ley, V., Andrews, N. W., Robbins, E. S., Nussenzweig, V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. Journal of Experimental Medicine. 168 (0022-1007 (Print)), 649-659 (1988).
- Bonfim-Melo, A., Ferreira, E. R., Florentino, P. T. V., Mortara, R. A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Frontiers in Microbiology. 9, 1341 (2018).
- Takagi, Y., Akutsu, Y., Doi, M., Furukawa, K. Utilization of proliferable extracellular amastigotes for transient gene expression, drug sensitivity assay, and CRISPR/Cas9-mediated gene knockout in Trypanosoma cruzi. PLOS Neglected Tropical Diseases. 13 (1), e0007088 (2019).
- Fernandes, J. F., Castellani, O. Growth characteristics and chemical composition of Trypanosoma cruzi. Experimental Parasitology. 18 (2), 195-202 (1966).
- Oberholzer, M., Lopez, M. A., Ralston, K. S., Hill, K. L. Approaches for Functional Analysis of Flagellar Proteins in African Trypanosomes. Methods in Cell Biology. 93, 21-57 (2009).
- van den Hoff, M. J. B., Moorman, A. F. M., Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Research. 20 (11), 2902-2902 (1992).
- Pacheco-Lugo, L., Díaz-Olmos, Y., Sáenz-García, J., Probst, C. M., DaRocha, W. D. Effective gene delivery to Trypanosoma cruzi epimastigotes through nucleofection. Parasitology International. 66 (3), 236-239 (2017).
- Coughlin, B. C., Teixeira, S. M., Kirchhoff, L. V., Donelson, J. E. Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3’-untranslated region position-dependent cis-element and an untranslated region-binding protein. The Journal of Biological Chemistry. 275 (16), 12051-1260 (2000).
- Shaw, A. K., Kalem, M. C., Zimmer, S. L. Mitochondrial Gene Expression Is Responsive to Starvation Stress and Developmental Transition in Trypanosoma cruzi. mSphere. 1 (2), (2016).
- Chao, D., Dusanic, D. G. Comparative studies of the isolation of metacyclic trypomastigotes of Trypanosoma cruzi by DEAE ion exchange chromatography. Zhonghua Minguo wei sheng wu ji mian yi xue za zhi (Chinese Journal of Microbiology and Immunology). 17 (3), 146-152 (1984).
- Nogueira, N., Bianco, C., Cohn, Z. Studies on the selective lysis and purification of Trypanosoma cruzi. The Journal of Experimental Medicine. 142 (1), 224-229 (1975).
- Minning, T. A., Weatherly, D. B., Atwood, J., Orlando, R., Tarleton, R. L., Tarleton, R. L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics. 10, 370 (2009).
- Rico, E., Jeacock, L., Kovářová, J., Horn, D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Scientific Reports. 8 (1), 7960 (2018).
- Fu, Y. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31 (9), 822-826 (2013).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Chiurillo, M. A., Lander, N., Bertolini, M. S., Storey, M., Vercesi, A. E., Docampo, R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of Trypanosoma cruzi. mBio. 8 (3), e00574-e00617 (2017).
Przedruki i uprawnienia
Zapytaj o uprawnienia na użycie tekstu lub obrazów z tego artykułu JoVE
Zapytaj o uprawnieniaThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Wszelkie prawa zastrzeżone