
Method Article
使用沉降机进行冷冻电镜和单颗粒分析
摘要
冷冻电子显微镜中的单颗粒分析是用于在高分辨率下确定生物集合体结构的主要技术之一。Scipion提供了创建整个管道的工具,以处理显微镜获取的信息并实现生物标本的3D重建。
摘要
冷冻电子显微镜已成为生物学研究中以近原子分辨率揭示大分子结构信息的最重要工具之一。在单颗粒分析中,玻化样品由电子束成像,显微镜柱末端的探测器产生该样品的电影。这些电影包含数千个随机方向的相同粒子的图像。数据需要经过具有多个步骤的图像处理工作流程才能获得最终的3D重建体积。图像处理工作流程的目标是确定采集参数,以便能够重建所研究的标本。Scipion提供了在集成框架中使用多个图像处理包创建此工作流程的所有工具,还允许结果的可追溯性。在本文中,Scipion中的整个图像处理工作流程通过来自真实测试用例的数据进行介绍和讨论,提供了从显微镜获得的电影到高分辨率最终3D重建所需的所有细节。此外,还讨论了使用共识工具的强大功能,这些工具允许组合方法,并在工作流程的每个步骤中确认结果,从而提高所获得结果的准确性。
引言
在冷冻电子显微镜(cryo-EM)中,玻璃化冷冻水合物标本的单颗粒分析(SPA)是生物大分子成像中最广泛使用和最成功的变体之一,因为它可以理解分子相互作用和生物集合的功能1。这要归功于这种成像技术的最新进展,该技术引发了"分辨率革命"2,并允许以近原子分辨率成功测定生物3D结构。目前,SPA冷冻电镜中达到的最高分辨率是1.15 Å的apoferritin3(EMDB条目:11668)。这些技术进步包括样品制备4、图像采集5和图像处理方法6的改进。本文将重点介绍最后一点。
简而言之,图像处理方法的目标是识别所有采集参数,以反转显微镜的成像过程并恢复所研究生物标本的3D结构。这些参数是相机的增益,光束诱导的运动,显微镜的像差(主要是散焦),每个粒子的3D角度方向和平移,以及具有构象变化的标本时的构象状态。然而,参数的数量非常高,cryo-EM需要使用低剂量图像以避免辐射损伤,这显着降低了采集图像的信噪比(SNR)。因此,问题不能明确解决,所有要计算的参数只能是估计值。在图像处理工作流程中,应识别正确的参数,丢弃剩余的参数,最终获得高分辨率的3D重建。
显微镜产生的数据被收集在框架中。简化一下,每当使用电子计数探测器时,帧都包含到达图像中特定位置(像素)的电子数量。在特定的视野中,收集多个帧,这称为电影。由于使用低电子剂量来避免可能破坏样品的辐射损伤,因此SNR非常低,并且需要对对应于同一电影的帧进行平均,以获得揭示样品结构信息的图像。然而,不仅应用了简单的平均值,由于需要补偿的光束诱导运动,样品在成像时间内可能会遭受偏移和其他类型的运动。移位补偿和平均帧源自显微照片。
一旦获得显微照片,我们需要估计显微镜为每个微照片引入的像差,称为对比度传递函数(CTF),它表示显微照片对比度的变化作为频率的函数。然后,可以选择和提取颗粒,这称为颗粒拾取。每个粒子都应该是一个小图像,只包含所研究标本的一个副本。有三个用于粒子拾取的算法系列:1)仅使用粒子外观的一些基本参数化来在整个显微照片(例如,粒度)中找到它们的算法,2)从用户或预先训练的集合中学习粒子的外观的算法,以及3)使用图像模板的算法。每个系列都有不同的属性,稍后将显示。
在显微照片中发现的提取的粒子集将用于2D分类过程,该过程有两个目标:1)通过丢弃包含纯噪声图像,重叠粒子或其他伪影的子集来清洁粒子集,以及2)代表每个类的平均粒子可以用作初始信息来计算3D初始体积。
3D初始体积计算是下一个关键步骤。获得3D结构的问题可以看作是多维解景观中的优化问题,其中全局最小值是表示原始结构的最佳3D体积,但可以找到几个表示次优解的局部最小值,并且很容易被困住。初始体积代表搜索过程的起点,因此错误的初始体积估计可能会阻止我们找到全局最小值。从初始体积开始,3D分类步骤将有助于发现不同的构象状态并再次清洁颗粒集;目标是获得结构上均匀的颗粒群。之后,3D细化步骤将负责细化每个粒子的角度和平移参数,以获得最佳的3D体积。
最后,在最后的步骤中,可以获得的3D重建可以进行锐化和抛光。锐化是提高重建体积的高频的过程,抛光是在颗粒水平上进一步细化某些参数的步骤,如CTF或光束诱导的运动补偿。此外,还可以使用一些验证过程来更好地了解在工作流结束时实现的分辨率。
在所有这些步骤之后,跟踪和对接过程7 将通过从头开始构建原子模型或拟合现有模型,帮助为获得的3D重建赋予生物学意义。如果达到高分辨率,这些过程将告诉我们生物结构的位置,甚至是不同原子在我们的结构中的位置。
Scipion8 允许以集成方式创建整个工作流程,将最相关的图像处理包组合在一起。Xmipp9,Relion10,CryoSPARC11,Eman12,Spider13,Cryolo14,Ctffind15,CCP416,Phenix17以及更多软件包可以包含在Scipion中。此外,它还集成了所有必要的工具,有利于集成、互操作性、可追溯性和可重复性,从而全面跟踪整个图像处理工作流程8。
Scipion允许我们使用的最强大的工具之一是共识,这意味着在处理的一个步骤中将获得的结果与几种方法进行比较,使不同方法传达的信息组合在一起,以产生更准确的输出。这可以帮助提高性能并提高估计参数中实现的质量。请注意,可以在不使用共识方法的情况下构建更简单的工作流程;但是,我们已经看到了这个工具的强大功能22,25 ,并且本手稿中介绍的工作流程将分几个步骤使用它。
前面段落中总结的所有步骤将在下一节中详细解释,并使用 Scipion 合并到一个完整的工作流中。此外,还将展示如何使用共识工具在生成的输出中实现更高的一致。为此,选择了 恶性疟原虫 80S核糖体的示例数据集(EMPIAR条目:10028,EMDB条目:2660)。该数据集由600部电影组成,这些电影的16帧大小为4096x4096像素,像素尺寸为1.34Å,使用FEI FALCON II相机在FEI POLARA 300上拍摄,EMDB上报告的分辨率为3.2Å18 。
研究方案
1. 在 Scipion 中创建项目并导入数据
- 打开 Scipion 并单击" 创建项目",指定项目的名称以及保存位置(补充图 1)。Scipion将打开项目窗口,显示一个画布,左侧是一个面板,其中包含可用方法的列表,每个方法代表一个可用于管理数据的图像处理工具。
注意:如果某个方法未出现在列表中, 则 Ctrl+F 可用于查找该方法。 - 要导入显微镜拍摄的短片 ,请选择pwem - 在 左侧面板上导入短片(或在按 Ctrl+F时键入)。
- 将打开一个新窗口(补充图2)。在那里,包括数据的路径和采集参数。在此示例中,请使用以下设置: 显微镜电压 300 kV、 球面像差 2.0 mm、 振幅对比度 0.1、 放大倍率 50000、 采样率模式 至 "从图像"和 像素大小 1.34 Å。填写表单中的所有参数后,单击" 执行 "按钮。
注意:当方法启动时,画布中会出现一个标记为 "正在运行"的黄色框。方法完成后,框将变为绿色,标签将更改为 "已完成"。如果在执行方法期间出现错误,则该框将显示为红色,标记为 失败。在这种情况下,请检查画布的底部,在" 输出日志 "选项卡中将显示错误说明。 - 方法完成后,在" 摘要 "选项卡中检查画布底部的结果。在这里,该方法生成的输出显示为电影集。在本例中为电影集。单击" 分析结果 "按钮,将出现一个新窗口,其中包含电影列表。
2. 短片对准:从短片到显微照片
- 使用方法 xmipp3 - 实现光流的光学对准19。使用以下参数填写表单(补充图3):输入影片是在步骤1中获得的,ALIGN帧中的范围是从2到13,其他选项保留默认值。执行程序。
注意:表单中以粗体显示的参数必须始终填写。其他的将具有默认值,或者不会强制要求。在表单窗口的上半部分,可以找到分布计算资源的字段,如线程、MPI 或 GPU。 - 单击" 分析结果" 以检查获得的显微照片和估计位移的轨迹(图1)。对于看到的每张显微照片:查看功率光谱密度(PSD),在笛卡尔和极坐标中对齐电影(每帧一个点)获得的轨迹,以及获得的显微照片的文件名(单击它,可以检查显微照片)。请注意,与电影的单帧相比,标本的颗粒在显微照片中更明显。
3. CTF估计:计算显微镜的像差
- 首先,使用 grigoriefflab - ctffind15 的方法。设置是:输入显微照片是步骤2的输出,手动CTF缩减采样因子设置为1.5,分辨率范围从0.06到0.42。此外,在"高级"选项(可通过在窗体的"专家级别"中选择此选项找到)中,将"窗口大小"设置为 256。其余参数保留默认值(补充图 4)。
注意:在 Scipion 中的大多数方法中, "高级 "选项显示更多配置参数。当要启动的程序完全已知并且理解参数的含义时,请仔细使用这些选项。如果不查看数据,某些参数可能难以填充;在这种情况下,Scipion 会在右侧显示一个魔杖,该魔杖将显示一个向导窗口(补充图 5)。例如,在此窗体的 "分辨率" 字段中特别有用,因为应选择这些值以大致覆盖从PSD的第一个零到最后一个明显环的区域。 - 方法完成后,单击" 执行 " 和"分析结果 "(图 2)。检查估计的 CTF 是否与实验 CTF 匹配。为此,查看PSD并将角落中的估计环与来自数据的环进行比较。还要检查获得的散焦值以找到任何意外值,并且可以丢弃或重新计算相应的显微照片。在这个例子中,可以使用整套显微照片。
注:使用窗口底部的按钮制作显微照片的子集(使用 "显微照片 "红色按钮)并重新计算CTF(使用 "重新计算CTF 红色"按钮),以备不时之需。 - 要优化前面的估计值,请使用 xmipp3 - ctf 估计值20。选择步骤 2 的输出作为 输入显微照片 ,选择选项 使用先前 CTF 估计中的 defoci,因为 以前的 CTF 估计 选择 grigoriefflab - ctffind 的输出,然后在 高级 级别中,将 窗口大小 更改为 256(补充图 6)。运行它。
- 单击" 分析结果" 以检查获得的 CTF。使用此方法时,将估计更多数据,并在一些额外的列中表示这些数据。由于它们都没有显示不正确的估计值,因此将在以下步骤中使用所有显微照片。
4. 颗粒拾取:在显微照片中查找颗粒
- 在开始拣选之前,请对显微照片进行预处理。打开 xmipp3 - 预处理显微照片,将步骤 2 中获得的微照片设置为输入显微照片,然后选择选项"删除坏像素?",将 Stddev 的倍数设置为 5,以及缩减采样系数为 2 的"缩减采样"微照片?(补充图 7)。单击"执行"并检查生成的显微照片的大小是否已减小。
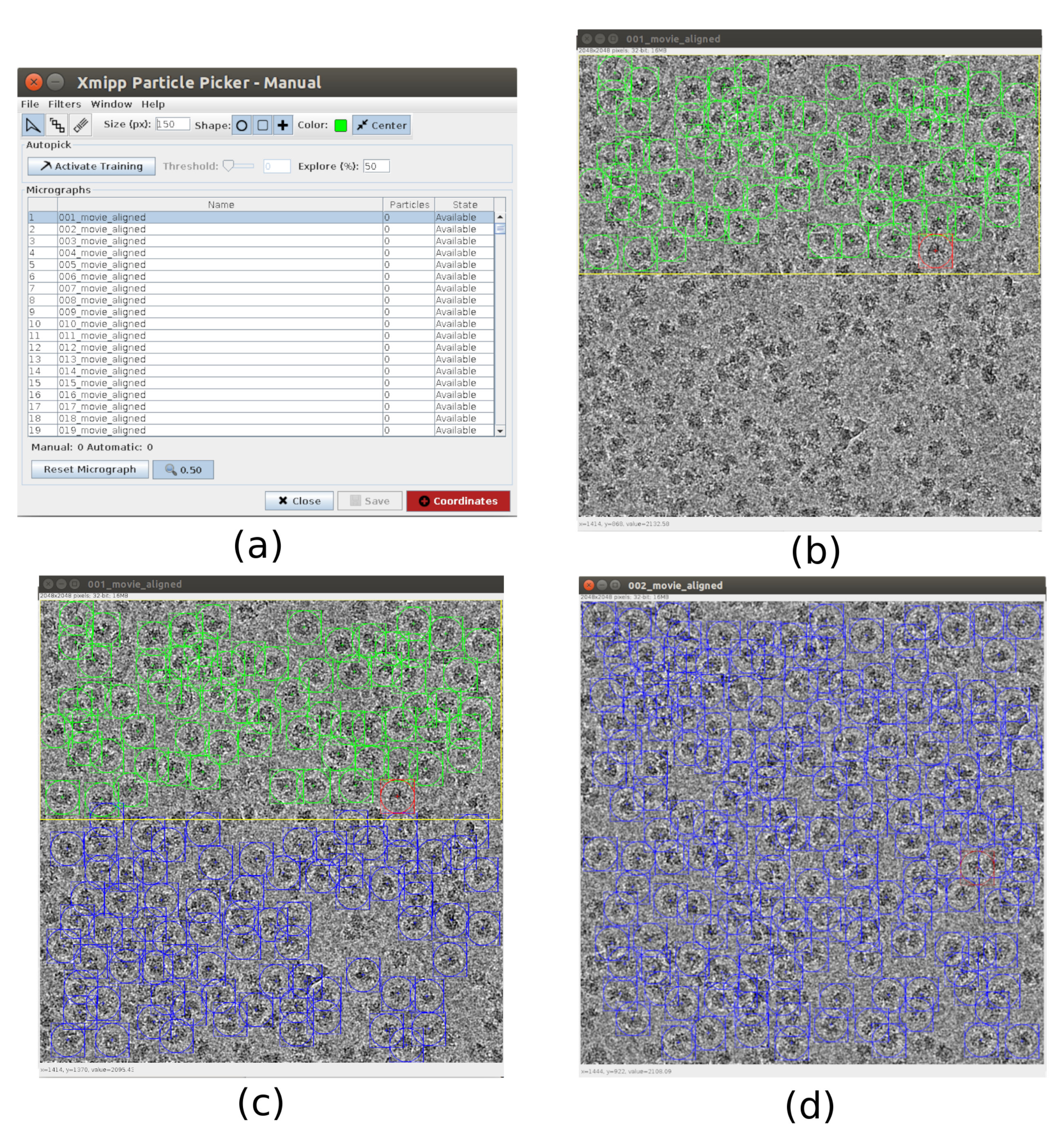
- 对于拣选,请使用 xmipp3 - 手动拣选(步骤 1)和 xmipp3 - 自动拣选(步骤 2)21。手动拣选允许手动准备一组颗粒,自动拣选步骤将学习并生成完整的颗粒集。首先,运行 xmipp3 - 手动拾取(步骤 1),将输入显微照片作为在上一个预处理中获得的显微照片。单击"执行",将出现一个新的交互式窗口(图 3)。
- 在此窗口中,显示了显微照片(图3a)和其他选项的列表。将 大小(px) 更改为150,这将是包含每个粒子的盒子的大小。选定的显微照片将显示在更大的窗口中。选择一个区域并选择其中的所有可见粒子(图3b)。然后,单击" 激活培训 "以开始学习。自动选取显微照片的其余区域(图3c)。检查拾取的粒子,然后单击它以包含更多粒子,或者如有必要,通过shift+单击删除不正确的粒子。
- 在第一个窗口中选择下一个显微照片。显微照片将被自动拾取。如有必要,请再次检查以包括或去除某些颗粒。用大约5张显微照片重复此步骤,以创建具有代表性的训练集。
- 完成此操作后,单击主窗口中的坐标以保存所有拾取粒子的坐标。粒子训练集已准备好进入自动拣选,以完成所有显微照片的过程。
- 打开 xmipp3 - 自动拾取(步骤 2) 指示在 Xmipp 颗粒拾取中运行 先前的手动拾取,并将 要拾取的显微照片 作为 与监督的相同。单击" 执行"。此方法将生成一组大约 100000 个坐标作为输出。
- 应用共识方法,因此执行第二种拾取方法来选择两种方法都同意的粒子。打开 sphire - cryolo picking14 并选择预处理的显微照片作为输入显微照片,使用通用模型?到是,置信度阈值为 0.3,盒尺寸为 150(补充图 8)。运行它。此方法还应生成大约 100000 个坐标。
- 运行 xmipp3 - 深度共识采摘22。由于 输入坐标 包括 sphire - cryolo 拾取 (步骤 4.7)和 xmipp3 - 自动拾取 (步骤 4.6)的输出, 因此将"选择模型类型" 设置为" 预训练",以及 "跳过训练并直接使用预训练模型进行评分? 为 "是" (补充图 9)。运行它。
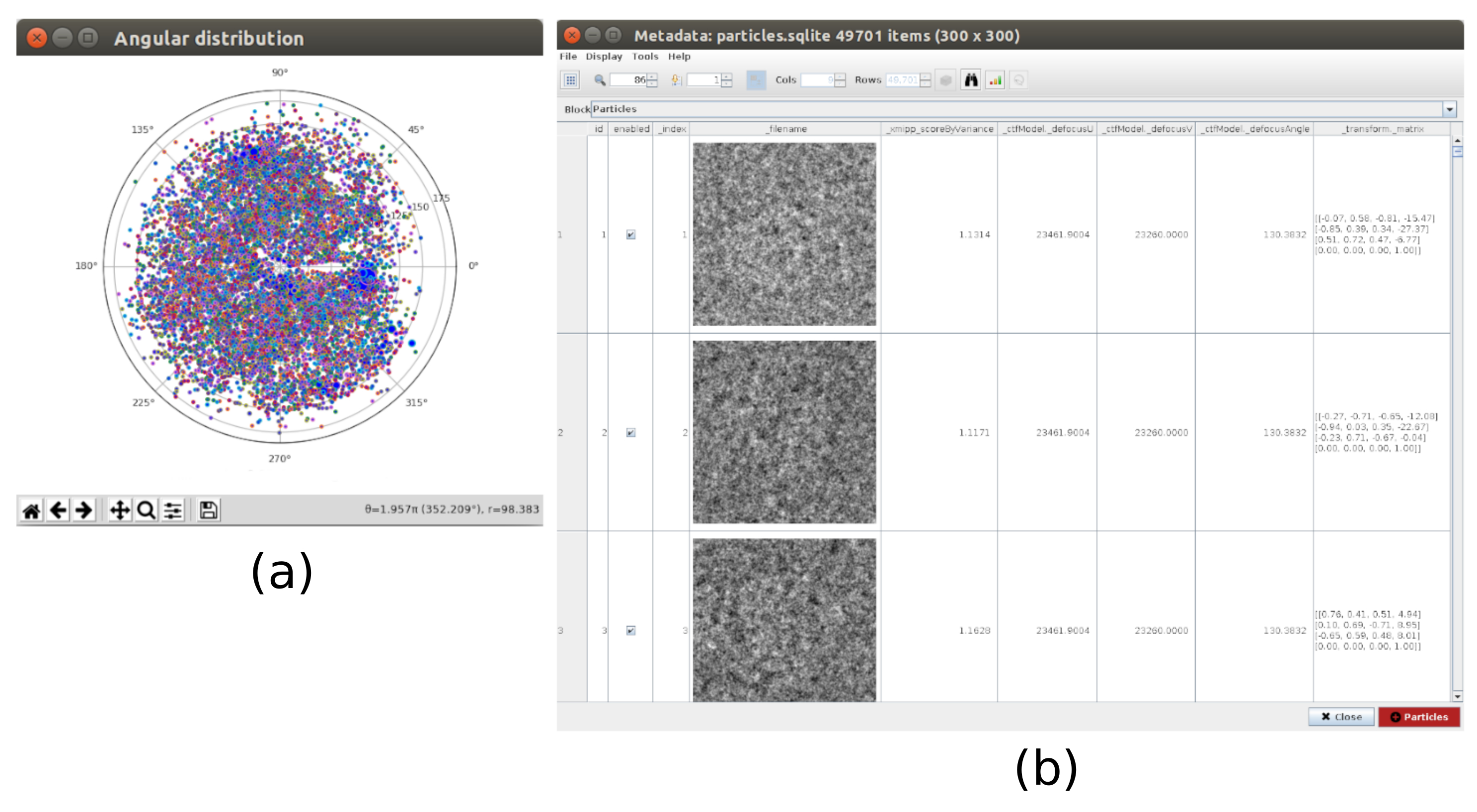
- 单击"分析结果",然后在新窗口中,单击"选择具有高"zScoreDeepLearning1"值的粒子/坐标旁边的眼睛图标。将打开一个新窗口,其中包含所有粒子的列表(图4)。列中的 zScore 值可以深入了解粒子的质量,低值表示质量差。
- 单击标签_xmipp_zScoreDeepLearning 以按从高到低 zScore 对粒子进行排序。选择 zScore 高于 0.75 的粒子 ,然后单击坐标 以创建新子集。这将创建一个具有大约 50000 个坐标的子集。
- 打开 xmipp3 - 深显微照片清洁剂。选择"输入坐标"作为上一步中获得的子集,"显微照片"源与坐标相同,并将阈值保持在0.75。运行它。在"摘要"选项卡中检查坐标数量是否已减少,尽管在这种情况下,只有少数坐标被删除。
注意:此步骤能够额外清洁坐标集,并且在清洁具有更多电影伪影(如碳区或大杂质)的其他数据集时可能非常有用。 - 运行 xmipp3 - 提取颗粒 (补充图10)。指示为 输入坐标 ,上一步后获得的坐标, 显微照片源 为 其他, 输入显微照片 为步骤2的输出, CTF估计 为 输出的xmipp3 - ctf估计, 缩减采样因子 为3, 粒子框大小 为100。在窗体的" 预处理 "选项卡中,选择"全部是 " 。运行它。
- 检查输出是否应包含缩小大小为 100x100 像素且像素大小为 4.02Å/px 的粒子。
- 再次运行 xmipp3 - 提取更改 以下参数的粒子: 缩减采样因子 为 1, 粒子盒大小 更改为 300。检查输出是否为同一组粒子,但现在处于全分辨率。
5. 二维分类:将相似的颗粒组合在一起
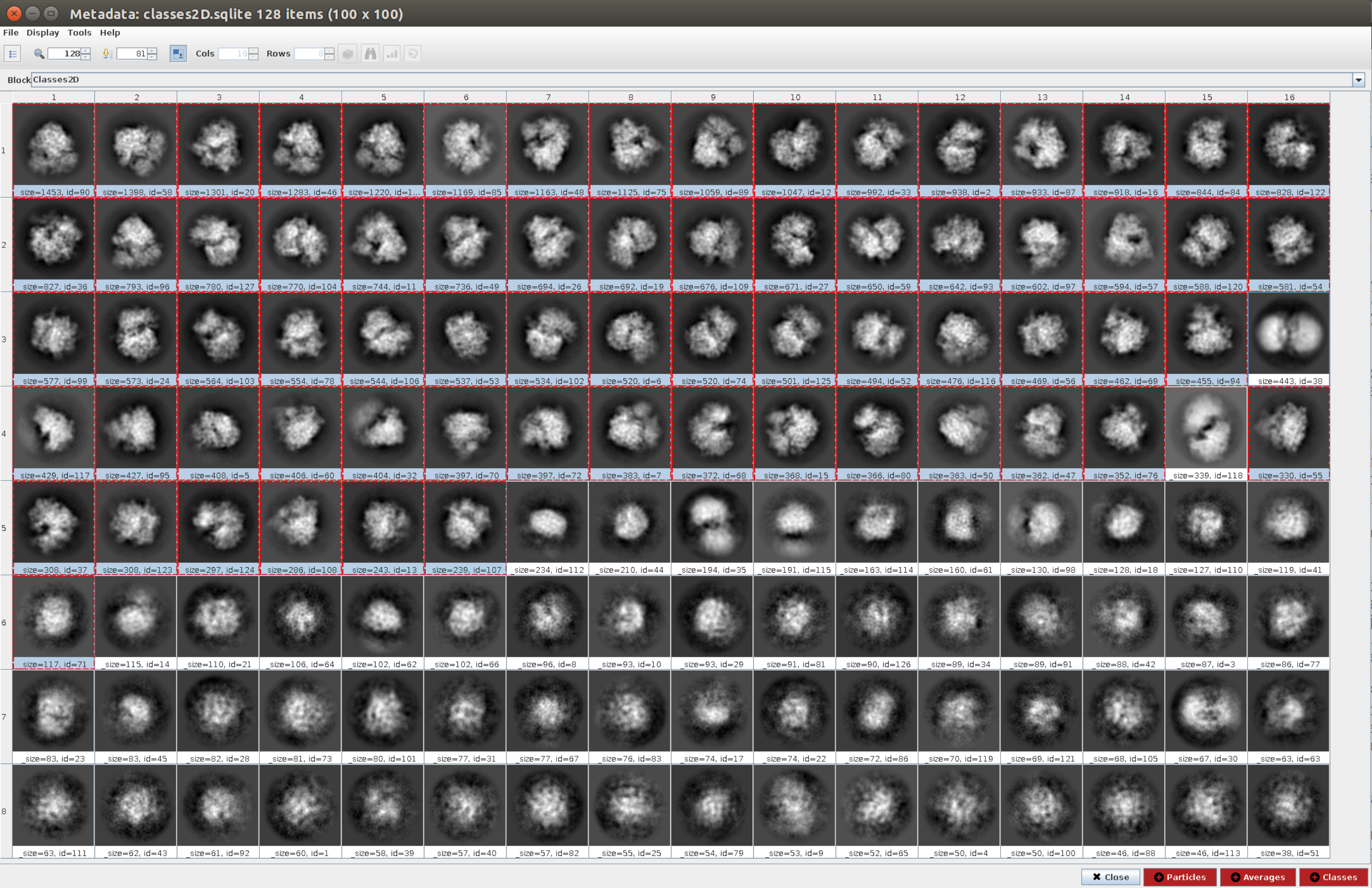
- 打开方法 cryosparc2 - 2d 分类11 , 输入粒子 与步骤 4.11 中获得的粒子相同,并在 "2D 分类 "选项卡中, 将类数 设置为 128,将所有其他参数保留为默认值。运行它。
- 单击" 分析结果" ,然后单击" 使用 Scipion 显示粒子类" 旁边的眼睛图标(图 5)。这种分类将有助于清理粒子集,因为有几个类会显得嘈杂或有伪影。选择包含良好视图的类。单击" 粒子 "(窗口下部的红色按钮)以创建更干净的子集。
- 现在,打开 xmipp3 - cl2d23 并将在上一步中获得的图像设置为输入图像,并将类数设置为 128。单击"执行"。
注:第二种分类用作颗粒集的附加清洁步骤。通常可用于尽可能多地去除噪声颗粒。但是,如果需要更简单的工作流程,则只能使用一种 2D 分类方法。 - 该方法完成后,通过单击" 分析结果 "和" 要显示的内容:类"来检查生成的 128 个类。大多数生成的类都显示具有一定程度细节的大分子投影。但是,其中一些看起来很嘈杂(在此示例中大约有 10 个类)。选择所有好的类,然后单击" 类 "按钮以生成一个新的子集,其中只有好的类。此子集将用作生成初始卷的方法之一的输入。对于相同的选定类,单击 "粒子 "以在删除属于不良类的类后创建一个更干净的子集。
- 打开 pwem - 以完整项目集作为 4.13 输出的子集(全尺寸的所有粒子),将随机子集设置为否,将其他设置为在上一步中创建的粒子子集,并将操作设置为交集。这将以全分辨率从粒子中提取前一个子集。
6. 初始体积估算:构建3D体积的第一个猜测
- 在此步骤中,使用不同的方法估计两个初始体积,然后使用共识工具生成最终估计的 3D 体积。打开 xmipp3 - 重构显著 24 方法,其中输入类为步骤 5 后获得的输入类,对称组为 c1,并将其余参数保留其默认值(补充图 11)。执行它。
- 单击 分析结果。检查是否获得了大小为 100x100x100 像素的低分辨率体积和大小为 4.02Å/px 的像素。
- 打开 xmipp3 - 使用在上一步中获得的卷作为输入卷的裁剪/调整大小卷(补充图 12),将卷大小调整为"是",将"调整大小"选项调整为"采样率",并将"将采样率调整为 1.34 Å/px"。运行它。在"摘要"选项卡中检查输出卷是否具有正确的大小。
- 现在,创建第二个初始卷。开放依赖 - 3D 初始模型10,因为输入粒子在全分辨率下使用良好的粒子(输出为 5.5)并将粒子掩模直径设置为 402Å,因此保留其余参数的默认值。运行它。
- 单击" 分析结果", 然后单击" 使用:切片显示卷"。检查是否获得了低分辨率体积,但具有结构的主要形状(补充图13)。
- 现在,打开 pwem - 连接集 以组合两个生成的初始卷,以创建共识方法的输入。只需将 "卷 "指示为 "输入类型 ",然后在" 输入集"中选择两个初始卷。运行它。输出应为包含两个项目且具有两个卷的集合。
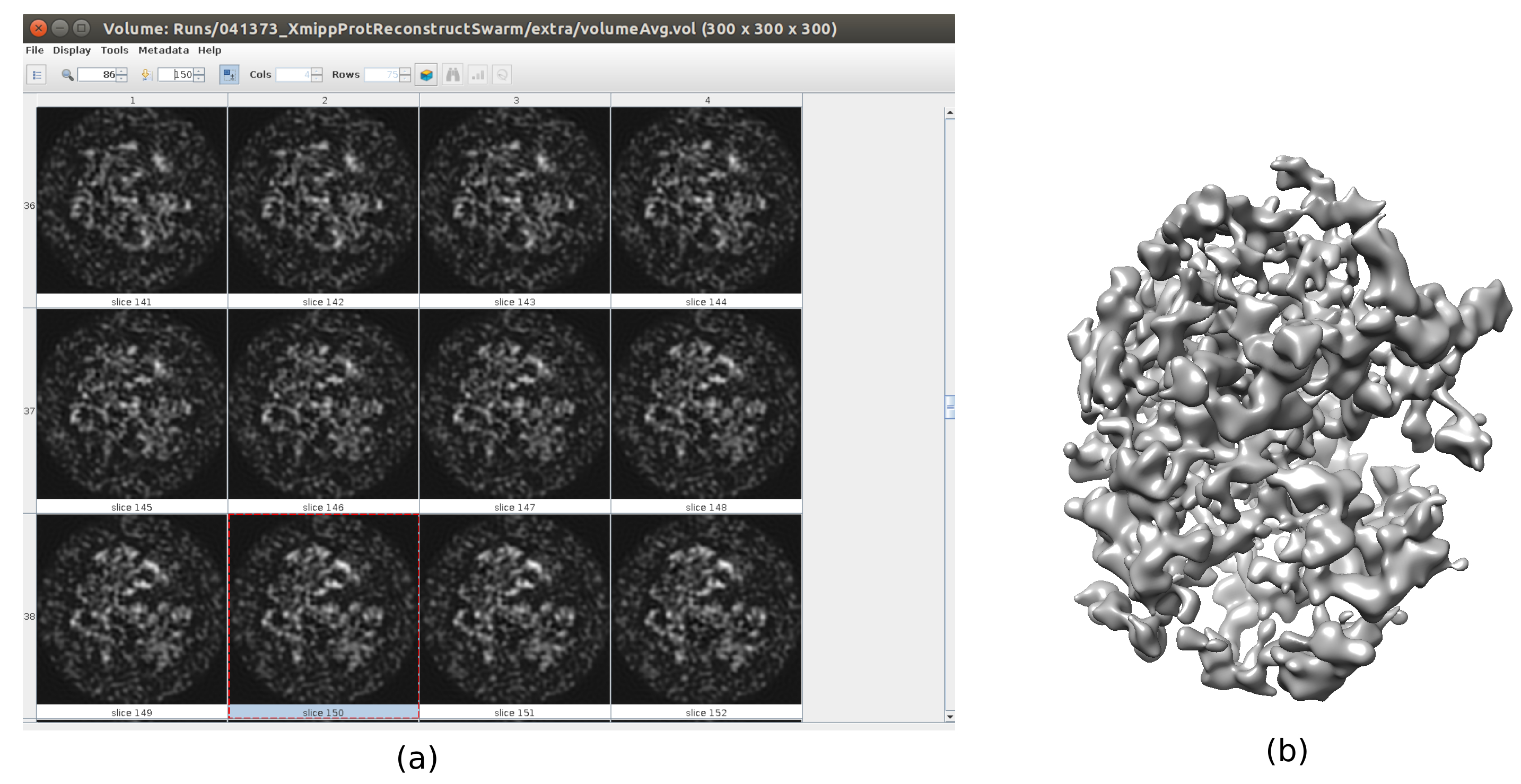
- 共识工具是包含在 xmipp3 - swarm consensus25 中的工具。打开。将全分辨率(输出为 5.5)的良好粒子用作全尺寸图像,将具有上一步中生成的两个项目的集合作为初始体积,并确保对称组为 c1。单击"执行"。
- 单击 分析结果。检查是否获得了更详细的输出音量(图6)。虽然结构周围有更多的噪音,但在结构图中有更多细节将有助于以下细化步骤,以避免局部最小值。
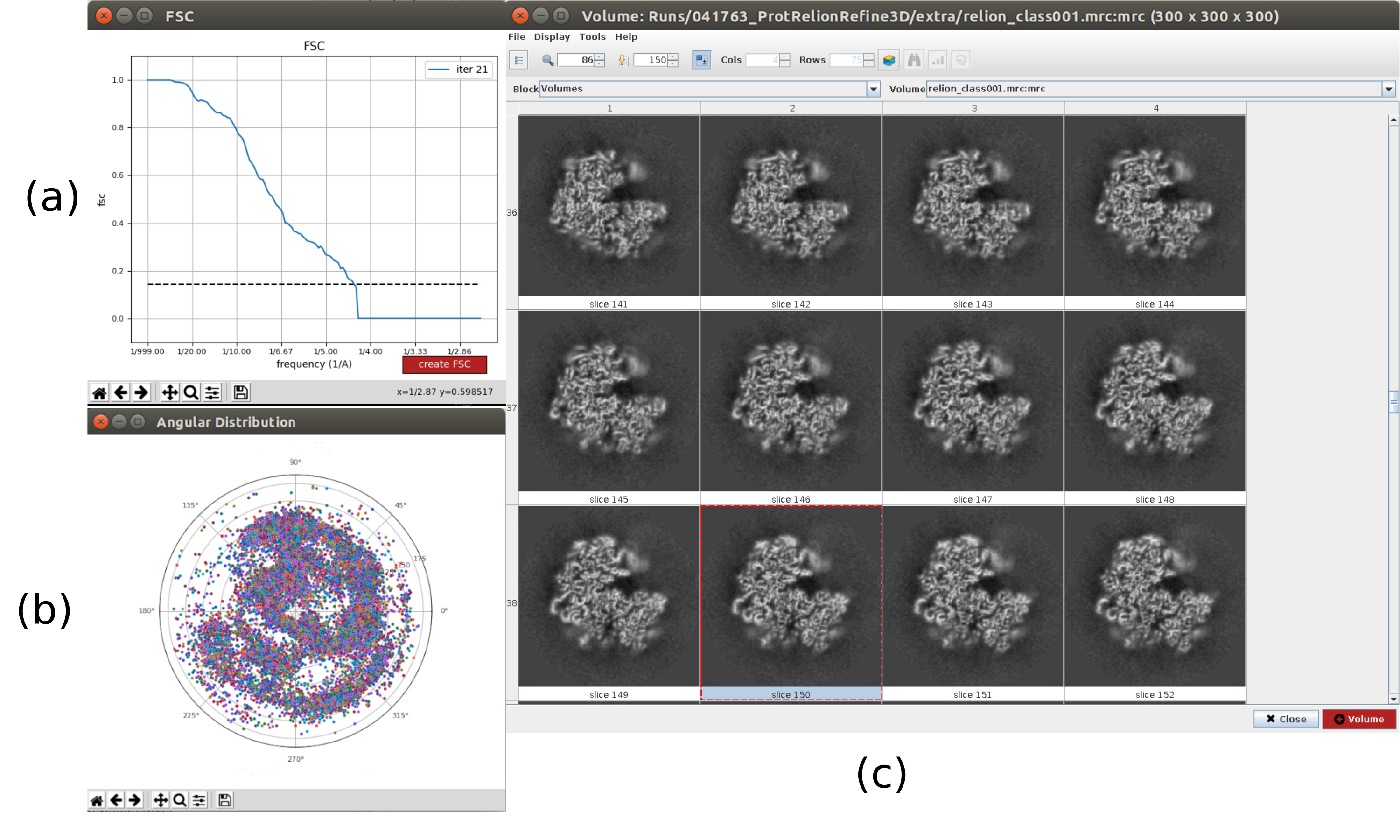
注:如果 UCSF Chimera26 可用,请使用窗口上部的最后一个图标对获得的体积进行 3D 可视化。 - 打开并执行 依赖 - 3D 自动细化10 ,对粒子进行首次 3D 角度分配。选择输出 5.5 作为 输入粒子 ,并将 粒子掩模直径 设置为 402Å。在 参考3D地图 选项卡中,选择在上一步中获得的 输入体积 , 对称性 为c1, 初始低通滤波器 为30Å(补充图14)。
- 单击分析结果。在新窗口中,选择"final"作为"卷"进行可视化,然后单击"显示卷与:切片"以查看获得的卷。还可以通过单击结果窗口中的显示分辨率图和"显示角度分布:2D 图"中的角度覆盖范围来检查傅里叶壳相关 (FSC)(图 7)。重建的体积包含更多细节(可能在结构的外部有一些模糊的区域),FSC在4.5Å附近越过了0.143的阈值。角度覆盖覆盖整个3D球体。
7.3D分类:发现构象状态
- 使用共识方法,如果数据中存在不同的构象状态,则可以发现。开放 依赖 - 3D 分类10 (补充图 15)。作为 输入粒子 ,请使用刚刚在 6.10 中获得的粒子,并将 粒子掩模直径 设置为 402Å。在 参考 3D 地图 选项卡中,将步骤 6.10 后获得的 体积用作输入体积 ,将 对称性 设置为 c1, 并将初始低通滤波器 设置为 15Å。最后,在 "优化"选项卡中,将 "类数" 设置为 3。运行它。
- 通过单击"分析结果"检查结果,选择"在 Scipion 中显示分类"。显示了三个生成的类和一些有趣的度量值。前两个类应具有相似数量的指定图像(大小列)并且看起来非常相似,而第三个类的图像较少,外观更模糊。此外,对于前两个类,rlnAccuracyRotations 和 rlnAccuracyTranslations 显然应该更好。选择两个最佳类,然后单击"类"按钮以生成包含它们的子集。
- 重复步骤 7.1 和 7.2 以生成第二组好类。两者都将成为共识工具的输入。
- 打开并运行 xmipp3 - 共识类 3D ,并将在前面步骤中生成的两个子集选为 输入类 。
- 单击 分析结果。给出了类之间的重合粒子数:第一个值是子集 1 的第一类和子集 2 的第一类中的重合粒子数,第二个值是子集 1 的第一类和子集 2 的第二类中的重合粒子数,依此类推。检查粒子是否随机分配到一类或二类,这意味着3D分类方法无法找到构象变化。鉴于这一结果,整个粒子集将用于继续处理。
8.3D细化:精炼齐次总体的角度分配
- 同样,在此步骤中应用共识方法。首先,打开并运行 pwem - 子集 , 其中"项目的完整集" 作为 6.9 的输出, "使随机子集" 变为 "是", "元素数" 设置为 5000。这样,将创建具有先前对齐方式的图像子集,以训练以下步骤中使用的方法。
- 打开 xmipp3 - 深度对齐,将 输入图像 设置为 5.5 中获得的良好粒子的输出, 将体积 设置为 6.10 之后获得的良好粒子的 输出,将输入训练集 设置为上一步中创建的 粒子,目标分辨率 设置为 10Å,并将其余参数保留默认值(补充图 16)。单击" 执行"。
- 单击" 分析结果" 以检查获得的角度分布,其中没有遗漏方向,并且角度覆盖率与6.10相比略有改善(图8)。
- 打开并执行 xmipp3 - 比较角度 ,并选择 输入粒子 1 作为 6.9 的输出和 输入粒子 2 的输出 8.2,确保 对称组 为 c1。此方法计算 xmipp3 - 深度对齐 和 依赖 - 3D 自动优化之间的一致性。
- 单击" 分析结果",将显示粒子列表,其中包含获得的位移和角度差异。单击窗口上部的条形图标,将打开另一个窗口,该窗口允许绘制计算变量的图。选择 _xmipp_angleDiff ,然后单击" 绘图 "以查看每个粒子的角度差异的表示形式。对_xmipp_shiftDiff执行相同的 操作。在这些数字中,大约一半的粒子两种方法都一致(图9)。选择角度差小于 10º 的粒子并创建新子集。
- 现在,打开 xmipp3 - highres27 以对指定的角度进行局部细化。首先,选择在上一步中获得的图像作为全尺寸图像,并将输出设置为 6.9 的初始体积,将粒子半径设置为 150 像素,并将对称组设置为 c1。在"角度分配"选项卡中,将"图像对齐方式"设置为"本地"、"迭代次数"设置为 1 和"最大"。目标分辨率为 5Å/px(补充图 17)。运行它。
- 在" 摘要 "选项卡中,检查输出音量是否小于 300x300x300 像素且像素大小略高。
- 单击" 分析结果" 以查看获得的结果。单击 "显示分辨率图" 以查看 FSC,单击" 显示体积:重建 "以查看获得的体积(补充图 18)。获得接近4-3.5Å的良好分辨率体积。
- 单击" 显示输出粒子 ",然后在包含粒子列表的窗口中,单击条形图标。在新窗口中,选择 "键入 直 方图",包含 100 个条柱,选择 _xmipp_cost标签,最后按 Plot (补充图 19)。这样,将呈现 成本 标签的直方图,其中包含粒子与为其选择的投影方向的相关性。在这种情况下,获得单峰密度函数,这是粒子集中没有不同群体的迹象。因此,所有这些将用于继续改进
注意:如果看到多模态密度函数,则应选择属于较高最大值的粒子集,以便仅使用它们继续工作流。 - 打开并再次执行 xmipp3 - 从上一次运行继续?到"是",设置为 8.5 版之后获得的"全尺寸图像",然后选择上一次运行以及之前执行的 Xmipp Highres。在"角度分配"选项卡中,将"图像对齐"设置为"局部",并将 1 次迭代和 2.6Å/px 作为目标分辨率(全分辨率)。
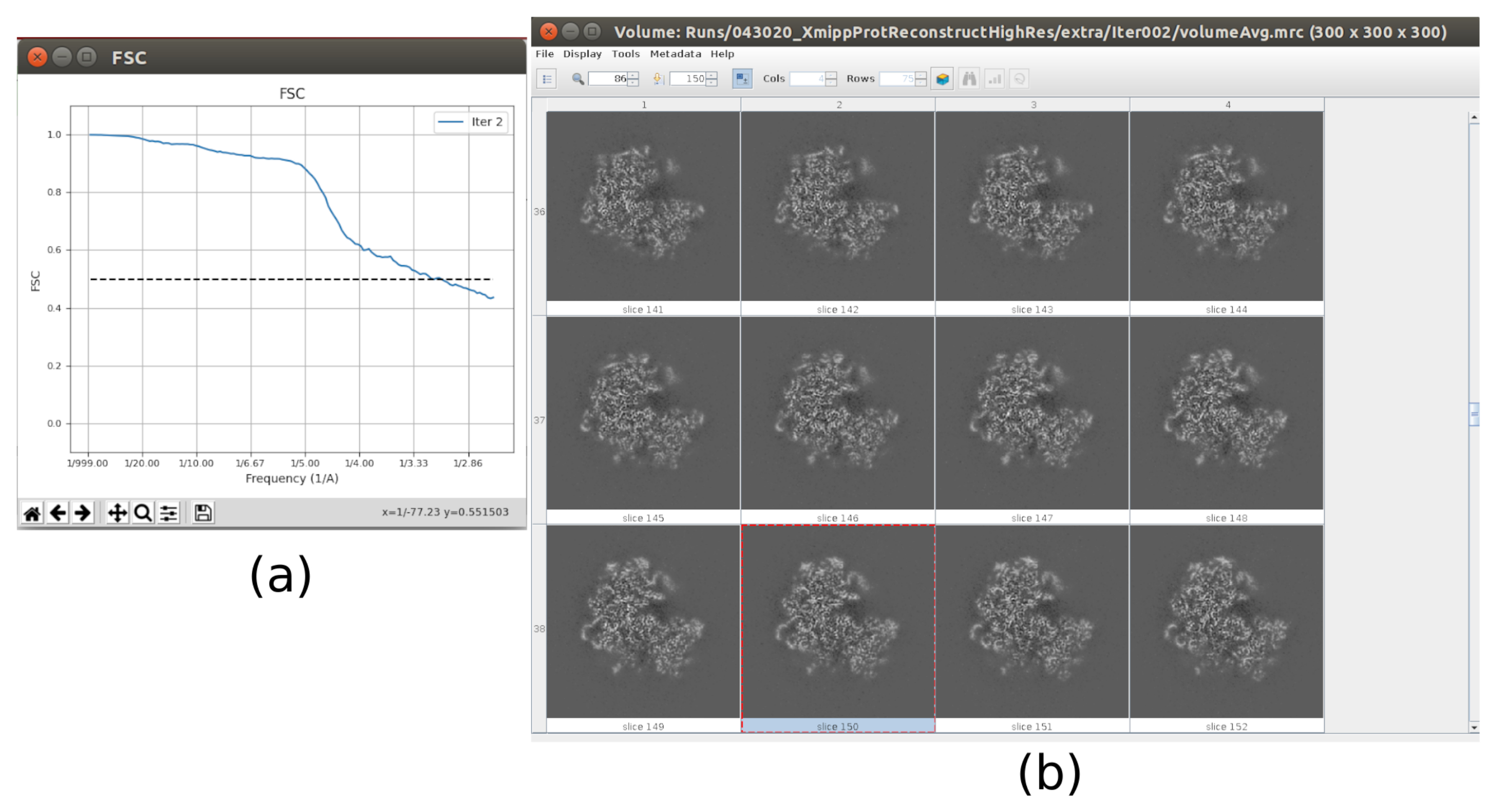
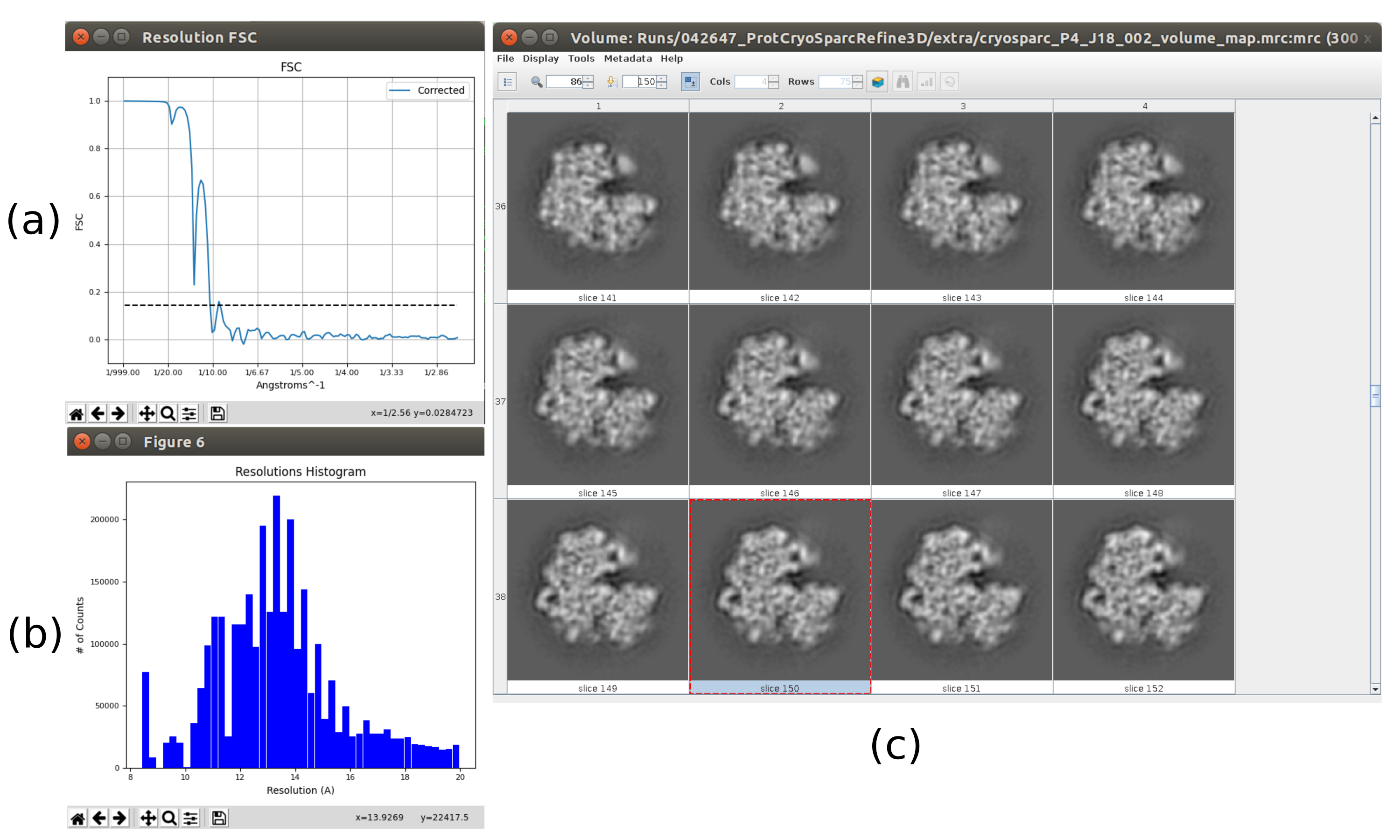
- 现在,输出应包含全分辨率(大小为 300x300x300 像素)的卷。单击" 分析结果" 以再次检查获得的体积和FSC,现在应该是3Å左右的高分辨率体积(图10)。
9. 评估和后处理
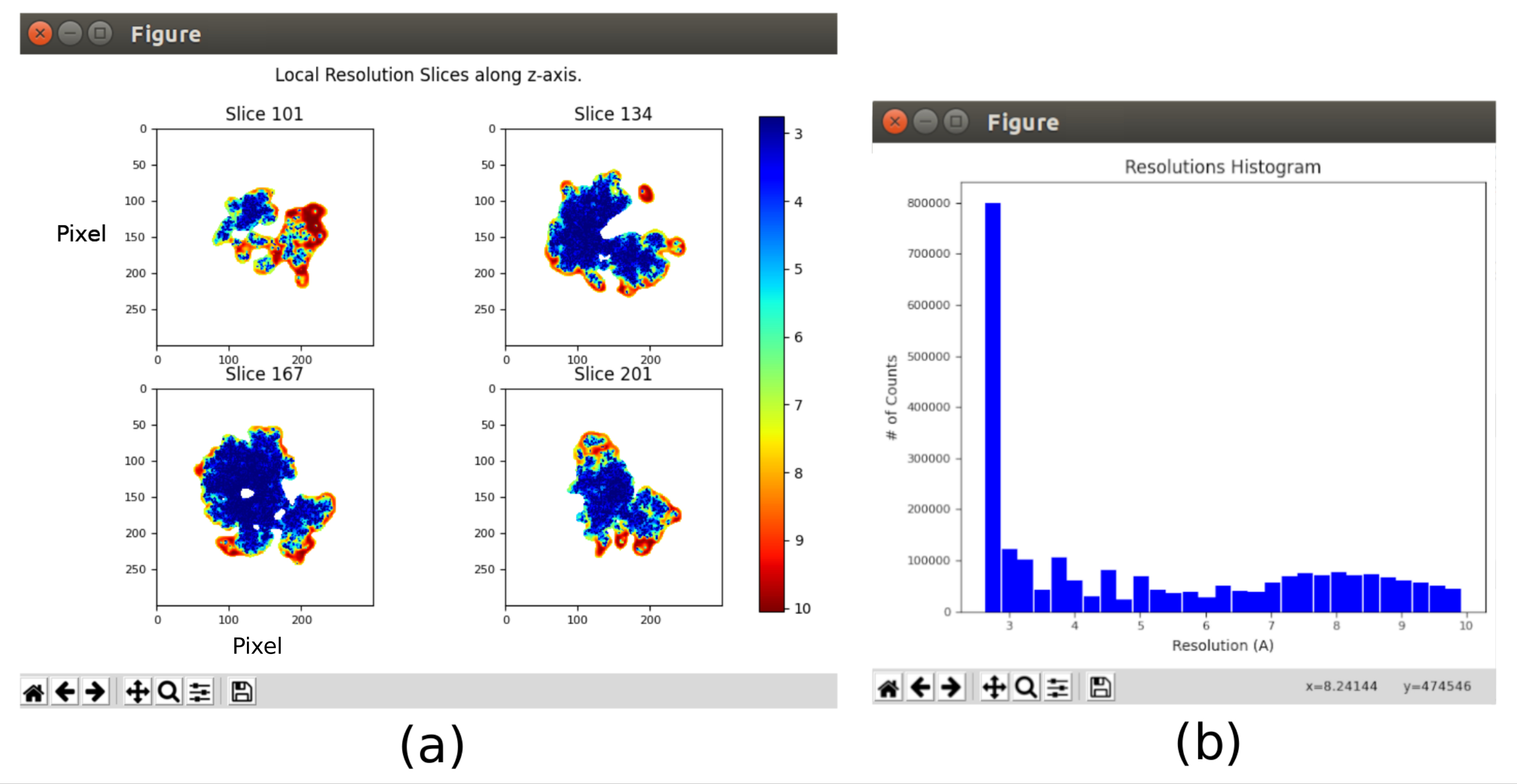
- 打开 xmipp3 - 本地 MonoRes28。此方法将在本地计算分辨率。将 8.10 之后获得的音量设置为"输入音量",将"是否要使用一半音量?"设置为"是",并将"分辨率范围从 1 到 10Å"。运行它。
- 单击 分析结果 ,然后选择 显示分辨率直方图 和 显示彩色切片 (图11)。图中显示了体积不同部分的分辨率。结构中央部分的大多数体素应该在3Å周围显示分辨率,而最差的分辨率则在外部实现。此外,还显示了每个体素的分辨率直方图,其峰值围绕(甚至低于)3Å。
- 打开并运行 xmipp3 - 局部锐化29 以应用锐化。选择在 8.10 中获得的作为 输入映射 ,并将在上一步中获得的 MonoRes 作为 分辨率映射 。
- 单击" 分析结果" 以检查获得的卷。打开最后一个,对应于算法的最后一次迭代。建议使用其他工具(如 UCSF Chimera26)打开体积,以便在 3D 模式下更好地查看体积的特征(图 12)。
- 最后,打开 xmipp3 中包含的验证工具 - 验证过拟合30 ,该工具将显示分辨率如何随粒子数量而变化。打开它,将步骤 8.5 中获得的粒子作为 输入 粒子包括在内,将 "计算分辨率的噪声" 设置为 "是", 将"初始 3D 参考体积 "设置为 8.10 的输出。在 "高级 "选项中,将 粒子数设置为"500 1000 1500 2000 3000 5000 10000 15000 20000"(补充图 20)。运行它。
- 单击" 分析结果"。随着分辨率的演变,将出现两个图(图13),在绿线中,随着重建中使用的粒子数量的增加。红线表示通过重建对齐的高斯噪声实现的分辨率。分辨率随着粒子数量的增加而提高,并且观察到粒子的重建与噪声的重建有很大差异,这是具有良好结构信息的粒子的指标。
- 从先前的结果来看,可以在后处理体积中对模型进行拟合,这将允许发现大分子的生物结构。
结果
我们使用 恶性疟原虫 80S核糖体(EMPIAR条目:10028,EMDB条目:2660)的数据集进行测试,并且使用上一节中介绍的Scipion协议,已经实现了该特定示例中大分子的高分辨率3D重建体积,从显微镜收集的信息开始,该信息由非常嘈杂的图像组成,其中包含标本任何方向的2D投影。
运行整个协议后获得的主要结果如图10、图11和 图12所示。图 10 显示了后处理前获得的 3D 体积。在图10a中,可以看到3 Å的FSC非常接近奈奎斯特极限(像素尺寸为1.34 Å的数据,奈奎斯特极限为2.6 Å)。图 10b 显示了重建的 3D 体积的一些切片,这些切片具有高水平的细节和明确定义的结构。在图11中,显示了本地分析后获得的3D体积的分辨率的结果。可以看出,结构中的大多数体素达到3 Å以下的分辨率,主要是那些位于结构中央部分的体素。然而,外部显示的分辨率较差,这与图10b切片中这些区域出现的模糊一致。图12显示了后处理后的相同3D地图,该地图能够突出显示体积的较高频率,揭示更多细节并改善表示,这在图12c中的3D演示中尤其可见。
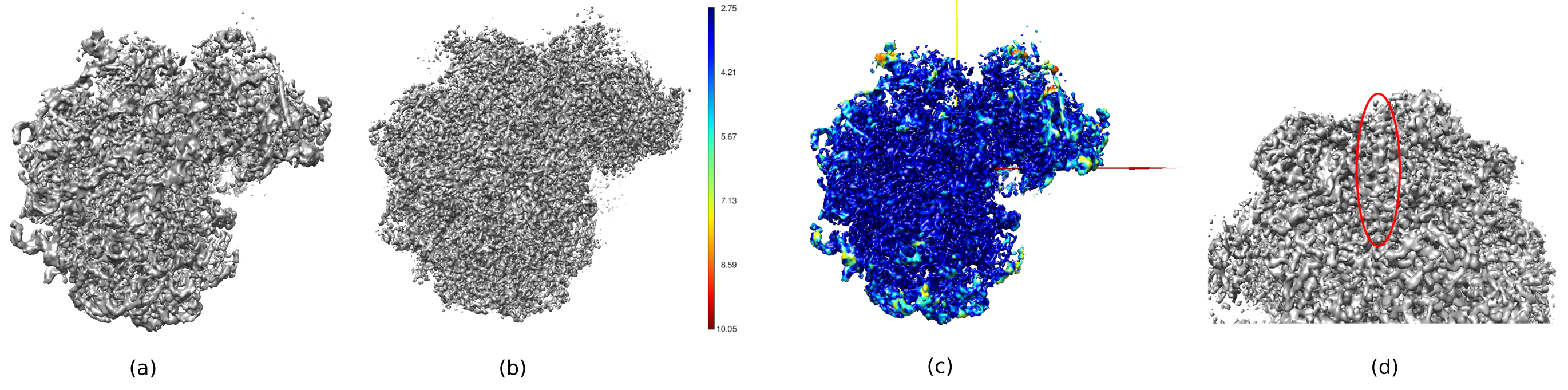
在图14中,Chimera26用于查看所获得体积(图14a),后处理(图14b)和分辨率图(图14c)的3D表示,并用局部分辨率的颜色代码着色。这可以提供有关所获得结构的更多信息。该工具对于深入了解所获得体积的质量非常有用,因为可以看到结构的整个3D环境中非常小的细节。当所达到的分辨率足够时,甚至可以找到结构的一些生化部分(例如,图14d中的α-螺旋。在此图中,必须突出显示在3D结构的所有中心部分实现的高分辨率,这可以看作是图14c中的深蓝色区域。
由于整个协议的良好性能,所有先前的结果都是实现的,但事实可能并非如此。有几种方法可以识别不良行为。在最一般的情况下,当获得的结构具有低分辨率并且无法演变为更好的结构时,就会发生这种情况。 图 15 显示了这方面的一个示例。模糊的体积(图15c)导致FSC较低,这可以在FSC曲线(图15a)和局部估计的直方图(图15b)中看到。此示例是使用输入数据不正确的 3D 细化方法生成的,因为它期望粒子的输入集中存在一些它们未满足的特定属性。可以看出,了解不同方法如何接收数据并正确准备数据始终非常重要。通常,当获得 如图 15 所示的输出时,处理工作流或基础数据中可能存在问题。
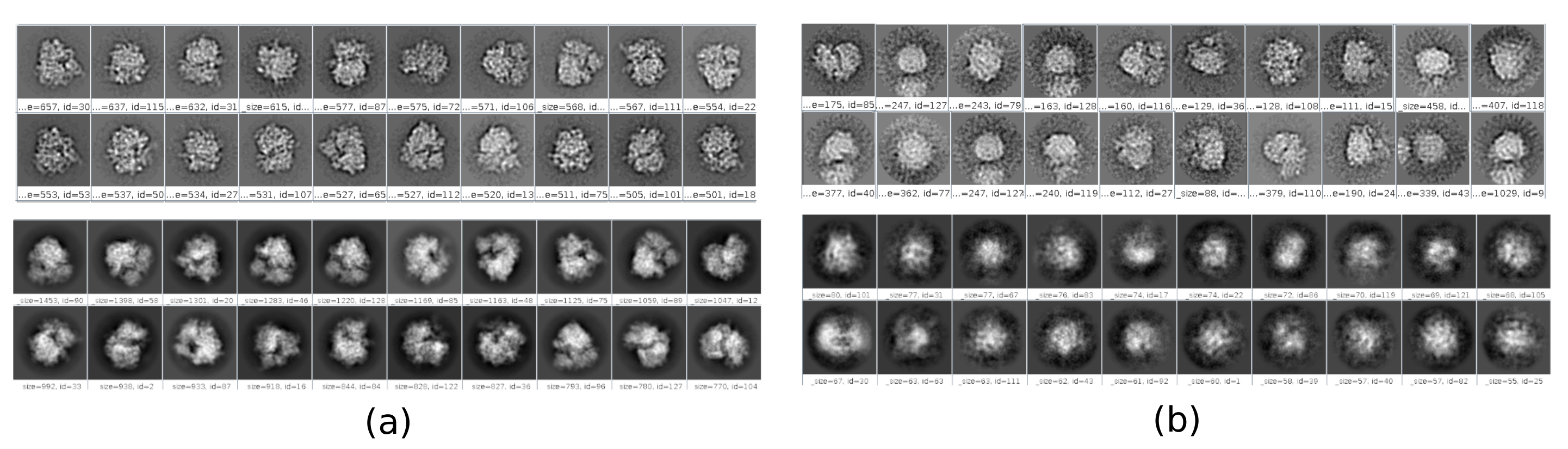
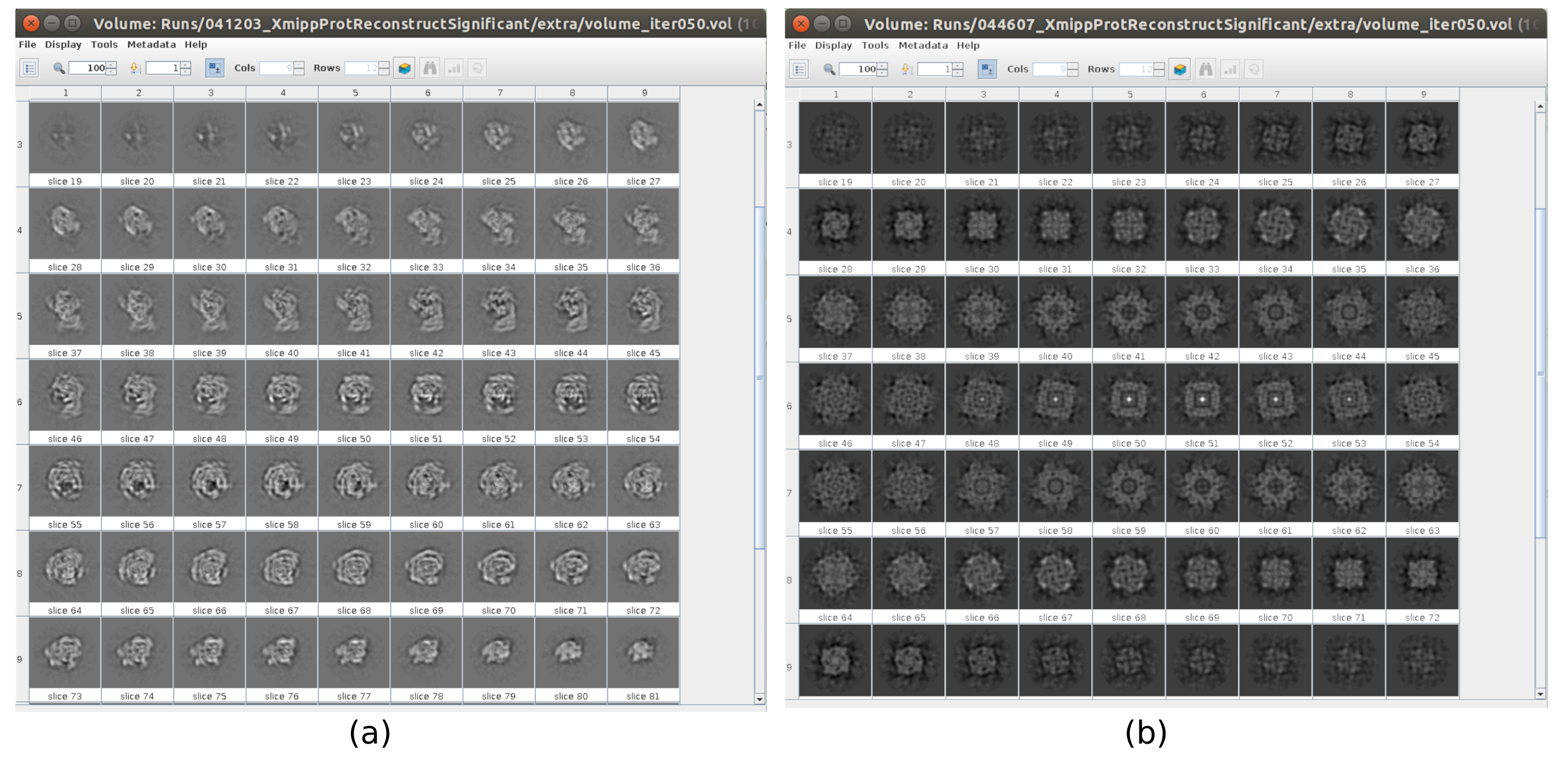
工作流中有几个检查点,可以对其进行分析,以了解协议是否正确发展。例如,在采摘之后,前面讨论的几种方法可以对粒子进行排名,并为每个粒子给出分数。在具有不良颗粒的情况下,这些方法允许识别和去除它们。此外,2D分类可以很好地指示具有一组不良的粒子。 图 16 显示了此类错误集的示例。在 图16a中,显示了包含结构的一些细节的良好类,而 图16b 显示了噪声或未居中的坏类,在最后一种情况下,可以看出拾取不正确,并且两个粒子似乎一起出现。另一个检查点是初始体积估计, 图17 显示了良好(图17a)和坏(图17b)初始估计的示例。错误估计是使用不正确的方法设置创建的。必须考虑到所有设置都应仔细完成,根据所分析的数据适当地选择每个参数。如果没有包含一些最少结构信息的地图,则以下细化将无法获得良好的重建。
当问题是不良的采集时,其中电影不保留结构信息,因此不可能从中提取好的颗粒并获得成功的处理。在这种情况下,应该收集更多的电影以获得高分辨率的3D重建。但是,如果不是这种情况,则有几种方法可以管理处理工作流中的问题。如果拣选不够好,有几种方法可以尝试修复它,例如,重复拣选,使用不同的方法,或尝试手动拾取更多粒子以帮助方法从中学习。在 2D 分类期间,如果只有几个类是好的,请考虑重复拾取过程。在初始体积估计中,如果其中一些方法给出的结果不准确,请尝试使用几种方法。这同样适用于3D细化。根据这一推理,在本手稿中,提出了几种共识工具,这些工具对于避免问题并继续使用准确的数据进行处理非常有用。由于在几种方法之间使用了共识,我们可以丢弃难以挑选,分类,对齐等的数据,这可能是不良数据的指标。但是,如果几种方法能够在生成的输出中达成一致,则这些数据可能包含可用于继续处理的有价值信息。
我们鼓励读者下载更多数据集,并尝试按照本手稿中提出的建议进行处理,并使用 Scipion 创建一个类似的工作流程,将处理包结合起来。尝试处理数据集是了解Cryo-EM中最先进的处理工具的强大功能,了解克服处理过程中可能出现的缺点的最佳规则,并提高每个特定测试用例中可用方法的性能的最佳方法。

图 1.影片对齐结果。(a) 结果的主窗口,列出所生成的所有显微照片和补充资料:功率谱密度、极坐标中估计对齐的轨迹、笛卡尔坐标中的相同轨迹、生成的显微照片的文件名。 (b) 以笛卡尔坐标表示的对齐轨迹。 (c) 生成的显微照片。 请点击此处查看此图的放大版本。
{kind=link}

图 2.CTF估计与Ctffind结果。 包含结果的主窗口包括一个数字,其中包含估计的PSD(在角落中)以及来自数据的PSD,以及几个散焦参数。 请点击此处查看此图的放大版本。
{kind=link}

图 3.带有 Xmipp 的手动拣选窗口。(a) 主窗口,其中有要处理的显微照片清单和其他一些参数。 (b) 在显微照片的某个区域内手工挑选颗粒。 (c) 和 (d) 自动拾取要监督的粒子,以便为 Xmipp 自动拾取方法创建一组训练粒子。 请点击此处查看此图的放大版本。
{kind=link}

图 4.与Xmipp结果的深度共识选择。 参数 zScoreDeepLearning 赋予了粒子的优劣权重,它是发现坏粒子的关键。 (a) 最低的 zScores 值与伪影相关联。 (b) 最高的zScores与含有大分子的颗粒有关。 请点击此处查看此图的放大版本。
{kind=link}

图 5.使用冷冻散炼结果进行 2D 分类。 显示生成的类(来自同一方向的粒子子集的平均值)。几个好的类以红色选择(具有一定程度的细节),一些坏类未选择(嘈杂和无中心类)。 请点击此处查看此图的放大版本。
{kind=link}

图 6.具有群体共识结果的3D初始体积。 运行共识工具 xmipp3 - 群共识后获得的 3D 初始体积视图,使用之前 Xmipp 和 Relion 的 3D 初始体积估计。 (a) 体积以切片表示。 (b) 体积的3D可视化。 请点击此处查看此图的放大版本。
{kind=link}

图 7.通过 Relion 结果细化 3D 初始体积。(a) 获得的FSC曲线,大约在4.5Å处越过阈值。 (b) 角度覆盖范围显示为 3D 球体的上视图。在这种情况下,由于没有对称性,分配的粒子应该覆盖整个球体。 (c) 以切片表示的精炼体积。 请点击此处查看此图的放大版本。
{kind=link}

图 8.基于深度学习的3D对齐与Xmipp结果。由xmipp3生成的结果 - 用于3D 对齐的深对齐方法。(a) 变换矩阵形式的每个粒子的角度分配。(b) 角度覆盖面。请点击此处查看此图的放大版本。
{kind=link}

图 9.3D 对齐共识结果。(a) 在偏移和角度参数方面所获得的差值的粒子清单。 (b) 每个粒子的角度差图。 (c) 每个粒子的移位差图。 请点击此处查看此图的放大版本。
{kind=link}

图 10.3D 优化结果的最终迭代。(a) FSC曲线。 (b) 通过切片以全分辨率获得体积。 请点击此处查看此图的放大版本。
{kind=link}

图 11.使用 Xmipp 结果进行局部分辨率分析。 该方法的结果 xmipp3 - 局部 MonoRes。 (a) 一些具有代表性的切片用每个体素的分辨率值着色,如颜色代码所示。 (b) 局部分辨率直方图。 请点击此处查看此图的放大版本。
{kind=link}

图 12.使用 Xmipp 结果锐化。xmipp3的结果 - 局部锐化方法。(a) 每次迭代获得的卷列表。(b) 在切片表示的最后一次迭代后获得的 3D 体积。(c) 最终体积的3D表示。请点击此处查看此图的放大版本。
{kind=link}

图 13.在 Xmipp 结果中验证过拟合工具。xmipp3 的结果 - 验证过拟合。绿线对应于从数据重建,红线对应于噪声。 (a) 平方分辨率与粒子数对数的倒数。(b) 与粒子数的分辨率。请点击此处查看此图的放大版本。
{kind=link}

图 14.所获得体积的几个 3D 表示。(a) 预处理量。 (b) 后处理量。 (c) 局部分辨率,深蓝色体素是分辨率较高(2.75Å)的体素,深红色体素是分辨率较低(10.05Å)的体素。 (d) 放大后处理的体积,可以看到一个α螺旋(红色椭圆形)。 请点击此处查看此图的放大版本。
{kind=link}

图 15.糟糕的 3D 重建示例。(a) FSC曲线急剧下降,并以低分辨率越过阈值。 (b) 局部分辨率直方图。 (c) 切片的3D体积。 请点击此处查看此图的放大版本。
{kind=link}

图 16.2D 类的示例。(a) 好的班级,显示一定程度的细节。 (b) 包含噪声和伪像的错误类别(上半部分使用 Xmipp 获得,下半部分使用 CryoSparc 获得)。 请点击此处查看此图的放大版本。
{kind=link}

图 17.具有不同质量的 3D 初始体积的示例。(a) 良好的初始体积,可以观察到大分子的形状。 (b) 初始体积不良,即获得的形状与预期的形状完全不同。 请点击此处查看此图的放大版本。
{kind=link}
补充图1。创建一个 Scipion 项目。 Scipion显示的窗口可以选择旧项目或创建新项目,并为该项目提供名称和位置。 请点击此处下载此文件。
补充图2。导入电影方法。 当 pwem - 导入影片 打开时,Scipion 显示的窗口。在这里,必须包括主要的采集参数,以便电影可以在Scipion中进行处理。 请点击此处下载此文件。
补充图3。影片对齐方法。 当使用 xmipp3 - 光学对准 时,Scipion 显示窗口。应填充输入影片、考虑对齐的帧范围以及用于处理影片的其他一些参数。 请点击此处下载此文件。
补充图4。CTF估计方法与Ctffind。 Scipion 中的窗体,其中包含运行程序 Ctffind 所需的所有字段。 请点击此处下载此文件。
补充图 5.巫师在西皮翁。 一个向导,用于帮助用户填写表单中的某些参数。在本例中,向导将完成 grigoriefflab - ctffind 方法中的分辨率字段。 请点击此处下载此文件。
补充图 6.使用Xmipp的CTF细化方法。xmipp3 - ctf 估计的形式,包含所有参数,以细化先前估计的 CTF。请单击此处下载此文件。
补充图 7.预处理显微照片方法。xmipp3的形式 - 预处理微照片,允许对它们执行一些操作。在此示例中,"删除坏像素"和"缩减像素图"是有用的一个。请点击此处下载此文件。
补充图 8.使用冷冻的拣选方法。 使用预训练网络运行 Cryolo 拣选方法的表单。 请点击此处下载此文件。
补充图 9.使用Xmipp的共识选择方法。xmipp3的形式-基于深度学习的深度共识采摘来计算坐标的共识,使用预先训练的网络在用不同的拣选方法获得的几组坐标上。请点击此处下载此文件。
补充图 10.提取颗粒法。 输入和 预处理 xmipp3 的选项卡 - 提取颗粒。 请点击此处下载此文件。
补充图11.3D Xmipp的初始体积方法。 该方法的形式 为xmipp3 - 重构, 获得初始3D地图。将显示" 输入 "和" 条件 "选项卡。 请点击此处下载此文件。
补充图 12.调整卷大小方法。 用于裁剪卷或调整体积块大小的形式。在此示例中,此方法用于在 xmipp3 - 重构显著性之后生成全尺寸卷。 请点击此处下载此文件。
补充图13.3D Relion结果的初始体积。通过切片获得的3D初始体积的视图 -3D初始模型方法。请点击此处下载此文件。
补充图 14.使用Relion改进了初始卷。方法依赖的形式 - 3D自动细化。在此示例中,它用于优化共识后估计的初始体积。将显示输入和参考 3D 地图选项卡。请点击此处下载此文件。
补充图15.3D分类方法。依赖形式 - 3D分类。将显示"输入"、"参考 3D 地图"和"优化"选项卡。请点击此处下载此文件。
补充图 16.3D基于深度学习方法的对齐方法。为方法 xmipp3 打开的窗体 - 深对齐。在这里,有必要使用训练集训练网络,然后该网络将预测每个粒子的角度分配。请点击此处下载此文件。
补充图17.3D细化方法。xmipp3 - 高分辨率方法的形式。将显示选项卡输入和角度分配。请点击此处下载此文件。
补充图 18.3D 优化结果的第一次迭代。(a) FSC曲线。 (b) 以切片表示的所得体积(小于全分辨率的尺寸)。 请点击此处下载此文件。
补充图 19.3D 细化相关性分析的第一次迭代。 通过单击窗口上部的条形图标(带有粒子列表)将出现一个新窗口。在 "绘图列 "窗口中,可以创建所需估计参数的直方图。 请点击此处下载此文件。
补充图 20.验证过拟合工具。xmipp3 的形式 - 验证过拟合方法。请点击此处下载此文件。
讨论
目前,冷冻电镜是揭示生物样品3D结构的关键工具。当用显微镜收集良好的数据时,可用的处理工具将使我们能够获得所研究大分子的3D重建。Cryo-EM数据处理能够实现近原子分辨率,这是理解大分子功能行为的关键,在药物发现中也至关重要。
Scipion是一款软件,允许以集成方式创建整个工作流程,将最相关的图像处理包组合在一起,这有助于整个图像处理工作流程的可追溯性和可重复性。Scipion提供了一套非常完整的工具来进行加工;然而,获得高分辨率重建完全取决于所获取数据的质量以及如何处理这些数据。
为了获得高分辨率的3D重建,首先需要从显微镜获得良好的电影,从而将结构信息保留到高分辨率。如果不是这种情况,工作流将无法从数据中提取高清信息。然后,成功的处理工作流程应该能够提取真正与结构相对应的粒子,并在3D空间中找到这些粒子的方向。如果工作流中的任何步骤失败,则重建卷的质量将降低。Scipion允许在任何处理步骤中使用不同的包,这有助于找到处理数据的最合适方法。此外,由于有许多可用的软件包,可以使用共识工具,通过在不同方法的估计输出中找到一致意见来提高准确性。此外,在"代表性成果"部分已经详细讨论了几种验证工具,以及如何在工作流程的每个步骤中识别准确和不准确的结果,以检测潜在问题以及如何尝试解决这些问题。协议中有几个检查点可以帮助确定协议是否正常运行。一些最相关的是:采摘,2D分类,初始体积估计和3D对齐。检查输入,使用其他方法重复步骤或使用共识,是Scipion中可用的选项,当问题出现时,用户可以使用这些选项来查找解决方案。
关于以前在冷冻电镜领域进行软件包集成的方法,Appion31 是唯一一种允许真正集成不同软件包的方法。然而,Appion与Leginon32紧密相连,Leginon32是一种从电子显微镜自动收集图像的系统。与 Scipion 的主要区别在于数据模型和存储的耦合程度较低。这样,要在Scipion中创建新协议,只需要开发一个Python脚本。但是,在 Appion 中,开发人员必须编写脚本并更改基础数据库。总之,Scipion的开发是为了简化维护和可扩展性。
我们在这份手稿中介绍了冷冻电镜处理的完整工作流程,使用了 恶性疟原虫 80S核糖体的真实案例数据集(EMPIAR条目:10028,EMDB条目:2660)。此处介绍和讨论的步骤可以概括为电影对齐、CTF 估计、粒子拾取、2D 分类、初始地图估计、3D 分类、3D 细化、评估和后处理。在其中几个步骤中使用了不同的软件包,并应用了共识工具。最终的3D重建体积达到了3 Å的分辨率,在后处理的体积中,可以区分一些二级结构,如α-螺旋,这有助于描述原子在空间中的排列方式。
本手稿中介绍的工作流程展示了如何使用 Scipion 以简单易用的方式组合不同的 Cryo-EM 软件包,以简化处理,同时获得更可靠的结果。
在未来,新方法和软件包的开发将继续增长,像Scipion这样的软件可以轻松集成所有这些方法和软件包对研究人员来说将更加重要。即使在那时,共识方法也将更加相关,届时将有大量具有不同基础的方法可用,有助于更准确地估计Cryo-EM重建过程中涉及的所有参数。跟踪和可重复性是研究过程中的关键,并且由于具有用于执行完整工作流程的通用框架,因此Scipion更容易实现。
披露声明
作者没有什么可透露的。
致谢
作者希望通过赠款感谢西班牙科学与创新部的经济支持:PID2019-104757RB-I00/AEI/10.13039/501100011033,通过赠款获得的"马德里自治委员会":S2017 / BMD-3817,Instituto de Salud Carlos III,PT17/0009/0010(ISCIII-SGEFI / ERDF),欧盟(EU)和Horizon 2020通过赠款:INSTRUCT - ULTRA(INFRADEV-03-2016-2017,提案:731005),EOSC Life(INFRAEOSC-04-2018,提案: 824087),iNEXT - Discovery(提案:871037)和HighResCells(ERC - 2018 - SyG,提案:810057)。产生这些成果的项目得到了"la Caixa"基金会(ID 100010434)的奖学金支持。奖学金代码为LCF /BQ/DI18/11660021。该项目已获得欧盟地平线2020研究和创新计划的资助,该计划根据玛丽·斯克沃多夫斯卡 - 居里赠款协议第713673号。作者感谢Landmark ESFRI项目Instruction的资源支持和使用。
材料
| Name | Company | Catalog Number | Comments |
| no material is used in this article | - | - | - |
参考文献
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Kühlbrandt, W. The Resolution Revolution. Science. 343 (6178), 1443-1444 (2014).
- . 1.15 A structure of human apoferritin obtained from Titan Mono- BCOR microscope Available from: https://www.rcsb.org/structure/7A6A (2021)
- Arnold, S. A., et al. Miniaturizing EM Sample Preparation: Opportunities, Challenges, and "Visual Proteomics". PROTEOMICS. 18 (5-6), 1700176 (2018).
- Faruqi, A. R., McMullan, G. Direct imaging detectors for electron microscopy. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 878, 180-190 (2018).
- Vilas, J. L., et al. Advances in image processing for single-particle analysis by electron cryomicroscopy and challenges ahead. Current Opinion in Structural Biology. 52, 127-145 (2018).
- Martinez, M., et al. Integration of Cryo-EM Model Building Software in Scipion. Journal of Chemical Information and Modeling. 60, 2533-2540 (2020).
- de la Rosa-Trevín, J. M., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195, 93-99 (2016).
- de la Rosa-Trevín, J. M., et al. Xmipp 3.0: an improved software suite for image processing in electron microscopy. Journal of Structural Biology. 184, 321-328 (2013).
- Scheres, S. H. W. . Methods in Enzymology. The Resolution Revolution: Recent Advances In cryoEM. , 125-157 (2016).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. 14, 290-296 (2017).
- Ludtke, S. J. 3-D structures of macromolecules using single-particle analysis in EMAN. Methods in Molecular Biology. 673, 157-173 (2010).
- Shaikh, T. R., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3, 1941-1974 (2008).
- Wagner, T., et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications Biology. 2, (2019).
- Mindell, J. A., Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology. 142, 334-347 (2003).
- Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography. 67, 235-242 (2011).
- Liebschner, D., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D. 75, 861-887 (2019).
- Wong, W., et al. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 3, 03080 (2014).
- Abrishami, V., et al. Alignment of direct detection device micrographs using a robust Optical Flow approach. Journal of Structural Biology. 189, 163-176 (2015).
- Sorzano, C. O. S., Jonic, S., Nunez Ramirez, R., Boisset, N., Carazo, J. M. Fast, robust and accurate determination of transmission electron microscopy contrast transfer function. Journal of Structural Biology. 160, 249-262 (2007).
- Abrishami, V., et al. A pattern matching approach to the automatic selection of particles from low-contrast electron micrographs. Bioinformatics. 29, 2460-2468 (2013).
- Sanchez-Garcia, R., Segura, J., Maluenda, D., Carazo, J. M., Sorzano, C. O. S. Deep Consensus, a deep learning-based approach for particle pruning in cryo-electron microscopy. IUCrJ. 5, 854-865 (2018).
- Sorzano, C. O. S., et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. Journal of Structural Biology. 171, 197-206 (2010).
- Sorzano, C. O. S., et al. A statistical approach to the initial volume problem in Single Particle Analysis by Electron Microscopy. Journal of Structural Biology. 189, 213-219 (2015).
- Sorzano, C. O. S., et al. Swarm optimization as a consensus technique for Electron Microscopy Initial Volume. Applied Analysis and Optimization. 2, 299-313 (2018).
- Pettersen, E. F., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 25, 1605-1612 (2004).
- Sorzano, C. O. S., et al. A new algorithm for high-resolution reconstruction of single particles by electron microscopy. Journal of Structural Biology. 204, 329-337 (2018).
- Vilas, J. L., et al. MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure. 26, 337-344 (2018).
- Ramirez-Aportela, E., et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics. 36, 765-772 (2020).
- Heymann, J. B. Validation of 3D EM Reconstructions: The Phantom in the Noise. AIMS Biophys. 2, 21-35 (2015).
- Lander, G. C., et al. Appion: An integrated, database-drive pipeline to facilitate EM image processing. Journal of Structural Biology. 166, 95-102 (2009).
- Suloway, C., et al. Automated molecular microscopy: The new Leginon system. Journal of Structural Biology. 151, 41-60 (2005).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。