
Method Article
Cryo-EM et analyse monoparticule avec Scipion
Dans cet article
Résumé
L’analyse d’une seule particule en cryo-microscopie électronique est l’une des principales techniques utilisées pour déterminer la structure des ensembles biologiques à haute résolution. Scipion fournit les outils pour créer l’ensemble du pipeline afin de traiter les informations acquises par le microscope et de réaliser une reconstruction 3D du spécimen biologique.
Résumé
La cryo-microscopie électronique est devenue l’un des outils les plus importants de la recherche biologique pour révéler l’information structurelle des macromolécules à une résolution quasi atomique. Dans l’analyse d’une seule particule, l’échantillon vitrifié est imagé par un faisceau d’électrons et les détecteurs à l’extrémité de la colonne du microscope produisent des films de cet échantillon. Ces films contiennent des milliers d’images de particules identiques dans des orientations aléatoires. Les données doivent passer par un flux de travail de traitement d’image avec plusieurs étapes pour obtenir le volume final reconstruit en 3D. L’objectif du flux de travail de traitement d’images est d’identifier les paramètres d’acquisition pour pouvoir reconstruire le spécimen à l’étude. Scipion fournit tous les outils pour créer ce workflow en utilisant plusieurs packages de traitement d’image dans un cadre intégratif, permettant également la traçabilité des résultats. Dans cet article, l’ensemble du flux de travail de traitement d’image dans Scipion est présenté et discuté avec des données provenant d’un cas de test réel, donnant tous les détails nécessaires pour passer des films obtenus par le microscope à une reconstruction 3D finale haute résolution. En outre, la puissance de l’utilisation d’outils de consensus qui permettent de combiner des méthodes et de confirmer les résultats à chaque étape du flux de travail, améliorant ainsi la précision des résultats obtenus, est discutée.
Introduction
En cryo-microscopie électronique (cryo-EM), l’analyse de particules uniques (SPA) d’échantillons vitrifiés congelés-hydratés est l’une des variantes d’imagerie les plus largement utilisées et les plus réussies pour les macromolécules biologiques, car elle permet de comprendre les interactions moléculaires et la fonction des ensembles biologiques1. C’est grâce aux progrès récents de cette technique d’imagerie qui ont donné lieu à la « révolution de la résolution »2 et ont permis la détermination réussie de structures 3D biologiques avec une résolution quasi atomique. Actuellement, la résolution la plus élevée atteinte dans SPA cryo-EM était de 1,15 Å pour l’apoferritine3 (entrée EMDB: 11668). Ces avancées technologiques comprennent des améliorations dans la préparation des échantillons4, l’acquisition d’images5 et les méthodes de traitement des images6. Cet article est axé sur ce dernier point.
En bref, l’objectif des méthodes de traitement d’image est d’identifier tous les paramètres d’acquisition pour inverser le processus d’imagerie du microscope et récupérer la structure 3D du spécimen biologique étudié. Ces paramètres sont le gain de la caméra, le mouvement induit par le faisceau, les aberrations du microscope (principalement la mise au point), l’orientation angulaire 3D et la translation de chaque particule, et l’état conformationnel en cas d’avoir un échantillon avec des changements conformationnels. Cependant, le nombre de paramètres est très élevé et cryo-EM nécessite l’utilisation d’images à faible dose pour éviter les dommages causés par les radiations, ce qui réduit considérablement le rapport signal/bruit (SNR) des images acquises. Ainsi, le problème ne peut pas être résolu sans équivoque et tous les paramètres à calculer uniquement peuvent être des estimations. Tout au long du flux de travail de traitement d’image, les paramètres corrects doivent être identifiés, en éliminant les paramètres restants pour finalement obtenir une reconstruction 3D haute résolution.
Les données générées par le microscope sont rassemblées dans des images. En simplifiant, une image contient le nombre d’électrons qui sont arrivés à une position particulière (pixel) dans l’image, chaque fois que des détecteurs de comptage d’électrons sont utilisés. Dans un champ de vision particulier, plusieurs images sont collectées et c’est ce qu’on appelle un film. Comme de faibles doses d’électrons sont utilisées pour éviter les dommages causés par le rayonnement qui pourraient détruire l’échantillon, le SNR est très faible et les images correspondant au même film doivent être moyennées pour obtenir une image révélant des informations structurelles sur l’échantillon. Cependant, non seulement une moyenne simple est appliquée, mais l’échantillon peut subir des décalages et d’autres types de mouvements pendant le temps d’imagerie en raison du mouvement induit par le faisceau qui doit être compensé. Les cadres compensés par décalage et moyennés sont à l’origine d’une micrographie.
Une fois les micrographies obtenues, nous devons estimer les aberrations introduites par le microscope pour chacune d’elles, appelées fonction de transfert de contraste (CTF), qui représente les changements de contraste de la micrographie en fonction de la fréquence. Ensuite, les particules peuvent être sélectionnées et extraites, ce qui s’appelle la cueillette de particules. Chaque particule doit être une petite image contenant une seule copie du spécimen étudié. Il existe trois familles d’algorithmes pour la sélection de particules: 1) ceux qui n’utilisent qu’un certain paramétrage de base de l’apparence de la particule pour les trouver dans l’ensemble des micrographies (par exemple, la taille des particules), 2) ceux qui apprennent à quoi ressemblent les particules de l’utilisateur ou d’un ensemble préentraîné, et 3) ceux qui utilisent des modèles d’image. Chaque famille a des propriétés différentes qui seront montrées plus tard.
L’ensemble extrait de particules trouvées dans les micrographies sera utilisé dans un processus de classification 2D qui a deux objectifs: 1) nettoyer l’ensemble de particules en éliminant le sous-ensemble contenant des images de bruit pur, des particules qui se chevauchent ou d’autres artefacts, et 2) les particules moyennes représentant chaque classe pourraient être utilisées comme informations initiales pour calculer un volume initial 3D.
Le calcul du volume initial 3D est la prochaine étape cruciale. Le problème de l’obtention de la structure 3D peut être considéré comme un problème d’optimisation dans un paysage de solutions multidimensionnelles, où le minimum global est le meilleur volume 3D qui représente la structure d’origine, mais plusieurs minima locaux représentant des solutions sous-optimales peuvent être trouvés, et où il est très facile de se faire piéger. Le volume initial représente le point de départ du processus de recherche, de sorte qu’une mauvaise estimation initiale du volume pourrait nous empêcher de trouver le minimum global. Dès le volume initial, une étape de classification 3D permettra de découvrir différents états conformationnels et de nettoyer à nouveau l’ensemble des particules ; l’objectif est d’obtenir une population structurellement homogène de particules. Après cela, une étape de raffinement 3D sera chargée d’affiner les paramètres angulaires et de traduction de chaque particule afin d’obtenir le meilleur volume 3D possible.
Enfin, dans les dernières étapes, la reconstruction 3D obtenue peut être affûtée et polie. L’affûtage est un processus d’augmentation des hautes fréquences du volume reconstruit, et le polissage est une étape pour affiner davantage certains paramètres, comme le CTF ou la compensation de mouvement induit par le faisceau, au niveau des particules. En outre, certaines procédures de validation pourraient être utilisées pour mieux comprendre la résolution obtenue à la fin du flux de travail.
Après toutes ces étapes, les processus de traçage et d’amarrage7 contribueront à donner un sens biologique à la reconstruction 3D obtenue, en construisant des modèles atomiques de novo ou en adaptant des modèles existants. Si une haute résolution est atteinte, ces processus nous indiqueront les positions des structures biologiques, même des différents atomes, dans notre structure.
Scipion8 permet de créer l’ensemble du flux de travail en combinant les packages de traitement d’image les plus pertinents de manière intégrative. Xmipp9, Relion10, CryoSPARC11, Eman12, Spider13, Cryolo14, Ctffind15, CCP416, Phenix17 et bien d’autres packages peuvent être inclus dans Scipion. En outre, il intègre tous les outils nécessaires pour bénéficier de l’intégration, de l’interopérabilité, de la traçabilité et de la reproductibilité afin de faire un suivi complet de l’ensemble du flux de travail de traitement d’image8.
L’un des outils les plus puissants que Scipion nous permet d’utiliser est le consensus, qui signifie comparer les résultats obtenus avec plusieurs méthodes en une seule étape du traitement, en combinant les informations véhiculées par différentes méthodes pour générer une sortie plus précise. Cela pourrait aider à augmenter les performances et à améliorer la qualité obtenue dans les paramètres estimés. Notez qu’un flux de travail plus simple peut être construit sans l’utilisation de méthodes consensuelles; cependant, nous avons vu la puissance de cet outil22,25 et le flux de travail présenté dans ce manuscrit l’utilisera en plusieurs étapes.
Toutes les étapes qui ont été résumées dans les paragraphes précédents seront expliquées en détail dans la section suivante et combinées dans un flux de travail complet à l’aide de Scipion. En outre, la façon d’utiliser les outils de consensus pour parvenir à un accord plus élevé dans les résultats générés sera montrée. À cette fin, l’exemple de jeu de données du ribosome Plasmodium falciparum 80S a été choisi (entrée EMPIAR: 10028, entrée EMDB: 2660). L’ensemble de données est formé par 600 films de 16 images de taille 4096x4096 pixels à une taille de pixel de 1,34Å prises à un FEI POLARA 300 avec une caméra FEI FALCON II, avec une résolution signalée à EMDB est de 3,2Å18 .
Protocole
1. Création d’un projet dans Scipion et importation des données
- Ouvrez Scipion et cliquez sur Créer un projet, spécifiez le nom du projet et l’emplacement où il sera enregistré (Figure supplémentaire 1). Scipion ouvrira la fenêtre du projet montrant un canevas avec, sur le côté gauche, un panneau avec une liste de méthodes disponibles, chacune d’entre elles représente un outil de traitement d’image qui peut être utilisé pour gérer les données.
Remarque : Ctrl + F peut être utilisé pour trouver une méthode si elle n’apparaît pas dans la liste. - Pour importer les films pris au microscope, sélectionnez le pwem - importer des films sur le panneau de gauche (ou tapez-le en appuyant sur Ctrl + F).
- Une nouvelle fenêtre s’ouvrira (Figure supplémentaire 2). Là, incluez le chemin d’accès aux données et les paramètres d’acquisition. Dans cet exemple, utilisez la configuration suivante : Tension du microscope 300 kV, Aberration sphérique 2,0 mm, Contraste d’amplitude 0,1, Taux de grossissement 50000, Mode Taux d’échantillonnage vers Image de l’expéditeur et Taille de pixel 1,34 Å. Lorsque tous les paramètres du formulaire sont remplis, cliquez sur le bouton Exécuter .
Remarque : Lorsqu’une méthode démarre, une zone apparaît dans la zone de travail en couleur jaune étiquetée comme en cours d’exécution. Lorsqu’une méthode se termine, la zone passe au vert et l’étiquette devient terminée. En cas d’erreur lors de l’exécution d’une méthode, la case apparaîtra en rouge, étiquetée comme ayant échoué. Dans ce cas, vérifiez la partie inférieure du canevas, dans l’onglet Journal de sortie , une explication de l’erreur apparaîtra. - Une fois la méthode terminée, vérifiez les résultats dans la partie inférieure du canevas dans l’onglet Résumé . Ici, les sorties générées par la méthode sont présentées, dans ce cas, l’ensemble des films. Cliquez sur le bouton Analyser les résultats et une nouvelle fenêtre apparaîtra avec la liste des films.
2. Alignement des films : des films aux micrographies
- Utilisez la méthode xmipp3 - alignement optique qui implémente le flux optique19. Utilisez les paramètres suivants pour remplir le formulaire (Figure supplémentaire 3) : les films d’entrée sont ceux obtenus à l’étape 1, la plage dans Images à ALIGN est comprise entre 2 et 13, les autres options restent avec les valeurs par défaut. Exécutez le programme.
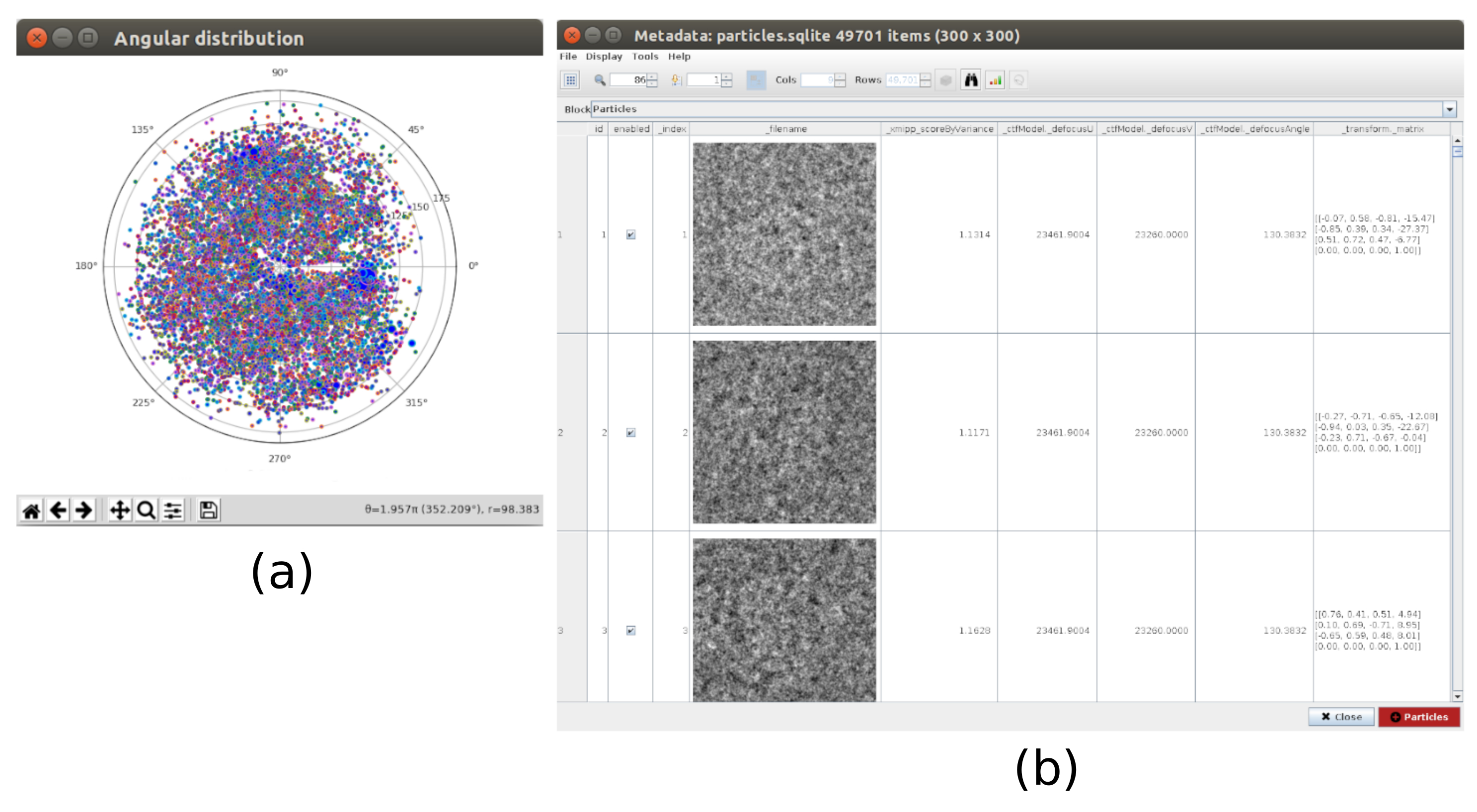
REMARQUE : Les paramètres en gras dans un formulaire doivent toujours être remplis. Les autres auront une valeur par défaut ou ne seront pas obligatoirement requis. Dans la partie supérieure de la fenêtre de formulaire, les champs où les ressources de calcul sont distribuées peuvent être trouvés, sous forme de threads, d’IPM ou de GPU. - Cliquez sur Analyser les résultats pour vérifier les micrographies obtenues et la trajectoire des décalages estimés (Figure 1). Pour chaque micrographie vue : regardez la densité spectrale de puissance (PSD), les trajectoires obtenues pour aligner le film (un point par image) en coordonnées cartésiennes et polaires, et le nom de fichier de la micrographie obtenue (en cliquant dessus, la micrographie peut être inspectée). Notez que les particules de l’échantillon sont beaucoup plus visibles dans la micrographie, par rapport à une seule image du film.
3. Estimation CTF : calcul des aberrations du microscope
- Tout d’abord, utilisez la méthode grigoriefflab - ctffind15. La configuration est la suivante: les micrographies d’entrée sont la sortie de l’étape 2, le facteur de sous-échantillonnage manuel CTF est défini sur 1,5 et la plage de résolution va de 0,06 à 0,42. De plus, dans les options Avancées (qui peuvent être trouvées en sélectionnant ce choix dans le niveau Expert du formulaire), définissez la taille de la fenêtre sur 256. Les paramètres restants restent avec les valeurs par défaut (figure supplémentaire 4).
REMARQUE: Dans la plupart des méthodes de Scipion, l’option Avancé affiche plus de paramètres de configuration. Utilisez ces options avec précaution, lorsque le programme à lancer est complètement connu et que la signification des paramètres est comprise. Certains paramètres peuvent être difficiles à remplir sans avoir à regarder les données; dans ce cas, Scipion montre une baguette magique sur le côté droit qui montrera une fenêtre d’assistant (Figure supplémentaire 5). Par exemple, dans le champ Résolution de ce formulaire est particulièrement utile, car ces valeurs doivent être sélectionnées pour couvrir approximativement la région du premier zéro au dernier anneau perceptible de la DSP. - Cliquez sur Exécuter et sur Analyser les résultats (Figure 2) lorsque la méthode est terminée. Vérifiez que le CTF estimé correspond à celui expérimental. À cette fin, examinez la DSP et comparez les anneaux estimés dans le coin avec ceux provenant des données. Vérifiez également les valeurs de défocalisation obtenues pour trouver des valeurs inattendues et les micrographies respectives peuvent être jetées ou recalculées. Dans cet exemple, l’ensemble des micrographies peut être utilisé.
REMARQUE: Utilisez les boutons dans la partie inférieure de la fenêtre pour créer un sous-ensemble de micrographies (avec le bouton rouge Micrographies ) et pour recalculer un CTF (avec le bouton rouge Recalculer les CTF ), en cas de besoin. - Pour affiner l’estimation précédente, utilisez xmipp3 - ctf estimation20. Sélectionnez comme Micrographies d’entrée la sortie de l’étape 2, sélectionnez l’option Utiliser defoci d’une estimation CTF précédente, comme Estimation CTF précédente choisissez la sortie de grigoriefflab - ctffind et, dans le niveau Avancé, modifiez la taille de la fenêtre à 256 (Figure supplémentaire 6). Exécutez-le.
- Cliquez sur Analyser les résultats pour vérifier les CTF obtenus. Avec cette méthode, plus de données sont estimées et représentées dans quelques colonnes supplémentaires. Comme aucune d’entre elles ne montre de valeurs estimées incorrectes, toutes les micrographies seront utilisées dans les étapes suivantes.
4. Prélèvement de particules : trouver des particules dans les micrographies
- Avant de commencer la cueillette, effectuez un prétraitement des micrographies. Ouvrez xmipp3 - prétraitez les micrographies, définissez comme micrographies d’entrée celles obtenues à l’étape 2 et sélectionnez les options Supprimer les pixels défectueux? avec Multiple de Stddev à 5 et Micrographies de sous-échantillonnage? avec un facteur de sous-échantillonnage de 2 (Figure supplémentaire 7). Cliquez sur Exécuter et vérifiez que la taille des micrographies résultantes a été réduite.
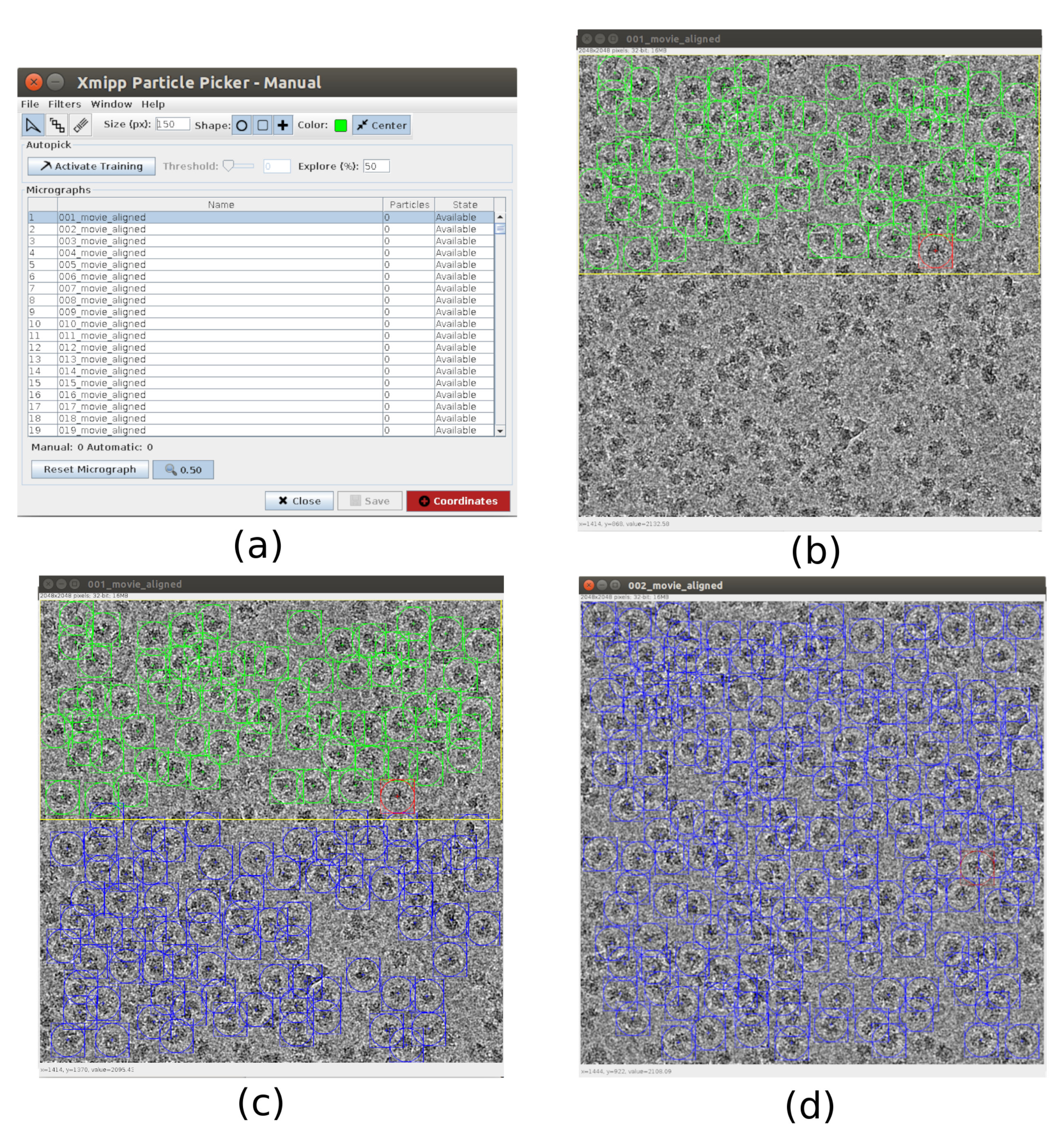
- Pour le picking, utilisez xmipp3 - manual-picking (étape 1) et xmipp3 - auto-picking (step 2)21. Le picking manuel permet de préparer manuellement un ensemble de particules avec lesquelles l’étape d’auto-picking apprendra et générera l’ensemble complet de particules. Tout d’abord, exécutez xmipp3 - prélèvement manuel (étape 1) avec des micrographies d’entrée comme micrographies obtenues dans le préprocessus précédent. Cliquez sur Exécuter et une nouvelle fenêtre interactive apparaîtra (Figure 3).
- Dans cette fenêtre, une liste des micrographies (Figure 3a) et d’autres options est présentée. Changez la taille (px) à 150, ce sera la taille de la boîte contenant chaque particule. La micrographie sélectionnée apparaît dans une fenêtre plus grande. Choisissez une région et choisissez toutes les particules visibles qu’elle contient (Figure 3b). Ensuite, cliquez sur Activer la formation pour commencer l’apprentissage. Les autres régions de la micrographie sont automatiquement prélevées (Figure 3c). Vérifiez les particules sélectionnées et incluez-en d’autres en cliquant dessus, ou supprimez les mauvaises avec Maj + clic, si nécessaire.
- Sélectionnez la micrographie suivante dans la première fenêtre. La micrographie sera automatiquement prélevée. Vérifiez à nouveau pour inclure ou enlever certaines particules, si nécessaire. Répétez cette étape avec environ 5 micrographies pour créer un ensemble de formation représentatif.
- Une fois cela fait, cliquez sur Coordonnées dans la fenêtre principale pour enregistrer les coordonnées de toutes les particules sélectionnées. L’ensemble de particules d’entraînement est prêt à passer au prélèvement automatique pour terminer le processus pour toutes les micrographies.
- Ouvrez xmipp3 - prélèvement automatique (étape 2) indiquant dans le prélèvement de particules Xmipp exécuter le prélèvement manuel précédent, et Micrographes à sélectionner comme Identique à supervisé. Cliquez sur Exécuter. Cette méthode générera en sortie un ensemble d’environ 100000 coordonnées.
- Appliquez une approche consensuelle, alors effectuez une deuxième méthode de prélèvement pour sélectionner les particules dans lesquelles les deux méthodes sont d’accord. Ouvrez sphire - cryolo picking14 et sélectionnez les micrographies prétraitées comme micrographies d’entrée, utilisez un modèle général ? à Oui, avec un seuil de confiance de 0,3 et une taille de boîte de 150 (figure supplémentaire 8). Exécutez-le. Cette méthode devrait également générer environ 100000 coordonnées.
- Exécutez xmipp3 - sélection par consensus profond22. Comme les coordonnées d’entrée incluent la sortie de sphire - cryolo picking (étape 4.7) et xmipp3 - auto-picking (étape 4.6), définissez Select model type sur Pretrained, et Skip training and score directly with pretrained model? À Oui (figure supplémentaire 9). Exécutez-le.
- Cliquez sur Analyser les résultats et, dans la nouvelle fenêtre, sur l’icône en forme d’œil à côté de Sélectionner les particules/coordonnées avec des valeurs élevées 'zScoreDeepLearning1'. Une nouvelle fenêtre s’ouvrira avec une liste de toutes les particules (Figure 4). Les valeurs zScore dans la colonne donnent un aperçu de la qualité d’une particule, des valeurs faibles signifient une mauvaise qualité.
- Cliquez sur l’étiquette_xmipp_zScoreDeepLearning pour commander les particules du zScore le plus élevé au plus bas. Sélectionnez les particules dont le zScore est supérieur à 0,75 et cliquez sur Coordonnées pour créer le nouveau sous-ensemble. Cela devrait créer un sous-ensemble avec environ 50000 coordonnées.
- Ouvrez xmipp3 - nettoyeur de micrographies profondes. Sélectionnez comme coordonnées d’entrée le sous-ensemble obtenu à l’étape précédente, Micrographies source identique aux coordonnées et maintenir le seuil à 0,75. Exécutez-le. Vérifiez dans l’onglet Résumé que le nombre de coordonnées a été réduit, bien que dans ce cas, seules quelques coordonnées soient supprimées.
REMARQUE: Cette étape est capable de nettoyer en outre l’ensemble des coordonnées et pourrait être très utile pour nettoyer d’autres ensembles de données avec plus d’artefacts de film comme des zones de carbone ou de grandes impuretés. - Exécutez xmipp3 - extraire des particules (Figure supplémentaire 10). Indiquez comme coordonnées d’entrée les coordonnées obtenues après l’étape précédente, la source des micrographies comme autre, les micrographies d’entrée comme sortie de l’étape 2, l’estimation CTF comme sortie de l’estimation xmipp3 - ctf, le facteur de sous-échantillonnage à 3 et la taille de la boîte de particules à 100. Dans l’onglet Prétraitement du formulaire, sélectionnez Oui à tous. Exécutez-le.
- Vérifiez que la sortie doit contenir les particules dans une taille réduite de 100x100 pixels et une taille de pixel de 4.02Å/px.
- Exécutez à nouveau xmipp3 - extrayez les particules en modifiant les paramètres suivants : Facteur de sous-échantillonnage à 1 et Taille de la boîte de particules à 300. Vérifiez que la sortie est le même ensemble de particules mais maintenant à la pleine résolution.
5. Classification 2D: regroupement de particules similaires
- Ouvrez la méthode cryosparc2 - classification 2d11 avec les particules d’entrée comme celles obtenues à l’étape 4.11 et, dans l’onglet Classification 2D, le Nombre de classes à 128, conservez tous les autres paramètres avec les valeurs par défaut. Exécutez-le.
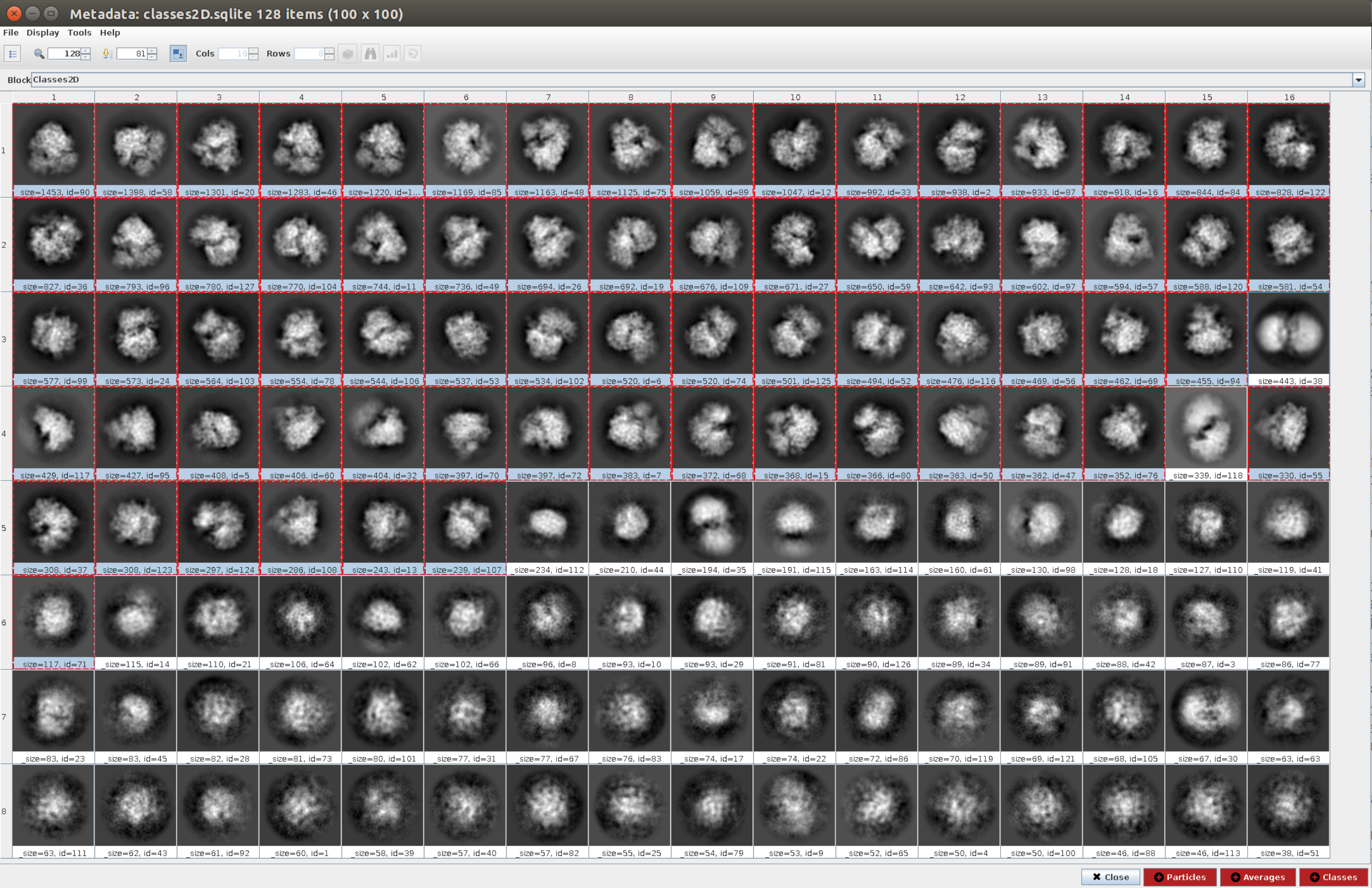
- Cliquez sur Analyser les résultats , puis sur l’icône en forme d’œil à côté de Afficher les classes de particules avec Scipion (Figure 5). Cette classification aidera à nettoyer l’ensemble des particules, car plusieurs classes apparaîtront bruyantes ou avec des artefacts. Sélectionnez les classes contenant de bonnes vues. Cliquez sur Particules (bouton rouge dans la partie inférieure de la fenêtre) pour créer le sous-ensemble nettoyeur.
- Maintenant, ouvrez xmipp3 - cl2d23 et définissez comme images d’entrée les images obtenues à l’étape précédente et le nombre de classes comme 128. Cliquez sur Exécuter.
REMARQUE: Cette deuxième classification est utilisée comme étape de nettoyage supplémentaire de l’ensemble de particules. Habituellement, il est utile d’éliminer autant de particules bruyantes que possible. Toutefois, si un flux de travail plus simple est souhaité, une seule méthode de classification 2D peut être utilisée. - Une fois la méthode terminée, vérifiez les 128 classes générées en cliquant sur Analyser les résultats et sur Ce qu’il faut afficher: classes. La plupart des classes générées montrent une projection de la macromolécule avec un certain niveau de détail. Cependant, certains d’entre eux semblent bruyants (dans cet exemple environ 10 classes). Sélectionnez toutes les bonnes classes et cliquez sur le bouton Classes pour générer un nouveau sous-ensemble avec uniquement les bonnes. Ce sous-ensemble sera utilisé comme entrée dans l’une des méthodes de génération d’un volume initial. Avec les mêmes classes sélectionnées, cliquez sur Particules pour créer un sous-ensemble plus propre après avoir supprimé celles appartenant aux mauvaises classes.
- Ouvrir pwem - sous-ensemble avec l’ensemble complet d’éléments comme sortie de 4.13 (toutes les particules en taille réelle), Définir le sous-ensemble aléatoire sur Non, Autre comme sous-ensemble de particules créé à l’étape précédente et Définir l’opération comme intersection. Cela extraira le sous-ensemble précédent des particules à pleine résolution.
6. Estimation initiale du volume : construire la première estimation du volume 3D
- Dans cette étape, estimez deux volumes initiaux avec des méthodes différentes, puis utilisez un outil de consensus pour générer le volume 3D estimé final. Ouvrez xmipp3 - reconstruisez la méthode significant24 avec des classes Input comme celles obtenues après l’étape 5, groupe de symétrie comme c1, et conservez les paramètres restants avec leurs valeurs par défaut (figure supplémentaire 11). Exécutez-le.
- Cliquez sur Analyser les résultats. Vérifiez qu’un volume basse résolution de taille 100x100x100 pixels et une taille de pixel de 4.02Å/px est obtenu.
- Ouvrir xmipp3 - recadrer/redimensionner les volumes (figure supplémentaire 12) en utilisant comme volumes d’entrée celui obtenu à l’étape précédente, Redimensionner les volumes ? en Oui, Redimensionner l’option vers Taux d’échantillonnage et Redimensionner le taux d’échantillonnage à 1,34 Å/px. Exécutez-le. Vérifiez dans l’onglet Résumé que le volume de sortie a la taille correcte.
- Maintenant, créez le deuxième volume initial. Open relion - Modèle initial 3D10, comme les particules d’entrée utilisent les bonnes particules à pleine résolution (sortie de 5,5) et définissent le diamètre du masque de particules sur 402Å, conservez les paramètres restants avec les valeurs par défaut. Exécutez-le.
- Cliquez sur Analyser les résultats , puis sur Afficher le volume avec : tranches. Vérifiez qu’un volume de faible résolution mais avec la forme principale de la structure est obtenu (Figure supplémentaire 13).
- Maintenant, ouvrez pwem - join sets pour combiner les deux volumes initiaux générés afin de créer l’entrée de la méthode de consensus. Indiquez simplement Volumes comme type d’entrée et sélectionnez les deux volumes initiaux dans Jeu d’entrées. Exécutez-le. La sortie doit être un ensemble contenant deux éléments avec les deux volumes.
- L’outil de consensus est celui inclus dans xmipp3 - swarm consensus25. Ouvre-le. Utilisez comme images pleine grandeur les bonnes particules en pleine résolution (sortie de 5,5), comme volumes initiaux l’ensemble avec deux éléments générés à l’étape précédente, et assurez-vous que le groupe symétrie est c1. Cliquez sur Exécuter.
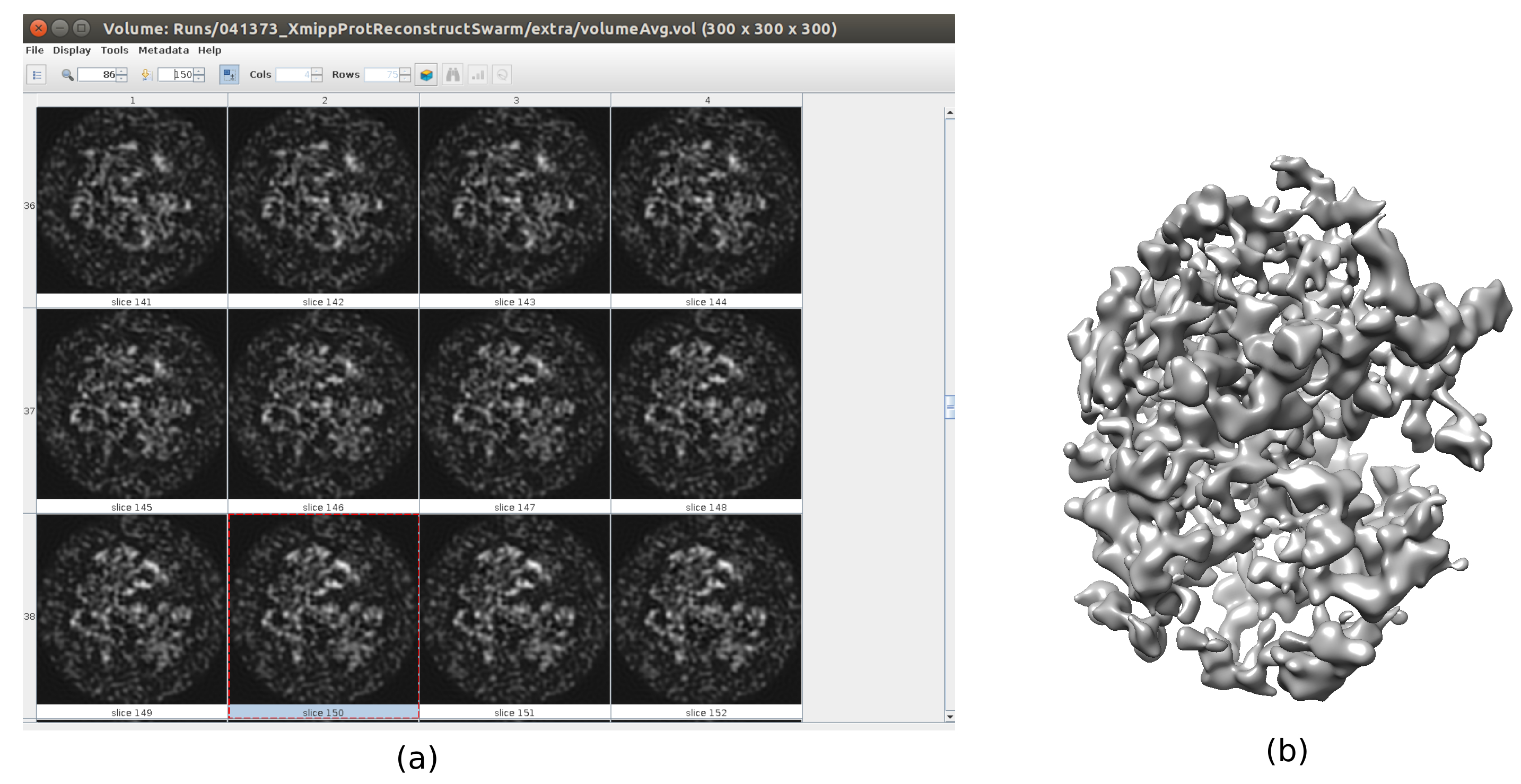
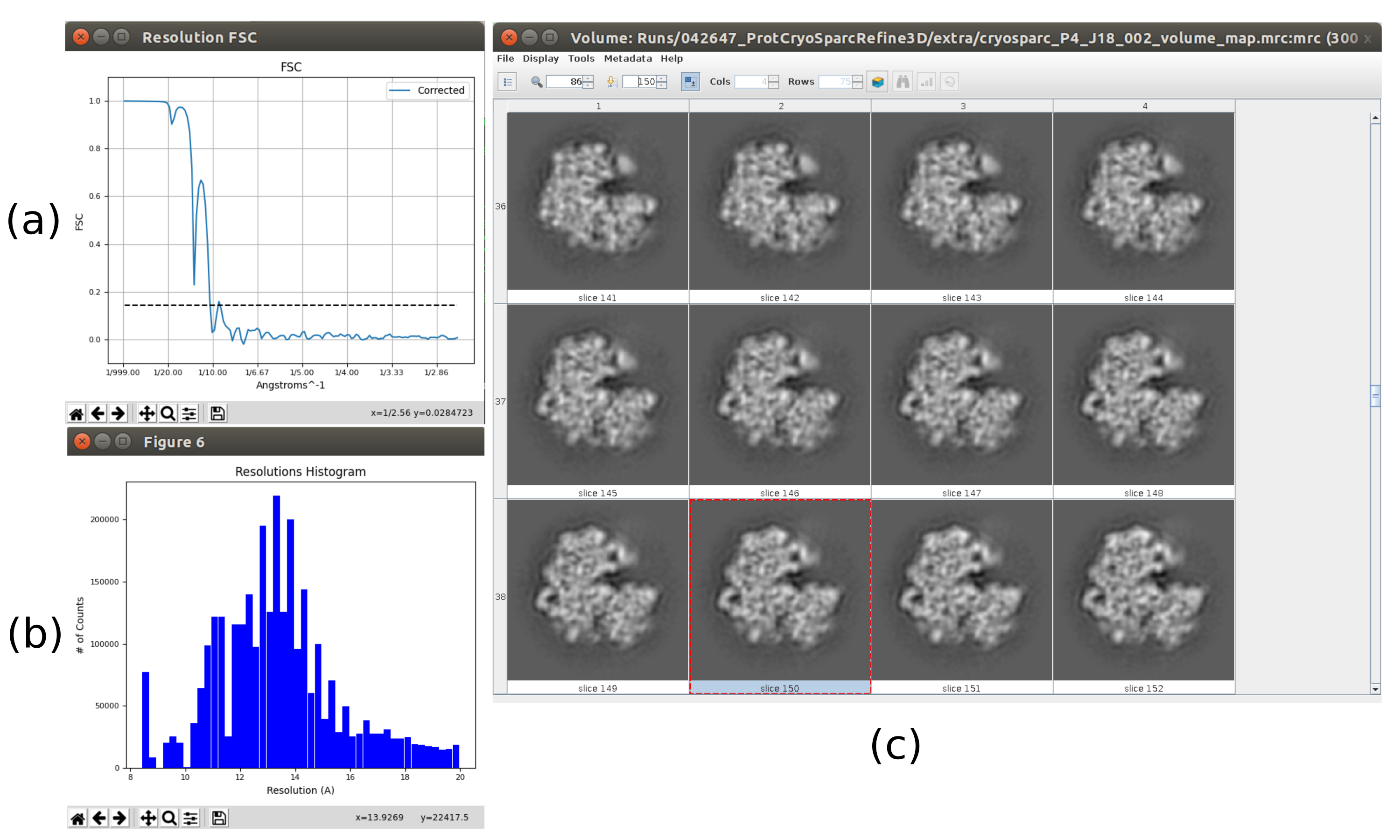
- Cliquez sur Analyser les résultats. Vérifiez qu’un volume de sortie plus détaillé est obtenu (Figure 6). Bien qu’il y ait plus de bruit autour de la structure, avoir plus de détails dans la carte de la structure aidera les étapes de raffinement suivantes pour éviter les minima locaux.
REMARQUE: Si UCSF Chimera26 est disponible, utilisez la dernière icône dans la partie supérieure de la fenêtre pour faire une visualisation 3D du volume obtenu. - Ouvrir et exécuter relion - 3D auto-refine10 pour faire une première affectation angulaire 3D des particules. Sélectionnez comme particules d’entrée la sortie de 5,5 et définissez le diamètre du masque de particules sur 402Å. Dans l’onglet Carte 3D de référence, sélectionnez comme volume d’entrée celui obtenu à l’étape précédente, Symétrie en c1 et Filtre passe-bas initial à 30Å (Figure supplémentaire 14).
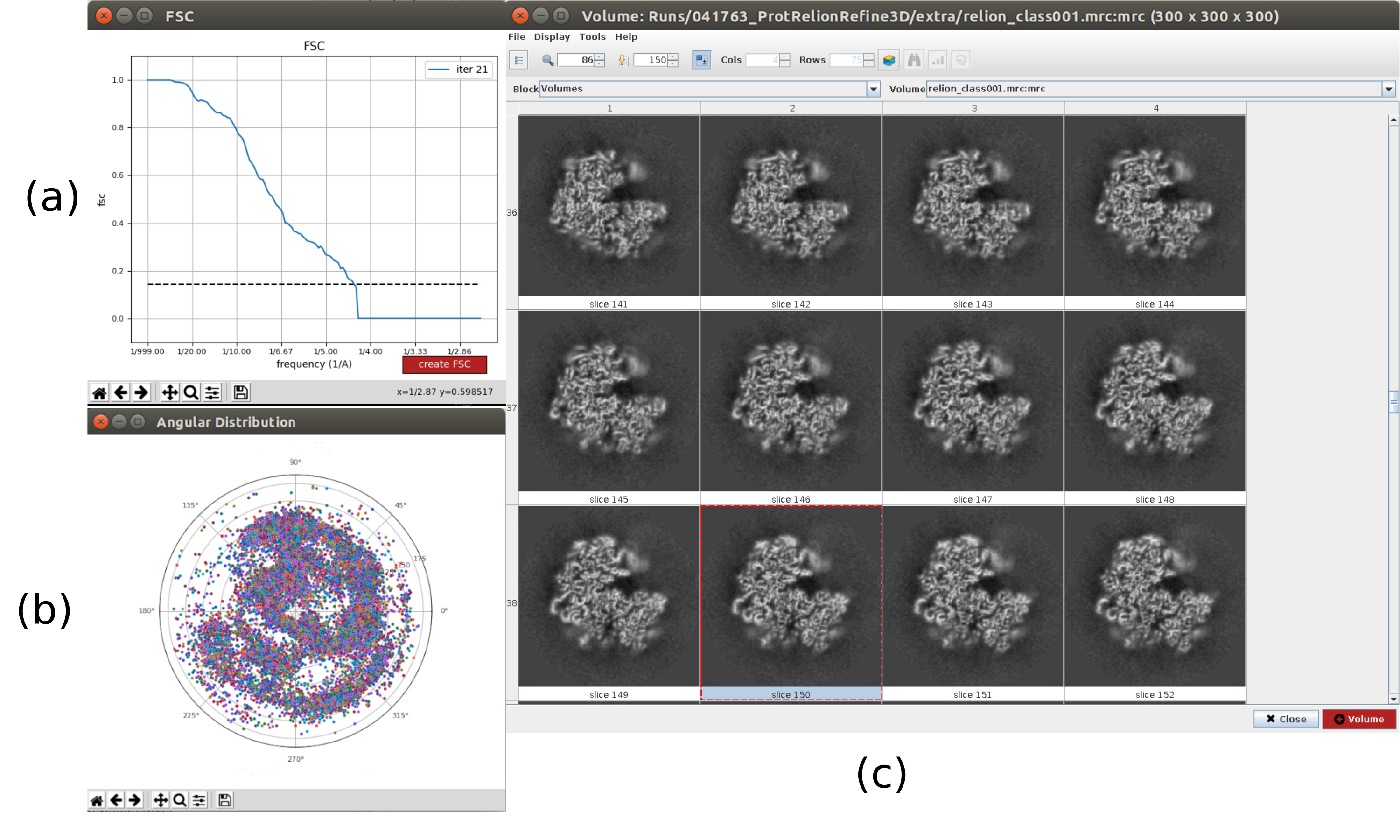
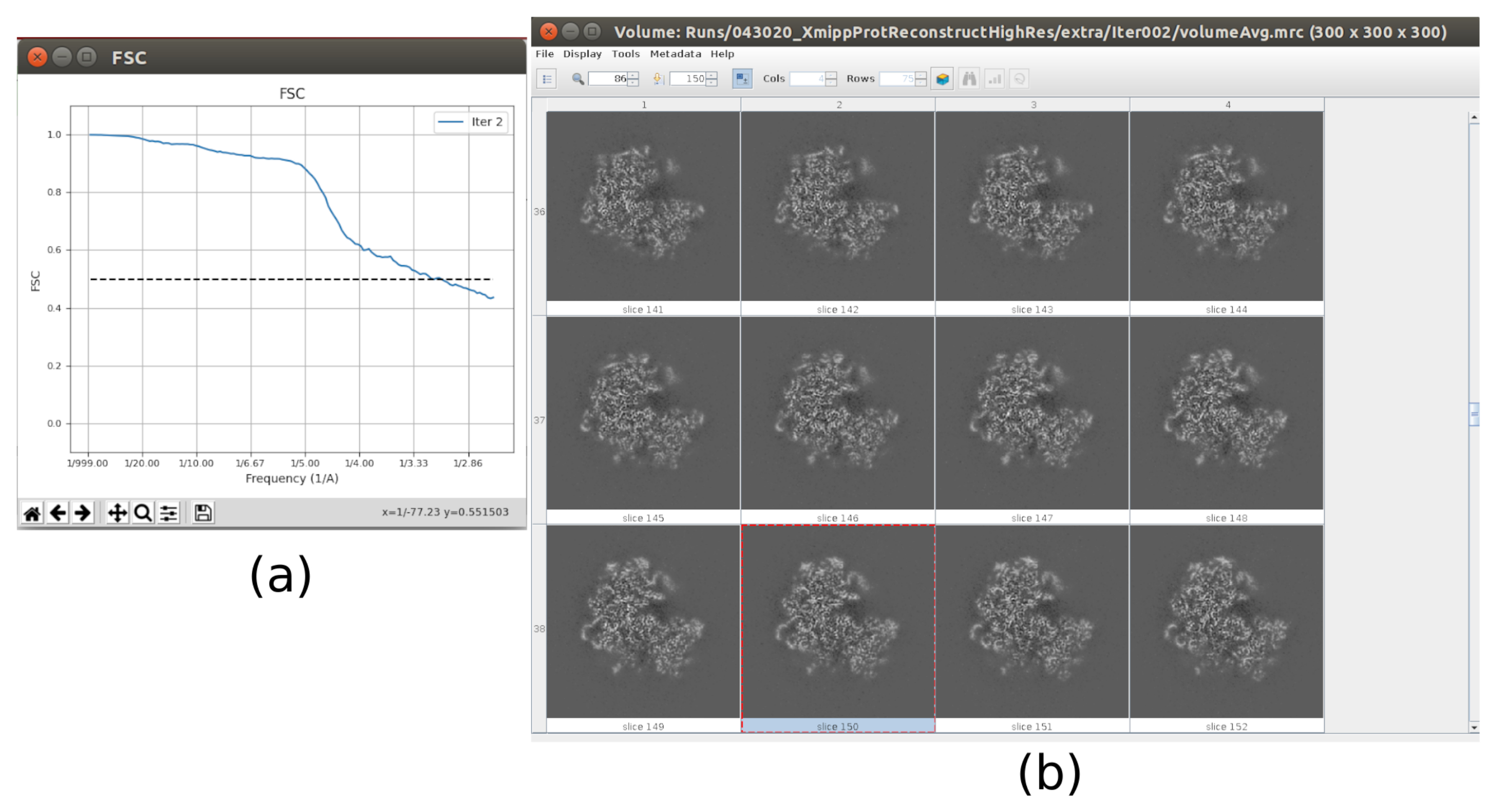
- Cliquez sur Analyser les résultats. Dans la nouvelle fenêtre, sélectionnez final comme Volume pour visualiser et cliquez sur Afficher le volume avec: tranches pour voir le volume obtenu. Vérifiez également la corrélation de coque de Fourier (FSC) en cliquant sur Afficher les tracés de résolution dans la fenêtre de résultats et sur la couverture angulaire dans Afficher la distribution angulaire : tracé 2D (Figure 7). Le volume reconstruit contient beaucoup plus de détails (probablement avec quelques zones floues dans la partie extérieure de la structure), et le FSC franchit le seuil de 0,143 autour de 4,5Å. La couverture angulaire couvre l’ensemble de la sphère 3D.
7.3D classification : découverte des états conformationnels
- En utilisant une approche consensuelle, si différents états conformationnels sont dans les données peut être découvert. Relion ouvert - Classification 3D10 (Figure supplémentaire 15). Comme particules d’entrée, utilisez celles qui viennent d’être obtenues en 6.10 et réglez le diamètre du masque de particules sur 402Å. Dans l’onglet Carte 3D de référence, utilisez comme volume d’entrée celui obtenu après l’étape 6.10, définissez Symétrie sur c1 et Filtre passe-bas initial sur 15Å. Enfin, dans l’onglet Optimisation, définissez le Nombre de classes sur 3. Exécutez-le.
- Vérifiez les résultats en cliquant sur Analyser les résultats, sélectionnez Afficher la classification dans Scipion. Les trois classes générées et quelques mesures intéressantes sont montrées. Les deux premières classes doivent avoir un nombre similaire d’images assignées (colonne de taille ) et se ressembler beaucoup, tandis que la troisième a moins d’images et une apparence plus floue. En outre, les rlnAccuracyRotations et rlnAccuracyTranslations devraient être nettement meilleures pour les deux premières classes. Sélectionnez les deux meilleures classes et cliquez sur le bouton Classes pour générer un sous-ensemble les contenant.
- Répétez les étapes 7.1 et 7.2 pour générer un deuxième groupe de bonnes classes. Les deux seront l’apport de l’outil de consensus.
- Ouvrez et exécutez xmipp3 - classes de consensus 3D et sélectionnez comme classes d’entrée les deux sous-ensembles générés dans les étapes précédentes.
- Cliquez sur Analyser les résultats. Le nombre de particules coïncidentes entre les classes est présenté : la première valeur est le nombre de particules coïncidentes dans la première classe du sous-ensemble 1 et la première classe du sous-ensemble 2, la deuxième valeur est le nombre de particules coïncidentes dans la première classe du sous-ensemble 1 et la deuxième classe du sous-ensemble 2, etc. Vérifiez que les particules sont assignées au hasard aux classes un ou deux, ce qui signifie que la méthode de classification 3D n’est pas en mesure de trouver des changements conformationnels. Compte tenu de ce résultat, l’ensemble des particules sera utilisé pour poursuivre le traitement.
8.3D raffinement : affiner les affectations angulaires d’une population homogène
- Encore une fois, appliquez une approche consensuelle à cette étape. Tout d’abord, ouvrez et exécutez pwem - sous-ensemble avec l’ensemble complet d’éléments comme sortie de 6.9, Faire un sous-ensemble aléatoire à Oui et Nombre d’éléments à 5000. Avec cela, un sous-ensemble d’images avec un alignement précédent pour entraîner la méthode utilisée dans l’étape suivante est créé.
- Ouvrez xmipp3 - alignement en profondeur, définissez les images d’entrée comme sortie de bonnes particules obtenues en 5,5 , Volume comme celui obtenu après 6,10, Jeu d’entraînement d’entrée comme celui créé à l’étape précédente, Résolution cible à 10Å et conservez les paramètres restants avec les valeurs par défaut (Figure supplémentaire 16). Cliquez sur Exécuter.
- Cliquez sur Analyser les résultats pour vérifier la distribution angulaire obtenue, où il n’y a pas de directions manquantes et la couverture angulaire s’améliore légèrement par rapport à celle de 6,10 (Figure 8).
- Ouvrir et exécuter xmipp3 - comparez les angles et sélectionnez comme particules d’entrée 1 la sortie de 6,9 et particules d’entrée 2 la sortie de 8,2, assurez-vous que le groupe de symétrie est c1. Cette méthode calcule l’accord entre xmipp3 - alignement profond et relion - affinement automatique 3D.
- Cliquez sur Analyser les résultats, la liste des particules, avec les différences obtenues dans les décalages et les angles, est affichée. Cliquez sur l’icône de la barre dans la partie supérieure de la fenêtre, une autre fenêtre s’ouvrira qui permet de faire des tracés des variables calculées. Sélectionnez _xmipp_angleDiff et cliquez sur Tracer pour voir une représentation des différences angulaires par particule. Faites de même avec _xmipp_shiftDiff. Dans ces chiffres, environ la moitié des particules concordent entre les deux méthodes (figure 9). Sélectionnez les particules dont les différences angulaires sont inférieures à 10º et créez un nouveau sous-ensemble.
- Maintenant, ouvrez xmipp3 - highres27 pour affiner localement les angles attribués. Tout d’abord, sélectionnez comme Images pleine grandeur les images obtenues à l’étape précédente, et comme volumes initiaux la sortie de 6,9, définissez Rayon de particule sur 150 pixels et Groupe symétrie sur c1. Dans l’onglet Affectation angulaire, définissez l’alignement de l’image sur Local, Nombre d’itérations sur 1 et Max. Résolution cible de 5Å/px (figure supplémentaire 17). Exécutez-le.
- Dans l’onglet Résumé , vérifiez que le volume de sortie est inférieur à 300x300x300 pixels et avec une taille de pixel légèrement supérieure.
- Cliquez sur Analyser les résultats pour voir les résultats obtenus. Cliquez sur Afficher les graphiques de résolution pour voir le FSC, et sur Afficher le volume : Reconstruit pour voir le volume obtenu (Figure supplémentaire 18). Un bon volume de résolution proche de 4-3,5Å est obtenu.
- Cliquez sur Afficher les particules de sortie et, dans la fenêtre avec la liste des particules, cliquez sur l’icône de la barre. Dans la nouvelle fenêtre, sélectionnez Taper comme histogramme, avec 100 bacs, sélectionnez _xmipp_cost’étiquette, puis appuyez sur Tracer (figure supplémentaire 19). De cette façon, l’histogramme de l’étiquette de coût est présenté, qui contient la corrélation de la particule avec la direction de projection sélectionnée pour elle. Dans ce cas, une fonction de densité unimodale est obtenue, ce qui est un signe de ne pas avoir de populations différentes dans l’ensemble des particules. Ainsi, tous seront utilisés pour poursuivre le raffinement
REMARQUE: En cas de voir une fonction de densité multimodale, l’ensemble des particules appartenant au maximum supérieur doit être sélectionné pour continuer le flux de travail uniquement avec elles. - Ouvrir et exécuter à nouveau xmipp3 - highres avec Continuer à partir d’une exécution précédente? sur Oui, définissez comme Images pleine grandeur celles obtenues après la version 8.5 et Sélectionnez l’exécution précédente avec l’exécution précédente de Xmipp Highres. Dans l’onglet Affectation angulaire , définissez l’alignement de l’image sur Local, avec 1 itération et 2,6Å/px comme résolution cible (pleine résolution).
- Maintenant, la sortie doit contenir un volume en pleine résolution (taille 300x300x300 pixels). Cliquez sur Analyser les résultats pour vérifier à nouveau le volume obtenu et le FSC, qui devrait maintenant être un volume haute résolution à environ 3Å (Figure 10).
9. Évaluation et post-traitement
- Ouvrez xmipp3 - MonoRes28 local. Cette méthode calculera la résolution localement. Définissez comme volume d’entrée celui obtenu après 8.10, définissez Souhaitez-vous utiliser des demi-volumes? sur Oui et Plage de résolution de 1 à 10Å. Exécutez-le.
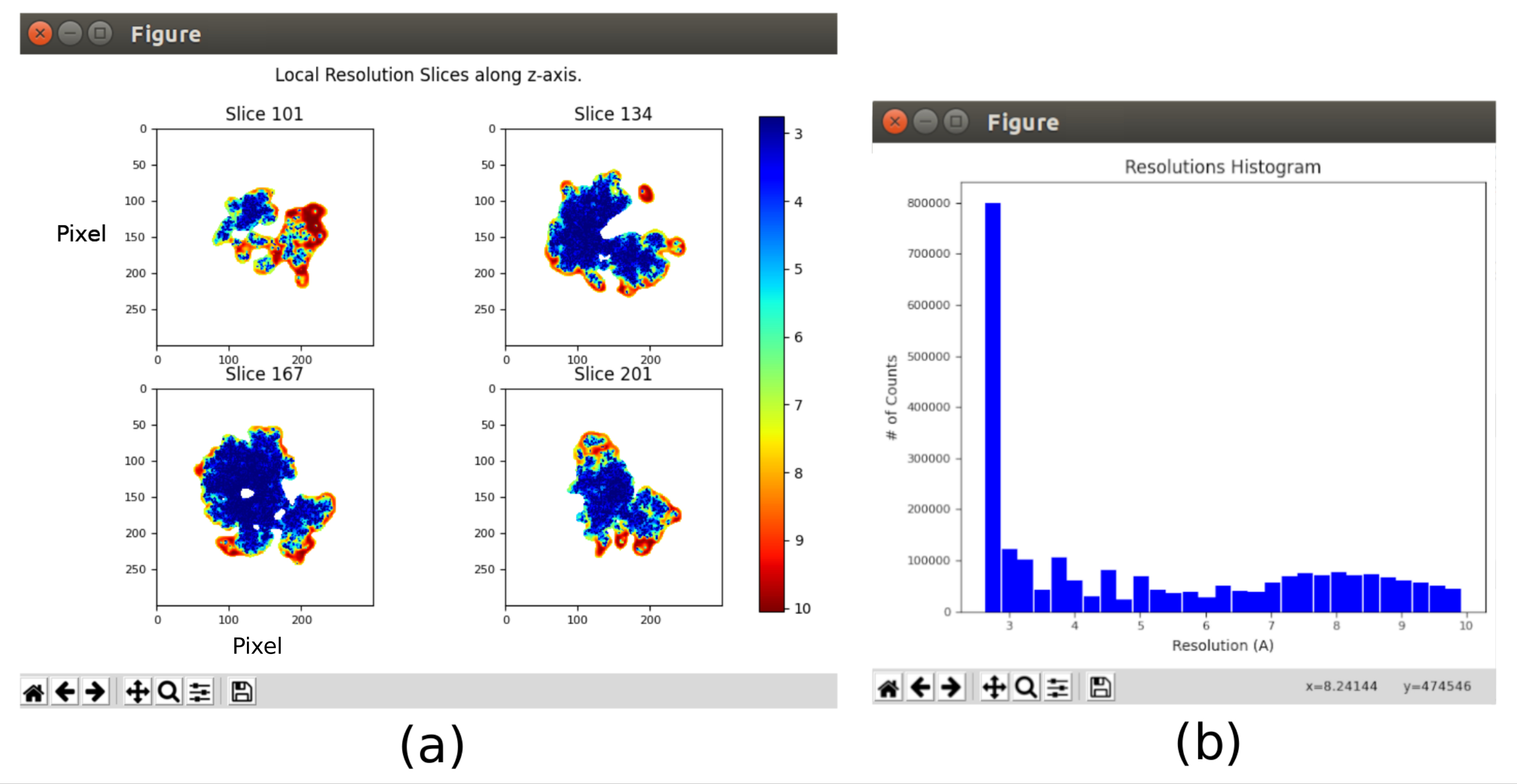
- Cliquez sur Analyser les résultats et sélectionnez Afficher l’histogramme de résolution et Afficher les tranches colorées (Figure 11). La résolution dans les différentes parties du volume est affichée. La plupart des voxels de la partie centrale de la structure doivent présenter des résolutions autour de 3Å, tandis que les pires résolutions sont atteintes dans les parties extérieures. En outre, un histogramme des résolutions par voxel est montré avec un pic autour de (même en dessous) de 3Å.
- Ouvrez et exécutez xmipp3 - affûtage localdeblur29 pour appliquer un affûtage. Sélectionnez comme carte d’entrée celle obtenue en 8.10, et comme carte de résolution celle obtenue à l’étape précédente avec MonoRes.
- Cliquez sur Analyser les résultats pour vérifier les volumes obtenus. Ouvrez le dernier, correspondant à la dernière itération de l’algorithme. Il est recommandé d’ouvrir le volume avec d’autres outils, tels que UCSF Chimera26, pour mieux voir les caractéristiques du volume en 3D (Figure 12).
- Enfin, ouvrez l’outil de validation inclus dans xmipp3 - validate overfitting30 qui montrera comment la résolution change avec le nombre de particules. Ouvrez-le et incluez comme particules d’entrée les particules obtenues à l’étape 8.5, réglez Calculer le bruit lié à la résolution? sur Oui, avec le volume de référence 3D initial comme sortie de 8.10. Dans Options avancées, définissez le Nombre de particules sur « 500 1000 1500 2000 3000 5000 10000 15000 20000 » (Figure supplémentaire 20). Exécutez-le.
- Cliquez sur Analyser les résultats. Deux tracés apparaîtront (Figure 13) avec l’évolution de la résolution, dans la ligne verte, au fur et à mesure que le nombre de particules utilisées dans la reconstruction augmentera. La ligne rouge représente la résolution obtenue avec une reconstruction du bruit gaussien aligné. La résolution s’améliore avec le nombre de particules et une grande différence de la reconstruction à partir de particules par rapport à celle du bruit est observée, ce qui est un indicateur d’avoir des particules avec de bonnes informations structurelles.
- À partir des résultats précédents, un ajustement d’un modèle dans le volume post-traité a pu être effectué, ce qui permettrait de découvrir les structures biologiques de la macromolécule.
Résultats
Nous avons utilisé l’ensemble de données du Plasmodium falciparum 80S Ribosome (entrée EMPIAR: 10028, entrée EMDB: 2660) pour effectuer le test et, avec le protocole Scipion présenté dans la section précédente, un volume reconstruit en 3D haute résolution de la macromolécule dans cet exemple particulier a été atteint, en commençant par les informations recueillies par le microscope qui consistent en des images très bruyantes contenant des projections 2D dans n’importe quelle orientation du spécimen.
Les principaux résultats obtenus après l’exécution de l’ensemble du protocole sont présentés à la Figure 10, à la Figure 11 et à la Figure 12. La figure 10 représente le volume 3D obtenu avant post-traitement. Dans la figure 10a, on peut voir un FSC de 3 Å, qu’il est très proche de la limite de Nyquist (avec des données avec une taille de pixel de 1,34 Å, la limite de Nyquist est de 2,6 Å). La figure 10b montre quelques tranches du volume 3D reconstruit avec des niveaux élevés de détails et des structures bien définies. Dans la figure 11, les résultats après analyse locale de la résolution du volume 3D obtenu sont présentés. On peut voir que la plupart des voxels de la structure atteignent une résolution inférieure à 3 Å, principalement ceux situés dans la partie centrale de la structure. Cependant, la partie extérieure affiche des résolutions pires, ce qui est cohérent avec le flou apparaissant dans ces zones dans les tranches de la figure 10b. La figure 12 montre la même carte 3D après post-traitement qui est capable de mettre en évidence les fréquences plus élevées du volume, révélant plus de détails et améliorant la représentation, ce qui peut être vu en particulier dans la présentation 3D de la figure 12c.
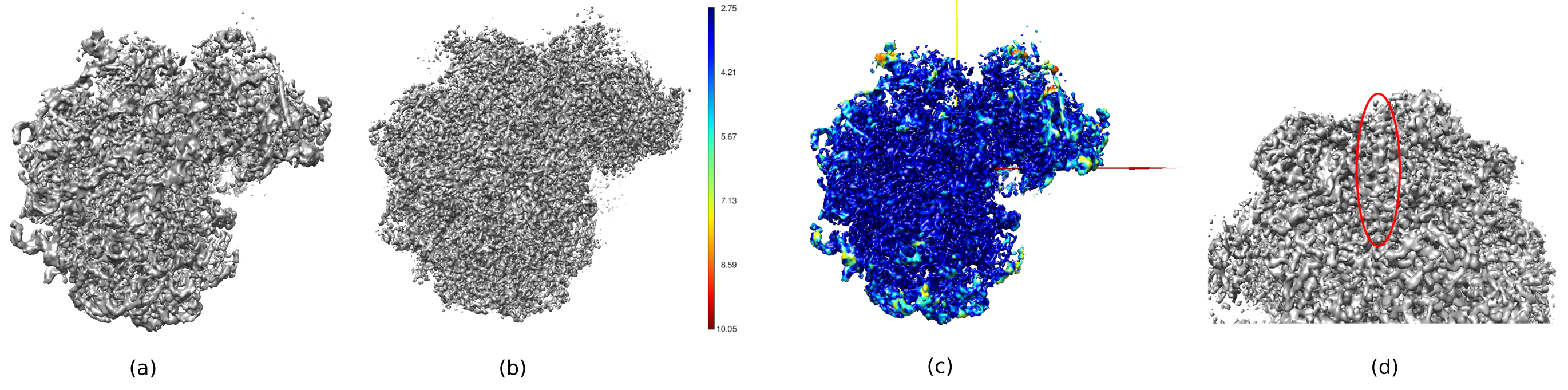
Dans la figure 14, Chimera26 a été utilisé pour voir une représentation 3D du volume obtenu (figure 14a), du post-traitement (figure 14b) et de la carte de résolution (figure 14c), colorée avec le code couleur des résolutions locales. Cela peut donner encore plus d’informations sur la structure obtenue. Cet outil est très utile pour avoir un aperçu de la qualité du volume obtenu, car de très petits détails dans l’ensemble du contexte 3D de la structure peuvent être vus. Lorsque la résolution obtenue est suffisante, même certaines parties biochimiques de la structure peuvent être trouvées (par exemple, les hélices alpha dans la figure 14d. Dans cette figure, il faut souligner la haute résolution obtenue dans toutes les parties centrales de la structure 3D, qui peuvent être vues comme les zones bleu foncé de la figure 14c.
Tous les résultats précédents ont été obtenus grâce à une bonne performance de l’ensemble du protocole, mais ce n’est peut-être pas le cas. Il existe plusieurs façons d’identifier un mauvais comportement. Dans le cas le plus général, cela se produit lorsque la structure obtenue a une faible résolution et qu’elle n’est pas en mesure d’évoluer vers une meilleure. La figure 15 en est un exemple. Un volume flou (Figure 15c) entraîne un faible FSC, ce qui peut être vu dans la courbe FSC (Figure 15a) et l’histogramme de l’estimation locale (Figure 15b). Cet exemple a été généré à l’aide d’une méthode d’affinement 3D avec des données d’entrée incorrectes, car il s’attendait à ce que certaines propriétés spécifiques dans l’ensemble d’entrée de particules ne remplissent pas. Comme on peut le voir, il est toujours très important de savoir comment les différentes méthodes s’attendent à recevoir les données et à les préparer correctement. En général, lorsqu’une sortie comme celle de la figure 15 est obtenue, il peut y avoir un problème dans le flux de travail de traitement ou les données sous-jacentes.
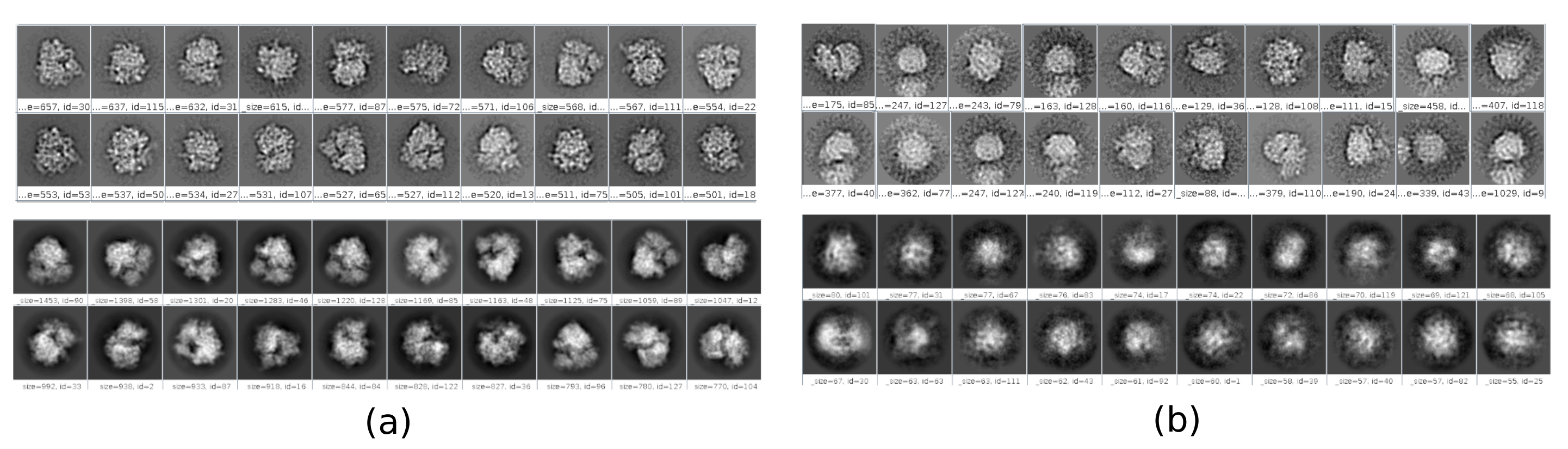
Il existe plusieurs points de contrôle le long du flux de travail qui peuvent être analysés pour savoir si le protocole évolue correctement ou non. Par exemple, juste après la cueillette, plusieurs des méthodes discutées précédemment peuvent classer les particules et donner un score pour chacune d’elles. Dans le cas d’avoir de mauvaises particules, ces méthodes permettent de les identifier et de les éliminer. En outre, la classification 2D peut être un bon indicateur d’avoir un mauvais ensemble de particules. La figure 16 montre un exemple d’un si mauvais ensemble. Dans la figure 16a, les bonnes classes contenant certains détails de la structure sont montrées, tandis que la figure 16b montre les mauvaises classes, qui sont bruyantes ou non centrées, dans ce dernier cas, on peut voir que le prélèvement était incorrect et que deux particules semblent apparaître ensemble. Un autre point de contrôle est l’estimation initiale du volume, la figure 17 montre un exemple de bonnes (figure 17a) et de mauvaises (figure 17b) estimations initiales. La mauvaise estimation a été créée à l’aide d’une configuration incorrecte pour la méthode. Il faut tenir compte du fait que toutes les configurations doivent être effectuées avec soin, en choisissant de manière appropriée chaque paramètre en fonction des données analysées. En cas de ne pas avoir de carte avec quelques informations structurelles minimales, le raffinement suivant ne sera pas en mesure d’obtenir une bonne reconstruction.
Lorsque le problème est une mauvaise acquisition, dans laquelle les films ne préservent pas les informations structurelles, il sera impossible d’en extraire de bonnes particules et d’obtenir un traitement réussi. Dans ce cas, plus de films devraient être collectés pour obtenir une reconstruction 3D haute résolution. Mais, si ce n’est pas le cas, il existe plusieurs façons de gérer les problèmes tout au long du flux de travail de traitement. Si la cueillette n’est pas assez bonne, il existe plusieurs façons d’essayer de la réparer, par exemple, répéter la cueillette, utiliser différentes méthodes ou essayer de choisir manuellement plus de particules pour aider les méthodes à en tirer des leçons. Pendant la classification 2D, si seulement quelques classes sont bonnes, pensez également à répéter le processus de prélèvement. Dans l’estimation initiale du volume, essayez d’utiliser plusieurs méthodes si certaines d’entre elles donnent des résultats inexacts. Il en va de même pour le raffinement 3D. Suivant ce raisonnement, dans ce manuscrit, plusieurs outils de consensus ont été présentés, ce qui pourrait être très utile pour éviter les problèmes et poursuivre le traitement avec des données précises. Grâce à l’utilisation d’un consensus entre plusieurs méthodes, nous pouvons éliminer les données difficiles à sélectionner, classer, aligner, etc., ce qui est probablement un indicateur de données médiocres. Cependant, si plusieurs méthodes sont en mesure de s’accorder dans la sortie générée, ces données contiennent probablement des informations précieuses avec lesquelles poursuivre le traitement.
Nous encourageons le lecteur à télécharger plus d’ensembles de données et à essayer de les traiter en suivant les recommandations présentées dans ce manuscrit et à créer un flux de travail similaire combinant des packages de traitement à l’aide de Scipion. Essayer de traiter un ensemble de données est le meilleur moyen d’apprendre la puissance des outils de traitement disponibles dans l’état de l’art dans Cryo-EM, de connaître les meilleures règles pour surmonter les inconvénients possibles apparaissant pendant le traitement et d’améliorer les performances des méthodes disponibles dans chaque cas de test spécifique.

Graphique 1. Résultat de l’alignement du film. (a) La fenêtre principale des résultats, avec une liste de toutes les micrographies générées et des informations supplémentaires: la densité spectrale de puissance, la trajectoire de l’alignement estimé en coordonnées polaires, la même en coordonnées cartésiennes, le nom de fichier de la micrographie générée. b) La trajectoire d’alignement représentée en coordonnées cartésiennes. c) La micrographie générée. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 2. Estimation CTF avec résultat Ctffind. La fenêtre principale avec les résultats comprend un chiffre avec le PSD estimé (dans un coin) avec le PSD provenant des données, et plusieurs paramètres de défocalisation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 3. Fenêtres de prélèvement manuel avec Xmipp. a) La fenêtre principale avec la liste des micrographies à traiter et d’autres paramètres. b) Prélèvement manuel de particules à l’intérieur d’une région d’une micrographie. (c) et (d) Particules prélevées automatiquement à superviser pour créer un ensemble de particules d’entraînement pour la méthode de prélèvement automatique Xmipp. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 4. Sélection consensuelle profonde avec le résultat Xmipp. Le paramètre zScoreDeepLearning donne du poids à la bonté d’une particule et il est la clé pour découvrir les mauvaises particules. (a) Les valeurs zScores les plus basses sont associées à des artefacts. b) Les zScores les plus élevés sont associés à des particules contenant la macromolécule. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 5. Classification 2D avec résultat Cryosparc. Les classes générées (moyennes de sous-ensembles de particules provenant de la même orientation) sont affichées. Plusieurs bonnes classes sélectionnées en rouge (avec un certain niveau de détail) et quelques mauvaises classes non sélectionnées (classes bruyantes et non centrées). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 6. Volume initial 3D avec résultat consensuel en essaim. Vue du volume initial 3D obtenu après l’exécution de l’outil de consensus xmipp3 - swarm consensus, en utilisant les estimations de volume initiales 3D précédentes de Xmipp et Relion. a) Le volume est représenté par des tranches. b) Visualisation 3D du volume. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 7. Raffinement d’un volume initial 3D avec résultat Relion. a) Courbe FSC obtenue, franchissant le seuil à 4,5Å, approximativement. b) Couverture angulaire représentée sous forme de vue supérieure de la sphère 3D. Dans ce cas, comme il n’y a pas de symétrie, les particules assignées doivent couvrir toute la sphère. c) Volume raffiné représenté par des tranches. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 8. Alignement 3D basé sur le deep learning avec résultat Xmipp. Les résultats générés par xmipp3 - méthode d’alignement profond pour l’alignement 3D. a) L’affectation angulaire de chaque particule sous forme de matrice de transformation. b) La couverture angulaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 9. Résultat du consensus sur l’alignement 3D. a) Liste des particules présentant les différences obtenues dans les paramètres de décalage et d’angle. b) Graphique des différences angulaires par particule. c) Graphique de la différence de décalage par particule. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 10. Itération finale du résultat du raffinement 3D. a) Courbe FSC. b) Volume obtenu en pleine résolution par tranches. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 11. Analyse de résolution locale avec résultat Xmipp. Résultats de la méthode xmipp3 - MonoRes local. (a) Certaines tranches représentatives colorées avec la valeur de résolution par voxel, comme indiqué dans le code couleur. b) Histogramme de résolution locale. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 12. Affûtage avec résultat Xmipp. Résultats de xmipp3 - méthode d’affûtage localdeblur. a) Liste des volumes obtenus par itération. b) Volume 3D obtenu après la dernière itération représenté par des tranches. c) Une représentation 3D du volume final. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 13. Valider l’outil de surajustement dans le résultat Xmipp. Résultats de xmipp3 - validation du surajustement. La ligne verte correspond à la reconstruction à partir des données, la ligne rouge au bruit. a) Inverse de la résolution au carré avec le logarithme du nombre de particules. b) Résolution avec le nombre de particules. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 14. Plusieurs représentations 3D du volume obtenu. a) Volume prétraité. b) Volume post-traité. (c) Résolution locale, les voxels bleu foncé sont ceux avec une résolution plus élevée (2,75Å) et les voxels rouge foncé sont ceux avec une résolution inférieure (10,05Å). d) Zoomez sur le volume post-traité où l’on peut voir une hélice alpha (ovale rouge). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 15. Exemple d’une mauvaise reconstruction 3D. a) Courbe FSC avec une chute brutale et franchissant le seuil à basse résolution. b) Histogramme de résolution locale. c) Volume 3D par tranches. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 16. Exemple de classes 2D. a) Bonnes classes montrant un certain niveau de détail. (b) Mauvaises classes contenant du bruit et des artefacts (partie supérieure obtenue avec Xmipp, partie inférieure avec CryoSparc). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 17. Exemple de volume initial 3D avec différentes qualités. a) Bon volume initial où la forme de la macromolécule peut être observée. b) Mauvais volume initial lorsque la forme obtenue est complètement différente de celle attendue. Veuillez cliquer ici pour voir une version agrandie de cette figure.
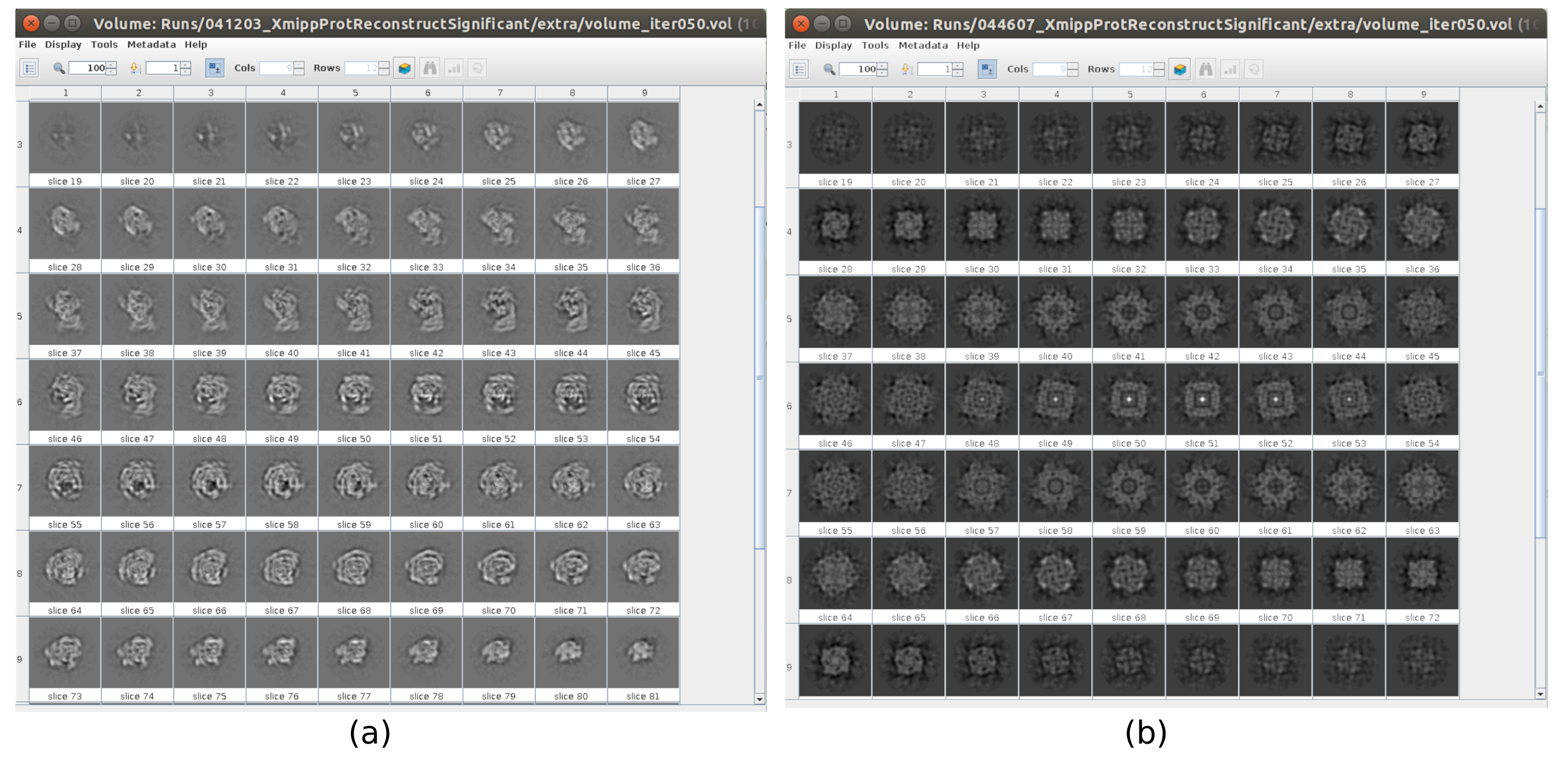{kind=link}
Figure supplémentaire 1. Création d’un projet Scipion. Fenêtre affichée par Scipion où un ancien projet peut être sélectionné ou un nouveau peut être créé en donnant un nom et un emplacement pour ce projet. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 2. Méthode d’importation de films. Fenêtre affichée par Scipion lorsque pwem - importer des films est ouvert. Ici, les principaux paramètres d’acquisition doivent être inclus pour permettre aux films disponibles d’être traités dans Scipion. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 3. Méthode d’alignement de film. Fenêtre affichée par Scipion lorsque xmipp3 - alignement optique est utilisé. Les films d’entrée, la plage d’images considérées pour l’alignement et certains autres paramètres pour traiter les films doivent être remplis. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 4. Méthode d’estimation CTF avec Ctffind. Le formulaire dans Scipion avec tous les champs nécessaires pour exécuter le programme Ctffind. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 5. Assistant dans Scipion. Un assistant pour aider l’utilisateur à remplir certains paramètres du formulaire. Dans ce cas, l’assistant doit remplir le champ de résolution dans la méthode grigoriefflab - ctffind . Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 6. Méthode de raffinement CTF avec Xmipp. La forme de xmipp3 - estimation ctf avec tous les paramètres pour faire un raffinement d’un CTF précédemment estimé. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 7. Méthode de micrographies préprocessus. La forme de xmipp3 - micrographies de prétraitement qui permet d’effectuer certaines opérations sur eux. Dans cet exemple, Supprimer les pixels défectueux et échantillonner les micrographies est utile. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 8. Méthode de cueillette avec Cryolo. Formulaire permettant d’exécuter la méthode de prélèvement Cryolo à l’aide d’un réseau préentraîné. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 9. Méthode de sélection de consensus avec Xmipp. La forme de xmipp3 - deep consensus picking basé sur le deep learning pour calculer un consensus de coordonnées, en utilisant un réseau préentraîné sur plusieurs ensembles de coordonnées obtenues avec différentes méthodes de picking. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 10. Méthode d’extraction de particules. Onglets d’entrée et de prétraitement de xmipp3 - particules d’extraction. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 11 .3D méthode du volume initial avec Xmipp. La forme de la méthode xmipp3 - reconstruire significatif pour obtenir une carte 3D initiale. Les onglets Entrée et Critères s’affichent. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 12. Méthode de redimensionnement du volume. Forme permettant de recadrer ou de redimensionner un volume. Dans cet exemple, cette méthode est utilisée pour générer un volume en taille réelle après xmipp3 - reconstruire significatif. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 13 .3D volume initial avec le résultat de Relion. Une vue du volume initial 3D obtenu avec relion - méthode de modèle initial 3D par tranches. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 14. Raffinement du volume initial avec Relion. La forme de la méthode relion - 3D auto-refine. Dans cet exemple, il a été utilisé pour affiner un volume initial estimé après consensus. Les onglets Carte 3D Entrée et Référence s’affichent. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 15 .3D méthode de classification. Forme de relion - classification 3D. Les onglets Entrée, Carte 3D de référence et Optimisation sont affichés. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 16 .3D alignement basé sur une méthode d’apprentissage profond. Formulaire ouvert pour la méthode xmipp3 - alignement profond. Ici, il est nécessaire d’entraîner un réseau avec un ensemble d’entraînement, puis ce réseau prédira l’affectation angulaire par particule. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 17 .3D méthode de raffinement. Forme de la méthode xmipp3 - highres . Onglets Entrée et Affectation angulaire sont affichés. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 18. Première itération du résultat du raffinement 3D. a) Courbe FSC. b) Volume obtenu (d’une taille inférieure à la résolution totale) représenté sous forme de tranches. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 19. Première itération de l’analyse de corrélation de raffinement 3D. Une nouvelle fenêtre apparaît en cliquant sur l’icône de la barre dans la partie supérieure de la fenêtre avec la liste des particules. Dans la fenêtre Colonnes du tracé , un histogramme du paramètre estimé souhaité peut être créé. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 20. Outil de surajustement de validation. Forme de xmipp3 - valider la méthode de surajustement . Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Actuellement, cryo-EM est un outil clé pour révéler la structure 3D des échantillons biologiques. Lorsque de bonnes données sont collectées au microscope, les outils de traitement disponibles nous permettront d’obtenir une reconstruction 3D de la macromolécule étudiée. Le traitement des données Cryo-EM est capable d’atteindre une résolution quasi atomique, ce qui est essentiel pour comprendre le comportement fonctionnel d’une macromolécule et est également crucial dans la découverte de médicaments.
Scipion est un logiciel qui permet de créer l’ensemble du flux de travail en combinant les packages de traitement d’image les plus pertinents de manière intégrative, ce qui contribue à la traçabilité et à la reproductibilité de l’ensemble du flux de travail de traitement d’image. Scipion fournit un ensemble très complet d’outils pour effectuer le traitement; cependant, l’obtention de reconstructions à haute résolution dépend entièrement de la qualité des données acquises et de la manière dont ces données sont traitées.
Pour obtenir une reconstruction 3D haute résolution, la première exigence est d’obtenir de bons films à partir du microscope, qui préservent les informations structurelles à haute résolution. Si ce n’est pas le cas, le workflow ne sera pas en mesure d’extraire des informations haute définition des données. Ensuite, un flux de travail de traitement réussi devrait être capable d’extraire des particules qui correspondent vraiment à la structure et de trouver les orientations de ces particules dans l’espace 3D. Si l’une des étapes du flux de travail échoue, la qualité du volume reconstruit sera dégradée. Scipion permet d’utiliser différents packages dans n’importe quelle étape de traitement, ce qui aide à trouver l’approche la plus adéquate pour traiter les données. De plus, grâce à la disponibilité de nombreux packages, des outils de consensus, qui augmentent la précision en trouvant un accord dans les résultats estimés de différentes méthodes, peuvent être utilisés. En outre, il a été discuté en détail dans la section Résultats représentatifs plusieurs outils de validation et comment identifier des résultats précis et inexacts à chaque étape du flux de travail, pour détecter les problèmes potentiels et comment essayer de les résoudre. Il existe plusieurs points de contrôle le long du protocole qui pourraient aider à réaliser si le protocole fonctionne correctement ou non. Certains des plus pertinents sont: le picking, la classification 2D, l’estimation initiale du volume et l’alignement 3D. La vérification des entrées, la répétition de l’étape avec une méthode différente ou l’utilisation du consensus sont des options disponibles dans Scipion que l’utilisateur peut utiliser pour trouver des solutions lorsque des problèmes apparaissent.
En ce qui concerne les approches précédentes de l’intégration de paquets dans le domaine Cryo-EM, Appion31 est le seul qui permet une intégration réelle de différents progiciels. Cependant, Appion est étroitement lié à Leginon32, un système de collecte automatisée d’images à partir de microscopes électroniques. La principale différence avec Scipion est que le modèle de données et le stockage sont moins couplés. De cette façon, pour créer un nouveau protocole dans Scipion, seul un script Python doit être développé. Toutefois, dans Appion, le développeur doit écrire le script et modifier la base de données sous-jacente. En résumé, Scipion a été développé pour simplifier la maintenance et l’extensibilité.
Nous avons présenté dans ce manuscrit un flux de travail complet pour le traitement Cryo-EM, en utilisant l’ensemble de données de cas réels du ribosome Plasmodium falciparum 80S (entrée EMPIAR: 10028, entrée EMDB: 2660). Les étapes couvertes et discutées ici peuvent être résumées comme l’alignement du film, l’estimation CTF, la sélection des particules, la classification 2D, l’estimation initiale de la carte, la classification 3D, le raffinement 3D, l’évaluation et le post-traitement. Différents packages ont été utilisés et des outils de consensus ont été appliqués dans plusieurs de ces étapes. Le volume final reconstruit en 3D a atteint une résolution de 3 Å et, dans le volume post-traité, certaines structures secondaires peuvent être distinguées, comme les hélices alpha, ce qui aide à décrire comment les atomes sont disposés dans l’espace.
Le flux de travail présenté dans ce manuscrit montre comment Scipion peut être utilisé pour combiner différents packages Cryo-EM de manière simple et intégrative afin de simplifier le traitement et d’obtenir des résultats plus fiables en même temps.
À l’avenir, le développement de nouvelles méthodes et de nouveaux packages continuera de croître et des logiciels comme Scipion pour les intégrer facilement seront encore plus importants pour les chercheurs. Les approches consensuelles seront plus pertinentes même dans ce cas, lorsque de nombreuses méthodes avec des bases différentes seront disponibles, ce qui aidera à obtenir des estimations plus précises de tous les paramètres impliqués dans le processus de reconstruction dans Cryo-EM. Le suivi et la reproductibilité sont essentiels dans le processus de recherche et plus faciles à réaliser avec Scipion grâce à un cadre commun pour l’exécution de flux de travail complets.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs souhaitent reconnaître le soutien économique du ministère espagnol de la Science et de l’Innovation par le biais de subventions: PID2019-104757RB-I00/AEI/10.13039/501100011033, de la « Comunidad Autónoma de Madrid » par le biais d’une subvention: S2017/BMD-3817, Instituto de Salud Carlos III, PT17/0009/0010 (ISCIII-SGEFI/ERDF), de l’Union européenne (UE) et d’Horizon 2020 par le biais d’une subvention: INSTRUCT - ULTRA (INFRADEV-03-2016-2017, Proposition: 731005), EOSC Life (INFRAEOSC-04-2018, Proposition: 824087), iNEXT - Discovery (Proposition : 871037) et HighResCells (ERC - 2018 - SyG, Proposition : 810057). Le projet qui a donné lieu à ces résultats a reçu le soutien d’une bourse de la Fondation « la Caixa » (ID 100010434). Le code de bourse est LCF/BQ/DI18/11660021. Ce projet a reçu un financement du programme de recherche et d’innovation Horizon 2020 de l’Union européenne dans le cadre de la convention de subvention Marie Skłodowska-Curie n° 713673. Les auteurs reconnaissent le soutien et l’utilisation des ressources d’Instruct, un projet Landmark ESFRI.
matériels
| Name | Company | Catalog Number | Comments |
| no material is used in this article | - | - | - |
Références
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Kühlbrandt, W. The Resolution Revolution. Science. 343 (6178), 1443-1444 (2014).
- . 1.15 A structure of human apoferritin obtained from Titan Mono- BCOR microscope Available from: https://www.rcsb.org/structure/7A6A (2021)
- Arnold, S. A., et al. Miniaturizing EM Sample Preparation: Opportunities, Challenges, and "Visual Proteomics". PROTEOMICS. 18 (5-6), 1700176 (2018).
- Faruqi, A. R., McMullan, G. Direct imaging detectors for electron microscopy. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 878, 180-190 (2018).
- Vilas, J. L., et al. Advances in image processing for single-particle analysis by electron cryomicroscopy and challenges ahead. Current Opinion in Structural Biology. 52, 127-145 (2018).
- Martinez, M., et al. Integration of Cryo-EM Model Building Software in Scipion. Journal of Chemical Information and Modeling. 60, 2533-2540 (2020).
- de la Rosa-Trevín, J. M., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195, 93-99 (2016).
- de la Rosa-Trevín, J. M., et al. Xmipp 3.0: an improved software suite for image processing in electron microscopy. Journal of Structural Biology. 184, 321-328 (2013).
- Scheres, S. H. W. . Methods in Enzymology. The Resolution Revolution: Recent Advances In cryoEM. , 125-157 (2016).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. 14, 290-296 (2017).
- Ludtke, S. J. 3-D structures of macromolecules using single-particle analysis in EMAN. Methods in Molecular Biology. 673, 157-173 (2010).
- Shaikh, T. R., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3, 1941-1974 (2008).
- Wagner, T., et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications Biology. 2, (2019).
- Mindell, J. A., Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology. 142, 334-347 (2003).
- Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography. 67, 235-242 (2011).
- Liebschner, D., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D. 75, 861-887 (2019).
- Wong, W., et al. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 3, 03080 (2014).
- Abrishami, V., et al. Alignment of direct detection device micrographs using a robust Optical Flow approach. Journal of Structural Biology. 189, 163-176 (2015).
- Sorzano, C. O. S., Jonic, S., Nunez Ramirez, R., Boisset, N., Carazo, J. M. Fast, robust and accurate determination of transmission electron microscopy contrast transfer function. Journal of Structural Biology. 160, 249-262 (2007).
- Abrishami, V., et al. A pattern matching approach to the automatic selection of particles from low-contrast electron micrographs. Bioinformatics. 29, 2460-2468 (2013).
- Sanchez-Garcia, R., Segura, J., Maluenda, D., Carazo, J. M., Sorzano, C. O. S. Deep Consensus, a deep learning-based approach for particle pruning in cryo-electron microscopy. IUCrJ. 5, 854-865 (2018).
- Sorzano, C. O. S., et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. Journal of Structural Biology. 171, 197-206 (2010).
- Sorzano, C. O. S., et al. A statistical approach to the initial volume problem in Single Particle Analysis by Electron Microscopy. Journal of Structural Biology. 189, 213-219 (2015).
- Sorzano, C. O. S., et al. Swarm optimization as a consensus technique for Electron Microscopy Initial Volume. Applied Analysis and Optimization. 2, 299-313 (2018).
- Pettersen, E. F., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 25, 1605-1612 (2004).
- Sorzano, C. O. S., et al. A new algorithm for high-resolution reconstruction of single particles by electron microscopy. Journal of Structural Biology. 204, 329-337 (2018).
- Vilas, J. L., et al. MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure. 26, 337-344 (2018).
- Ramirez-Aportela, E., et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics. 36, 765-772 (2020).
- Heymann, J. B. Validation of 3D EM Reconstructions: The Phantom in the Noise. AIMS Biophys. 2, 21-35 (2015).
- Lander, G. C., et al. Appion: An integrated, database-drive pipeline to facilitate EM image processing. Journal of Structural Biology. 166, 95-102 (2009).
- Suloway, C., et al. Automated molecular microscopy: The new Leginon system. Journal of Structural Biology. 151, 41-60 (2005).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.