
Method Article
Scipion을 가진 극저온 EM 및 단일 입자 분석
요약
극저온 전자 현미경 검사법의 단일 입자 분석은 고해상도에서 생물학적 앙상블의 구조를 결정하는 데 사용되는 주요 기술 중 하나입니다. Scipion은 현미경에 의해 획득된 정보를 처리하고 생물학적 표본의 3D 재구성을 달성하기 위해 전체 파이프라인을 만드는 도구를 제공합니다.
초록
극저온 전자 현미경 검사는 거의 원자 분해능에서 거대 분자의 구조적 정보를 밝히기 위한 생물학적 연구에서 가장 중요한 도구 중 하나가 되었습니다. 단일 입자 분석에서, 유리화 된 샘플은 전자 빔에 의해 심화되고 현미경 기둥의 끝에 검출기는 그 샘플의 영화를 생성한다. 이 영화에는 임의 방향으로 동일한 파티클의 수천 개의 이미지가 포함되어 있습니다. 데이터는 최종 3D 재구성 볼륨을 얻기 위해 여러 단계로 이미지 처리 워크플로우를 거쳐야 합니다. 이미지 처리 워크플로우의 목표는 연구 중인 시편을 재구성할 수 있는 획득 매개 변수를 식별하는 것입니다. Scipion은 통합 프레임워크에서 여러 이미지 처리 패키지를 사용하여 이 워크플로우를 만드는 모든 도구를 제공하여 결과의 추적성을 제공합니다. 이 문서에서Scipion의 전체 이미지 처리 워크플로우가 실제 테스트 사례에서 나오는 데이터와 함께 제시되고 논의되어 현미경에서 얻은 영화에서 고해상도 최종 3D 재구성으로 이동하는 데 필요한 모든 세부 정보를 제공합니다. 또한, 결합 방법을 허용하는 합의 도구를 사용하고 워크플로의 모든 단계에서 결과를 확인하여 얻은 결과의 정확성을 향상시키는 힘에 대해 논의합니다.
서문
극저온 전자 현미경 검사법(cryo-EM)에서, 유리화 된 냉동 수화 표본의 단일 입자 분석 (SPA)은 분자 상호 작용및 생물학적 앙상블의 기능을 이해할 수 있기 때문에 생물학적 거대 분자에 대한 이미징의 가장 널리 사용되고 성공적인 변형 중 하나입니다1. 이것은 "해상도 혁명"2 를 초래하고 거의 원자 해상도를 가진 생물학 3D 구조물의 성공적인 결정을 허용한 이 화상 진찰 기술의 최근 어드밴스 덕분에 입니다. 현재 SPA 극저온EM에서 달성한 가장 높은 해상도는 아포페리틴3 (EMDB 입력: 11668)의 경우 1.15Å였습니다. 이러한 기술적 진보는 샘플 제제4, 이미지 수집5 및 이미지 처리 방법의 개선을 포함한다6. 이 문서는 이 마지막 점에 중점을 두습니다.
간단히, 영상 처리 방법의 목표는 현미경의 이미징 과정을 반전시키고 연구 하에 생물학적 표본의 3D 구조를 복구하기 위해 모든 획득 파라미터를 식별하는 것이다. 이들 파라미터는 카메라의 게인, 빔 유도 운동, 현미경의 수차(주로 디포커스), 각 입자의 3D 각도 방향 및 번역, 및 형태 적 변화를 갖는 시편이 있는 경우 의 형성 상태입니다. 그러나, 파라미터의 수는 매우 높고 극저온-EM은 방사선 손상을 피하기 위해 저용량 이미지를 사용하여 획득된 이미지의 신호 대 잡음 비율(SNR)을 현저히 감소시킨다. 따라서 문제를 명백히 해결할 수 없으며 계산할 모든 매개 변수는 추정일 수 있습니다. 이미지 처리 워크플로우를 따라 올바른 매개 변수를 식별하고 나머지 매개 변수를 제거하여 고해상도 3D 재구성을 마지막으로 얻어야 합니다.
현미경에 의해 생성된 데이터는 프레임으로 수집됩니다. 단순화, 프레임은 전자 계수 검출기를 사용할 때마다 이미지의 특정 위치(픽셀)에 도착한 전자의 수를 포함합니다. 특정 시야에서 여러 프레임이 수집되고 이를 영화라고 합니다. 낮은 전자 용량은 샘플을 파괴 할 수있는 방사선 손상을 방지하기 위해 사용되기 때문에, SNR은 매우 낮으며 동일한 영화에 대응하는 프레임은 샘플에 대한 구조적 정보를 밝히는 이미지를 얻기 위해 평균될 필요가있다. 그러나, 단순한 평균뿐만 아니라, 시료는 보상이 필요한 빔 유도 운동으로 인해 이미징 시간 동안 교대 및 기타 종류의 움직임을 겪을 수 있다. 시프트 보정 및 평균 프레임은 현미경 그래프에서 발생합니다.
일단 현미경이 얻어지면, 우리는 주파수의 함수로 현미경의 대조에 있는 변경을 나타내는 대조 전송 기능 (CTF)에게 불린 그들 각각에 대한 현미경에 의해 소개된 수차를 추정할 필요가 있습니다. 그런 다음 파티클을 선택하고 추출할 수 있으며, 이는 입자 따기라고 합니다. 모든 입자는 연구 중인 표본의 복사본을 하나만 포함하는 작은 이미지여야 합니다. 입자 따기 알고리즘의 세 제품군이 있습니다: 1) 입자의 모양의 일부 기본 매개 변수를 사용하여 전체 현미경 사진 세트(예: 파티클 크기), 2) 파티클이 사용자 또는 미리 학습된 집합에서 어떻게 보이는지 배우는 것, 그리고 3) 이미지 템플릿을 사용하는 알고리즘이 있습니다. 각 패밀리에는 나중에 표시되는 속성이 다릅니다.
마이크로그래프에서 발견되는 추출된 입자 세트는 2D 분류 공정에 사용되며, 1) 순수한 노이즈 이미지, 겹치는 입자 또는 기타 아티팩트를 포함하는 하위 집합을 폐기하여 입자 세트를 청소하고, 2) 각 클래스를 나타내는 평균 입자는 3D 초기 부피를 계산하기 위해 초기 정보로 사용될 수 있다.
3D 초기 볼륨 계산은 다음 중요한 단계입니다. 3D 구조를 획득하는 문제는 다차원 솔루션 환경에서 최적화 문제로 볼 수 있으며, 여기서 글로벌 최소값은 원래 구조를 나타내는 최고의 3D 볼륨이지만, 최적이 아닌 솔루션을 나타내는 여러 로컬 미니마를 찾을 수 있으며, 갇히기 매우 쉬운 곳. 초기 볼륨은 검색 프로세스의 시작점을 나타내므로 초기 볼륨 추정이 좋지 않으므로 전역 최소값을 찾을 수 없습니다. 초기 부피에서 3D 분류 단계는 다른 형성 상태를 발견하고 입자 세트를 다시 청소하는 데 도움이 됩니다. 목표는 입자의 구조적으로 균일한 인구를 얻는 것입니다. 그 후, 3D 정제 단계는 가능한 최고의 3D 볼륨을 얻기 위해 모든 입자에 대한 각 및 번역 매개 변수를 정제담당합니다.
마지막으로, 마지막 단계에서, 얻은 3D 재건은 선명하고 연마될 수 있다. 선명하게 하는 공정은 재구성된 부피의 높은 주파수를 증폭시키는 과정이며, 연마는 입자 수준에서 CTF 또는 빔 유도 이동 보정으로서 일부 파라미터를 더욱 정제하는 단계이다. 또한 일부 유효성 검사 프로시저는 워크플로가 끝날 때 달성된 해상도를 더 잘 이해하는 데 사용될 수 있습니다.
이러한 모든 단계 후, 추적 및 도킹 프로세스7 은 원자 모델을 구축하거나 기존 모델을 피팅하여 얻어진 3D 재건에 생물학적 의미를 부여하는 데 도움이 될 것입니다. 고해상도가 달성되면 이러한 프로세스는 생물학적 구조의 위치를 알려줍니다.
Scipion8을 사용하면 가장 관련성이 뛰어난 이미지 프로세싱 패키지를 통합방식으로 결합한 전체 워크플로우를 만들 수 있습니다. Xmipp9, Relion10, CryoSPARC11, Eman12, Spider13, Cryolo14, Ctffind15, CCP416, Phenix17 및 더 많은 패키지가 시피온에 포함될 수 있습니다. 또한 통합, 상호 운용성, 추적성 및 재현성을 이점하는 데 필요한 모든 도구를 통합하여 전체 이미지 처리 워크플로우8을 완전히 추적합니다8.
Scipion이 사용할 수 있는 가장 강력한 도구 중 하나는 컨센서스로, 이는 보다 정확한 출력을 생성하기 위해 다른 방법으로 전달되는 정보의 조합을 만드는 처리의 한 단계에서 여러 가지 방법과 비교하여 얻은 결과를 비교하는 것을 의미합니다. 이렇게 하면 성능을 향상시키고 예상 매개 변수에서 달성된 품질을 개선하는 데 도움이 될 수 있습니다. 컨센서스 메서드를 사용하지 않고도 더 간단한 워크플로를 빌드할 수 있습니다. 그러나 이 도구의 힘을 보았습니다22,25 이 원고에 제시된 워크플로우는 여러 단계에서 사용할 것입니다.
이전 단락에 요약된 모든 단계는 다음 섹션에서 자세히 설명하고 Scipion을 사용하여 전체 워크플로에서 결합됩니다. 또한, 생성된 출력에서 더 높은 합의를 달성하기 위해 컨센서스 도구를 사용하는 방식이 표시됩니다. 이를 위해 플라스모듐 팔시파룸 80S 리보솜의 예제 데이터 집합이 선택되었습니다(EMPIAR 항목: 10028, EMDB 항목: 2660). 데이터 세트는 FEI FALCON II 카메라로 FEI POLARA 300에서 촬영한 1.34Å 크기의 16프레임 크기 4096x4096 픽셀의 600개 영화에 의해 형성되며 EMDB에서 보고된 해상도는 3.2Å18 입니다.
프로토콜
1. Scipion에서 프로젝트 생성 및 데이터 가져오기
- Scipion을 열고 프로젝트 만들기를 클릭하고 프로젝트의 이름과 프로젝트가 저장될 위치를 지정합니다(추가 그림 1). Scipion은 캔버스를 보여주는 프로젝트 창을 열며, 왼쪽에는 사용 가능한 메서드 목록이 있는 패널이 있으며, 각각은 데이터를 관리하는 데 사용할 수 있는 하나의 이미지 처리 도구를 나타냅니다.
참고: Ctrl+F 를 사용하여 목록에 나타나지 않는 경우 메서드를 찾을 수 있습니다. - 현미경으로 촬영한 동영상을 가져오려면 pwem을 선택합니다 - 왼쪽 패널에서 영화를 가져오거나 Ctrl+F를 누를 때 입력합니다.
- 새 창이 열립니다(추가 그림 2). 여기에는 데이터에 대한 경로 및 수집 매개 변수가 포함됩니다. 이 예에서는 현미경 전압 300 kV, 구형 수차 2.0mm, 진폭 대비 0.1, 배율 50000, 이미지에서 샘플링 속도 모드, 픽셀 크기 1.34 Å 등의 설정을 사용합니다. 양식의 모든 매개 변수가 채워지면 실행 단추를 클릭합니다.
참고: 메서드가 시작되면 캔버스에 실행으로 레이블이 지정된 노란색으로 상자가 나타납니다. 메서드가 완료되면 상자가 녹색으로 변경되고 레이블이 완료됩니다. 메서드를 실행하는 동안 오류가 발생하면 상자가 빨간색으로 표시되어 실패로 표시됩니다. 이 경우 출력 로그 탭에서 캔버스의 아래쪽 부분을 확인하여 오류에 대한 설명이 나타납니다. - 메서드가 완료되면 요약 탭에서 캔버스의 하단 부분에 있는 결과를 확인합니다. 여기서 메서드에 의해 생성된 출력이 이 경우 영화 집합이 표시됩니다. 결과 분석 버튼을 클릭하면 영화 목록에 새 창이 나타납니다.
2. 영화 정렬 : 영화에서 마이크로 그래프로
- 광학 유량19을 구현하는 광학 정렬 - 방법 xmipp3을 사용합니다. 양식을 채우기 위해 다음 매개 변수를 사용합니다(추가 그림 3): 입력 동영상은 1단계에서 얻은 것, 프레임에서 정렬범위는 2에서 13사이이며, 다른 옵션은 기본 값과 함께 유지됩니다. 프로그램을 실행합니다.
참고: 양식의 굵게 표시된 매개 변수는 항상 채워져야 합니다. 다른 값은 기본 값을 가지거나 의무적으로 필요하지 않습니다. 폼 창의 위쪽 부분에서 계산 리소스가 분산되는 필드를 스레드, MPI 또는 GPU로 찾을 수 있습니다. - 결과 분석 을 클릭하여 얻은 현미경 그래프와 예상 교대의 궤적을 확인합니다(그림 1). 본 모든 현미경 그래프에 대해: 전력 스펙트럼 밀도(PSD), 카르테시안 및 극성 좌표에서 영화(프레임당 1점)를 정렬하기 위해 얻은 궤적, 그리고 얻어진 현미경 그래프의 파일 이름(클릭, 현미경 그래프를 검사할 수 있음)을 살펴보십시오. 영화의 단일 프레임과 비교하여 표본의 입자가 현미경 그래프에서 훨씬 더 많이 표시됩니다.
3. CTF 추정 : 현미경의 수차 계산
- 첫째, 방법 grigoriefflab을 사용 - ctffind15. 설정은 입력 현미경 그래프가 2단계의 출력이고 수동 CTF 다운샘플링 계수는 1.5로 설정되고 해상도 범위는 0.06에서 0.42로 바릅니다. 또한 고급 옵션(양식의 전문가 수준에서 이 옵션을 선택하여 찾을 수 있음)에서 창 크기를 256으로 설정합니다. 나머지 매개 변수는 기본 값(추가 그림 4)과 함께 유지됩니다.
참고: Scipion의 대부분의 메서드에서 고급 옵션은 더 많은 구성 매개 변수를 표시합니다. 시작할 프로그램이 완전히 알려지고 매개 변수의 의미를 이해할 때 이러한 옵션을 신중하게 사용합니다. 일부 매개 변수는 데이터를 살펴보지 않고도 채우기 어려울 수 있습니다. 이 경우 Scipion은 오른쪽에 마법사 창(추가 그림 5)을 표시하는 마법의 지팡이를 보여줍니다. 예를 들어 이 양식의 해상도 필드에서는 이러한 값을 선택하여 첫 번째 0에서 PSD의 마지막 눈에 띄는 링까지 영역을 대략 커버하도록 선택해야 하므로 특히 유용합니다. - 메서드가 완료되면 실행 및 분석 결과 (그림 2)를 클릭합니다. 예상 CTF가 실험용 CTF와 일치하는지 확인합니다. 이를 위해 PSD를 살펴보고 모서리의 예상 링과 데이터에서 오는 링을 비교합니다. 또한 얻은 디포커스 값을 확인하여 예기치 않은 값을 찾고 각 현미경그래프를 폐기하거나 다시 계산할 수 있습니다. 이 예제에서는 전체 현미경 그래프 집합을 사용할 수 있습니다.
참고: 창 하단의 버튼을 사용하여 마이크로그래프의 하위 집합( 마이크로그래프 빨간색 버튼 사용)을 만들고 CTF(CTF 의 빨간색 버튼 재계산)를 다시 계산해야 하는 경우 다시 계산합니다. - 이전 추정을 구체화하려면 xmipp3 - ctf 추정20을 사용합니다. 입력 현미경 2의 출력으로 선택하고, 이전 CTF 추정이 grigoriefflab - ctffind의 출력을 선택하고, 고급 수준에서, 256 (보충 도 6)의 출력을 선택으로, 이전 CTF 추정에서 defoci를 사용합니다. 실행합니다.
- 결과 분석 분석을 클릭하여 얻은 CTF를 확인합니다. 이 방법을 사용하면 더 많은 데이터가 추정되고 일부 추가 열에 표시됩니다. 그 중 어느 것도 잘못된 추정 값을 나타내지 않기 때문에 모든 현미경 사진은 다음 단계에서 사용됩니다.
4. 입자 따기 : 현미경 그래프에서 입자찾기
- 따기를 시작하기 전에 현미경 사진의 사전 프로세스를 수행합니다. 오픈 xmipp3 - 사전 처리 현미경, 입력 현미경으로 설정 2 단계에서 얻은 것과 옵션을 선택 나쁜 픽셀을 제거? Stddev의 배수 5, 다운 샘플 현미경 사진? 2의 다운 샘플링 계수 (보충 도 7). 실행을 클릭하고 결과 현미경 사진의 크기가 감소되었는지 확인합니다.
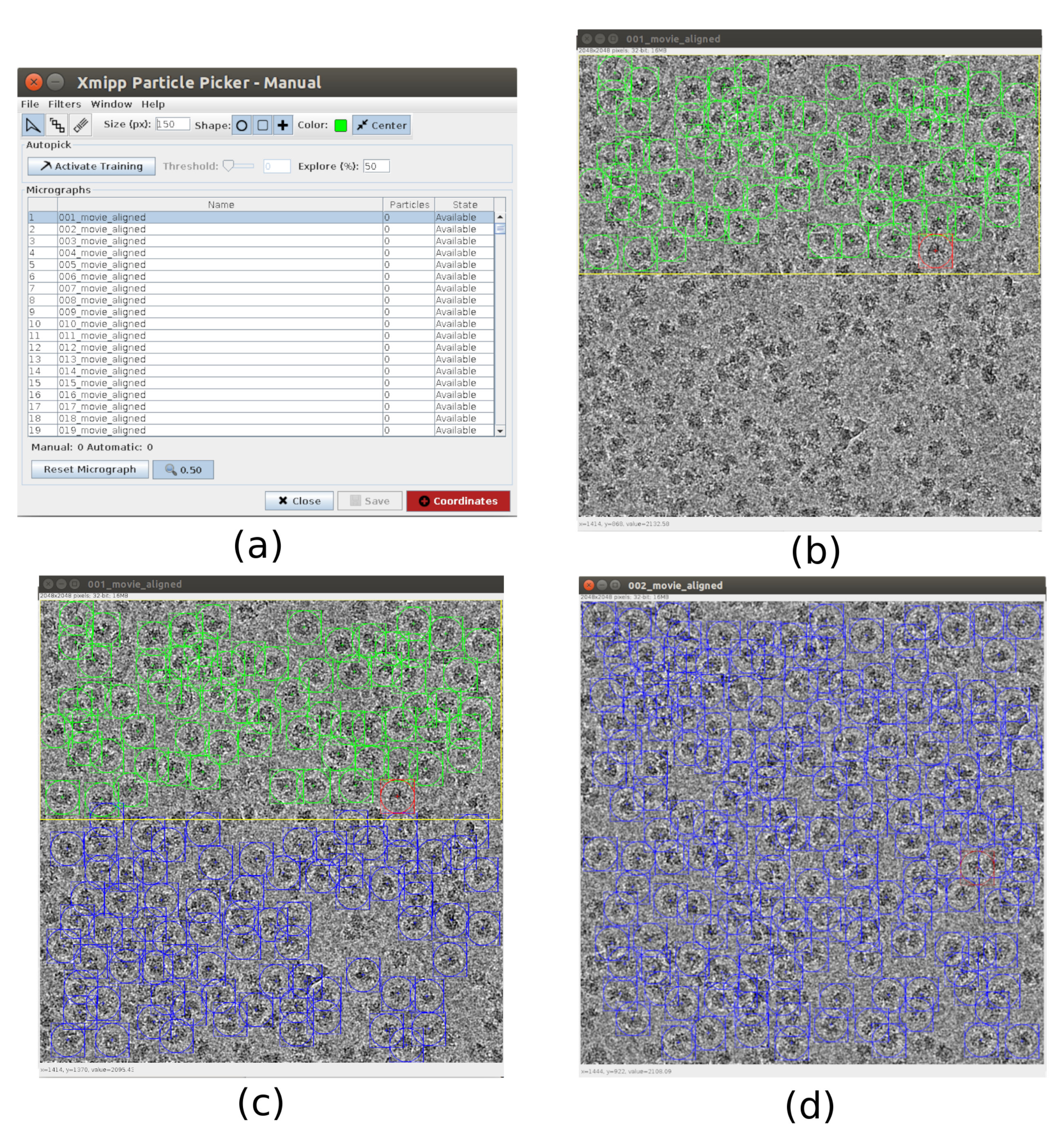
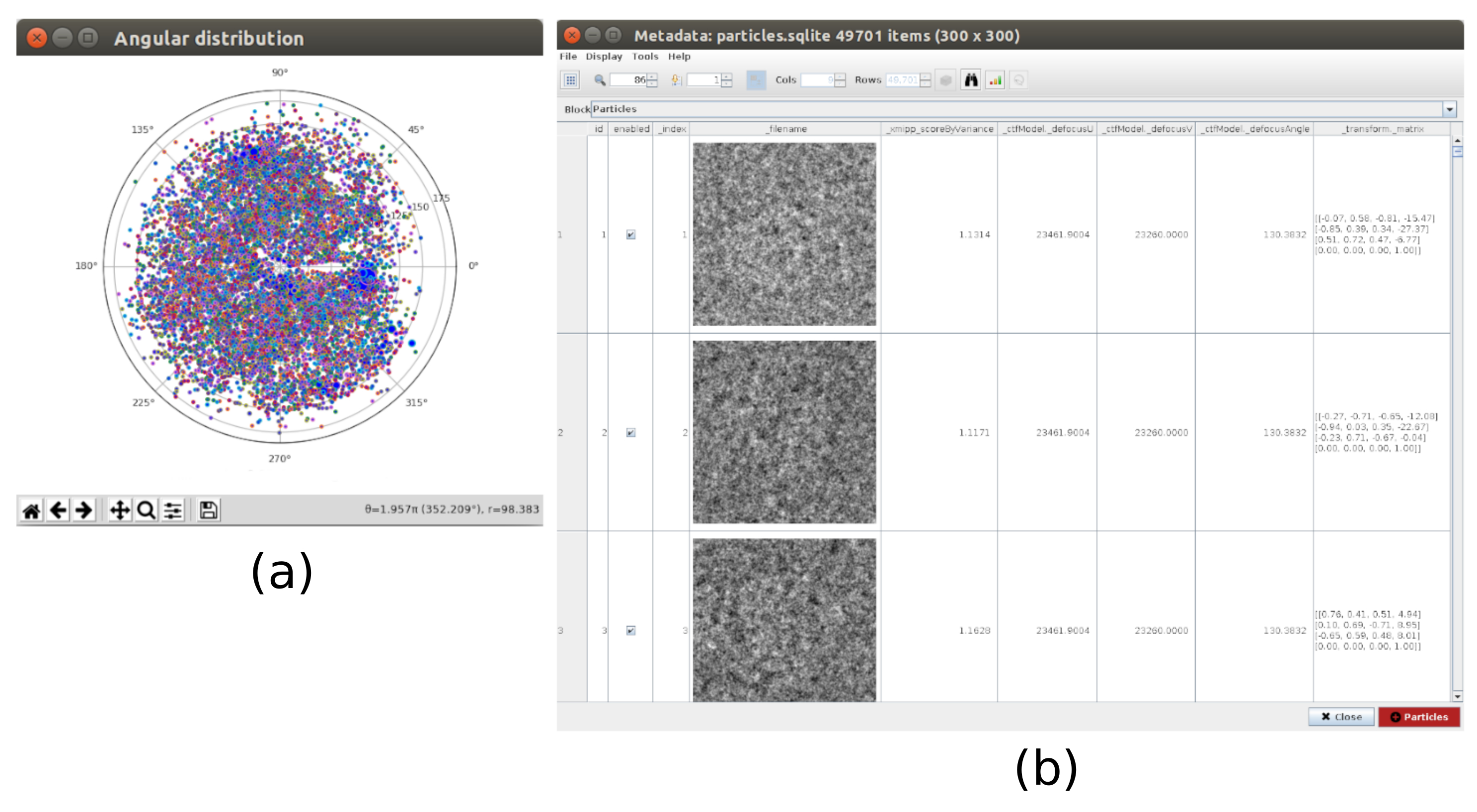
- 피킹 용 xmipp3 - 수동 따기 (1 단계) 및 xmipp3 - 자동 따기 (단계 2)21. 수동 피킹을 사용하면 자동 따기 단계가 전체 파티클 집합을 학습하고 생성하는 파티클 집합을 수동으로 준비할 수 있습니다. 첫째, xmipp3 실행 - 입력 현미경으로 수동 따기 (1 단계)를 이전 전공정에서 얻은 현미경으로. 실행을 클릭하면 새 대화형 창이 나타납니다(그림 3).
- 이 창에서 현미경 사진(그림 3a) 및 기타 옵션 목록이 표시됩니다. 크기(px) 를 150으로 변경하면 각 파티클이 포함된 상자의 크기가 됩니다. 선택한 현미경이 더 큰 창에 나타납니다. 영역을 선택하고 그 안에 있는 모든 표시되는 파티클을 선택합니다(그림 3b). 그런 다음 학습 활성화 를 클릭하여 학습을 시작합니다. 현미경 그래프의 나머지 영역은 자동으로 선택됩니다(그림 3c). 선택한 파티클을 확인하고 클릭하여 더 많이 포함하거나 필요한 경우 shift+클릭으로 잘못된 입자를 제거합니다.
- 첫 번째 창에서 다음 현미경 그래프를 선택합니다. 현미경 그래프는 자동으로 선택됩니다. 필요한 경우 일부 파티클을 포함하거나 제거하려면 다시 확인하십시오. 약 5개의 현미경으로 이 단계를 반복하여 대표 교육 세트를 만듭니다.
- 이 작업이 완료되면 주 창의 좌표를 클릭하여 선택한 모든 파티클의 좌표를 저장합니다. 파티클의 교육 세트는 모든 현미경 에 대한 프로세스를 완료하기 위해 자동 따기로 이동 할 준비가되어 있습니다.
- 오픈 xmipp3 - Xmipp 입자 따기에서 나타내는 자동 따기 (2 단계)는 이전 수동 피킹을 실행하고, 마이크로 그래프는 감독과 동일하게 선택합니다. 실행을 클릭합니다. 이 메서드는 약 100000 좌표 집합을 출력으로 생성합니다.
- 컨센서스 접근 방식을 적용하므로 두 가지 방법이 모두 동의하는 파티클을 선택하는 두 번째 선택 방법을 수행하십시오. 오픈 스퍼퍼 - cryolo picking14 및 입력 현미경으로 미리 처리 된 현미경 그래프를 선택, 일반 모델을 사용하려면 예, 신뢰 임계값 0.3, 상자 크기 150 (보충 도 8). 실행합니다. 이 메서드는 약 100000 좌표를 생성해야 합니다.
- 실행 xmipp3 - 깊은 합의 따기22. 입력 좌표에는 스파이어의 출력이 포함되기 때문에 크라이올로 피킹 (단계 4.7) 및 xmipp3 - 자동 따기 (단계 4.6), 사전 학습된 모델 유형 선택 설정, 사전 학습 된 모델로 직접 학습 및 점수를 건너 뛰기? 예 (보충 그림 9). 실행합니다.
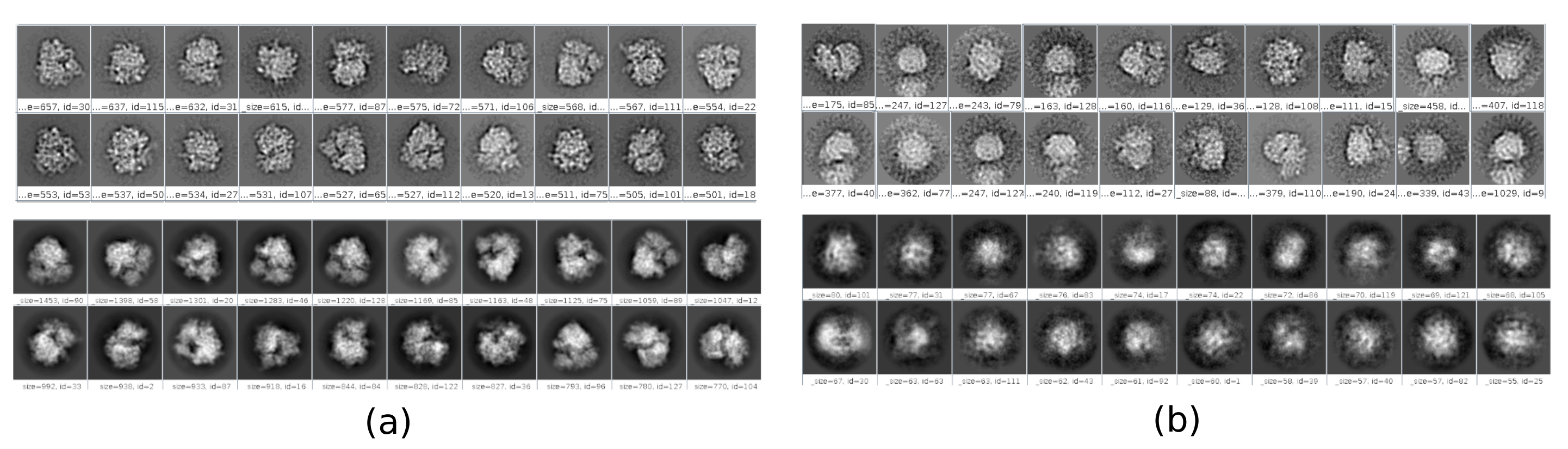
- 결과 분석을 클릭하고 새 창에서 높은 'zScoreDeepLearning1' 값을 가진 파티클/좌표 선택 옆에 있는 눈 아이콘을 클릭합니다. 새 창은 모든 파티클 목록(그림 4)과 함께 열립니다. 열의 zScore 값은 파티클의 품질에 대한 통찰력을 제공하며 값이 낮으면 품질이 좋지 않습니다.
- 레이블을 클릭_xmipp_zScoreDeepLearning 가장 높은 zScore에 파티클을 주문합니다. zScore 가 0.75보다 높은 파티클 을 선택하고 좌표를 클릭하여 새 하위 집합을 만듭니다. 이렇게 하면 약 50000개의 좌표가 있는 하위 집합이 생성되어야 합니다.
- 오픈 xmipp3 - 깊은 현미경 사진 클리너. 입력이 이전 단계에서 얻은 하위 집합을 좌표로 선택하고, 마이크로그래프는 좌표와 동일하게 소스로 설정하고 임계값을 0.75로 유지합니다. 실행합니다. 요약 탭에서 좌표 수가 줄었는지는 확인하지만 이 경우 좌표만 제거됩니다.
참고: 이 단계는 좌표 세트를 추가로 청소할 수 있으며 탄소 영역 이나 큰 불순물로 더 많은 영화 아티팩트로 다른 데이터 집합을 청소하는 데 매우 유용 할 수 있습니다. - xmipp3 실행 - 추출 입자 (보충 도 10). 입력이 이전 단계 이후 얻은 좌표로 나타내고, 마이크로그래프는 다른 단계의 출력으로, 입력 현미경은 단계 2의 출력으로, CTF 추정은 xmipp3의 출력으로 - ctf 추정, 3까지의 다운샘플링 계수, 및 파티클 박스 크기가 100으로 나타낸다. 양식의 사전 처리 탭에서 모두에게 예를 선택합니다. 실행합니다.
- 출력에 100x100 픽셀의 크기가 감소된 파티클과 4.02Å/px의 픽셀 크기가 포함되어야 하는지 확인합니다.
- 다시 xmipp3 실행 - 다음 매개 변수를 변경 추출 입자 : 1로 다운 샘플링 계수 , 파티클 상자 크기 는 300로. 출력이 동일한 파티클 집합이지만 이제 전체 해상도로 확인합니다.
5. 2D 분류: 유사한 파티클을 함께 그룹화
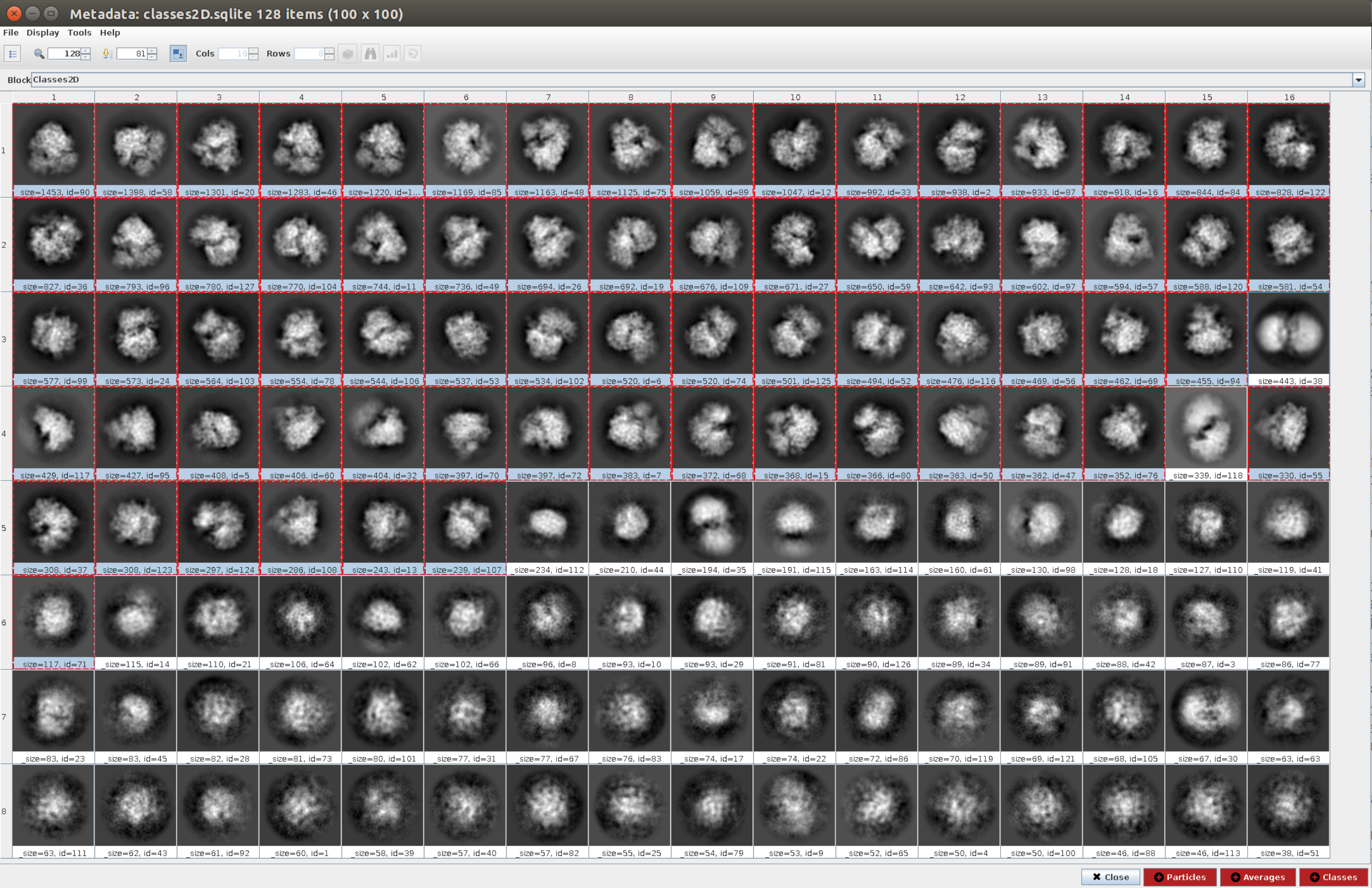
- 4.11 단계에서 얻은 것과 같이 입력 입자가 있는 2d 분류11을 열고, 2D 분류 탭에서 클래스 수를 128로 열어 기본 값으로 다른 모든 매개 변수를 유지합니다. 실행합니다.
- 결과 분석을 클릭한 다음 Scipion을 사용하여 파티클 클래스 표시 옆에 있는 눈 아이콘을 클릭합니다(그림 5). 이 분류는 여러 클래스가 시들게 표시되거나 아티팩트로 표시되기 때문에 파티클 집합을 청소하는 데 도움이 됩니다. 좋은 뷰가 포함된 클래스를 선택합니다. 파티클(창 아래쪽의 빨간색 버튼)을 클릭하여 더 깨끗한 하위 집합을 만듭니다.
- 이제 xmipp3 - cl2d23을 열고 입력 이미지로 설정하여 이전 단계에서 얻은 이미지와 클래스 수를 128로 설정합니다. 실행을 클릭합니다.
참고: 이 두 번째 분류는 파티클 집합의 추가 세척 단계로 사용됩니다. 일반적으로 가능한 한 많은 시끄러운 입자를 제거하는 것이 유용합니다. 그러나 더 간단한 워크플로우를 원하는 경우 하나의 2D 분류 방법만 사용할 수 있습니다. - 메서드가 완료되면 결과 분석 및 표시 할 내용 : 클래스를 클릭하여 생성 된 128 클래스를 확인합니다. 생성된 클래스의 대부분은 어느 정도의 세부 사항을 가진 거대 분자의 투영을 보여줍니다. 그러나 일부 사람들은 시끄러운 것처럼 보입니다(이 예에서는 약 10개의 클래스). 모든 좋은 클래스를 선택하고 클래스 버튼을 클릭하여 좋은 클래스만 사용하여 새 하위 집합을 생성합니다. 이 하위 집합은 초기 볼륨을 생성하는 방법 중 하나에 대한 입력으로 사용됩니다. 동일한 선택된 클래스를 사용하면 파티클 을 클릭하여 잘못된 클래스에 속한 클래스를 제거한 후 더 깨끗한 하위 집합을 만듭니다.
- 오픈 pwem - 4.13 (전체 크기의 모든 파티클)의 출력으로 항목의 전체 세트와 하위 집합, 아니오로 임의의 하위 집합을 확인, 다른 세트는 이전 단계에서 만든 파티클의 하위 집합으로, 교차로 작업을 설정합니다. 이렇게 하면 전체 해상도의 파티클에서 이전 하위 집합이 추출됩니다.
6. 초기 볼륨 추정 : 3D 볼륨의 첫 번째 추측을 구축
- 이 단계에서는 서로 다른 방법으로 두 개의 초기 볼륨을 추정한 다음 컨센서스 도구를 사용하여 최종 추정 된 3D 볼륨을 생성합니다. 열기 xmipp3 - 5단계 이후 얻은 것과 같이 입력 클래스로 중요한24 메서드를 재구성하고 대칭 그룹을 c1로 재구성하고 나머지 매개 변수를 기본 값(보충 도 11)으로 유지합니다. 실행합니다.
- 결과 분석을 클릭합니다. 크기 100x100x100 픽셀의 낮은 해상도 볼륨과 4.02Å/px의 픽셀 크기를 얻을 수 있는지 확인합니다.
- 오픈 xmipp3 - 이전 단계에서 얻은 입력 볼륨으로 사용하는 자르기/크기 조정 볼륨(추가 그림 12) 볼륨 크기를 조정하고, 볼륨 크기를 조정하려면 예, 샘플링 속도로 크기를 조정하고 샘플링 속도를 1.34 Å/px로 조정합니다. 실행합니다. 출력 볼륨에 올바른 크기가 있는 요약 탭을 확인합니다.
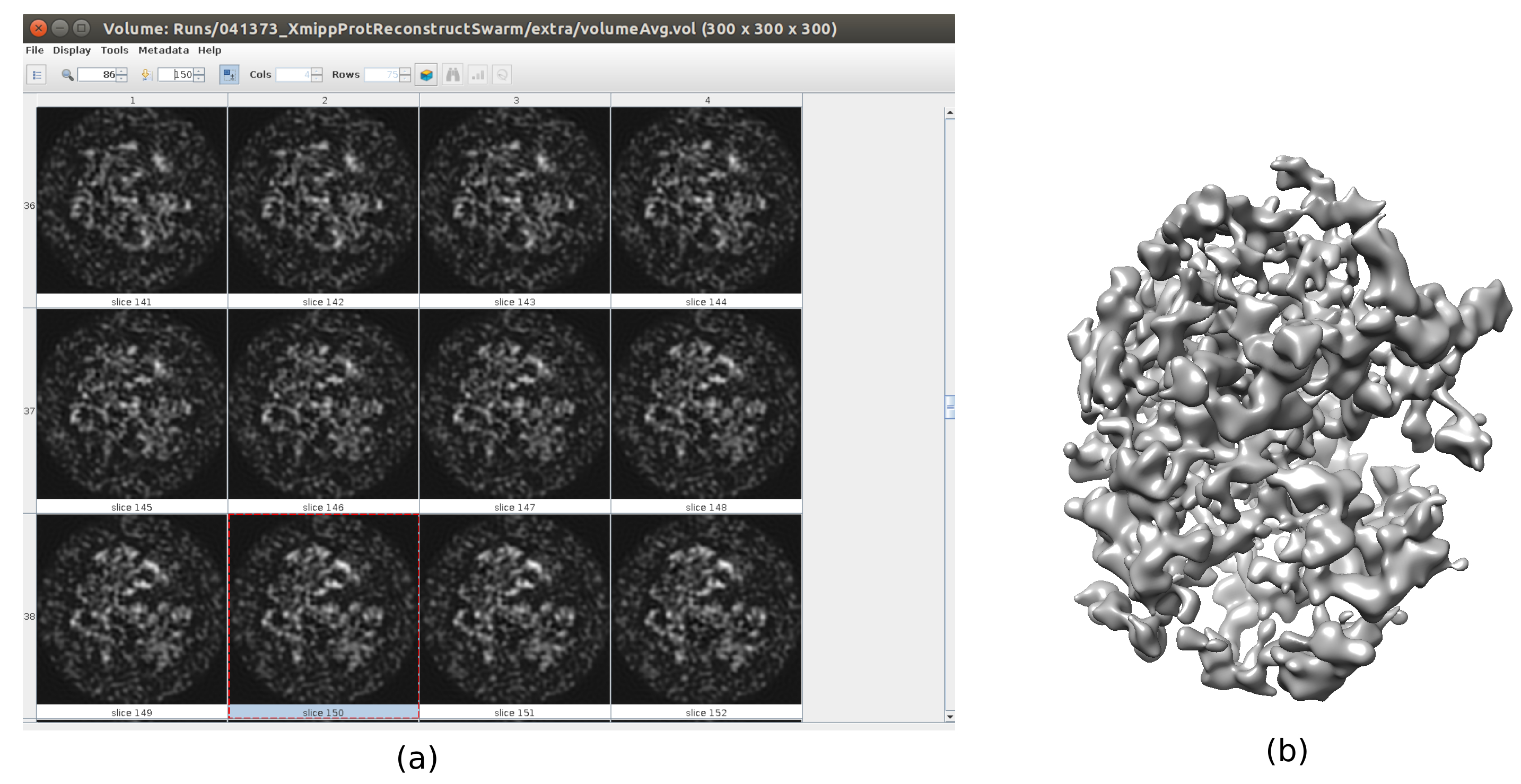
- 이제 두 번째 초기 볼륨을 만듭니다. 열기 리온 - 3D 초기 model10, 입력 입자가 전체 해상도 (5.5의 출력)에서 좋은 입자를 사용하고 입자 마스크 직경을 402Å로 설정하여 나머지 매개 변수를 기본 값으로 유지합니다. 실행합니다.
- 결과 분석 한 다음 표시 볼륨에서 슬라이스를 클릭합니다. 낮은 해상도 볼륨하지만 구조의 주요 형상이 얻어지는지 확인합니다(보충 도 13).
- 이제 열기 pwem - 결합 방식에 대한 입력을 만들기 위해 두 개의 생성된 초기 볼륨을 결합하는 집합을 조인합니다. 볼륨을 입력 유형으로 표시하고 입력 집합의 두 개의 초기 볼륨을 선택하기만 하면 됩니다. 실행합니다. 출력은 두 볼륨을 모두 가진 두 개의 항목을 포함하는 집합이어야 합니다.
- 합의 도구는 xmipp3에 포함 된 것입니다 - 군단 합의25. 그거 여세요. 전체 크기 이미지 로 사용하면 초기 볼륨이 이전 단계에서 생성된 두 개의 항목으로 세트를 볼륨 하고 대칭 그룹이 c1인지 확인하므로 전체 해상도(출력 5.5)에서 좋은 파티클을 사용합니다. 실행을 클릭 합니다.
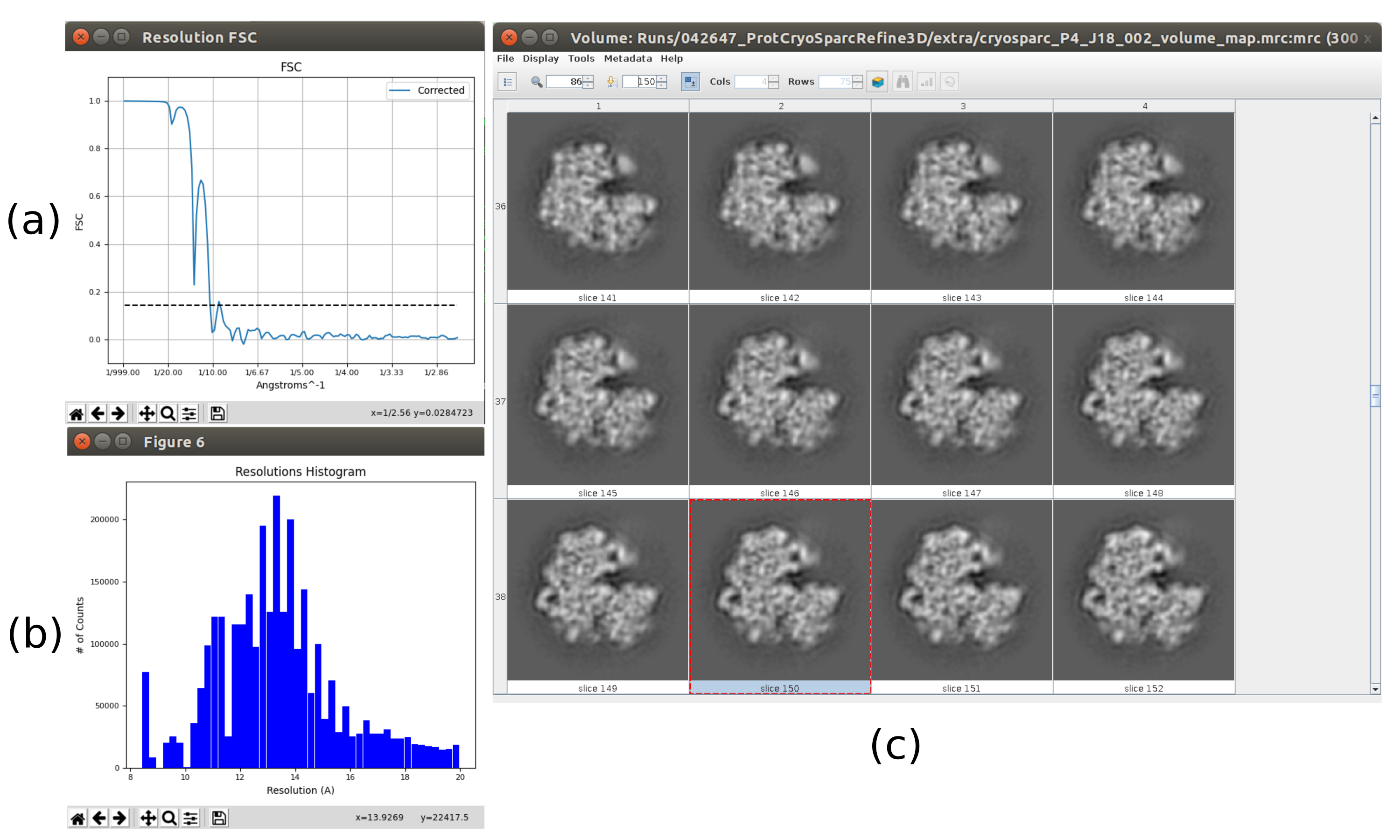
- 결과 분석을 클릭합니다. 보다 자세한 출력 볼륨이 얻어지는지 확인합니다(그림 6). 구조물을 둘러싼 소음이 더 많지만 구조 맵에 자세한 내용을 두면 로컬 미니마를 피하기 위한 다음 구체화 단계에 도움이 됩니다.
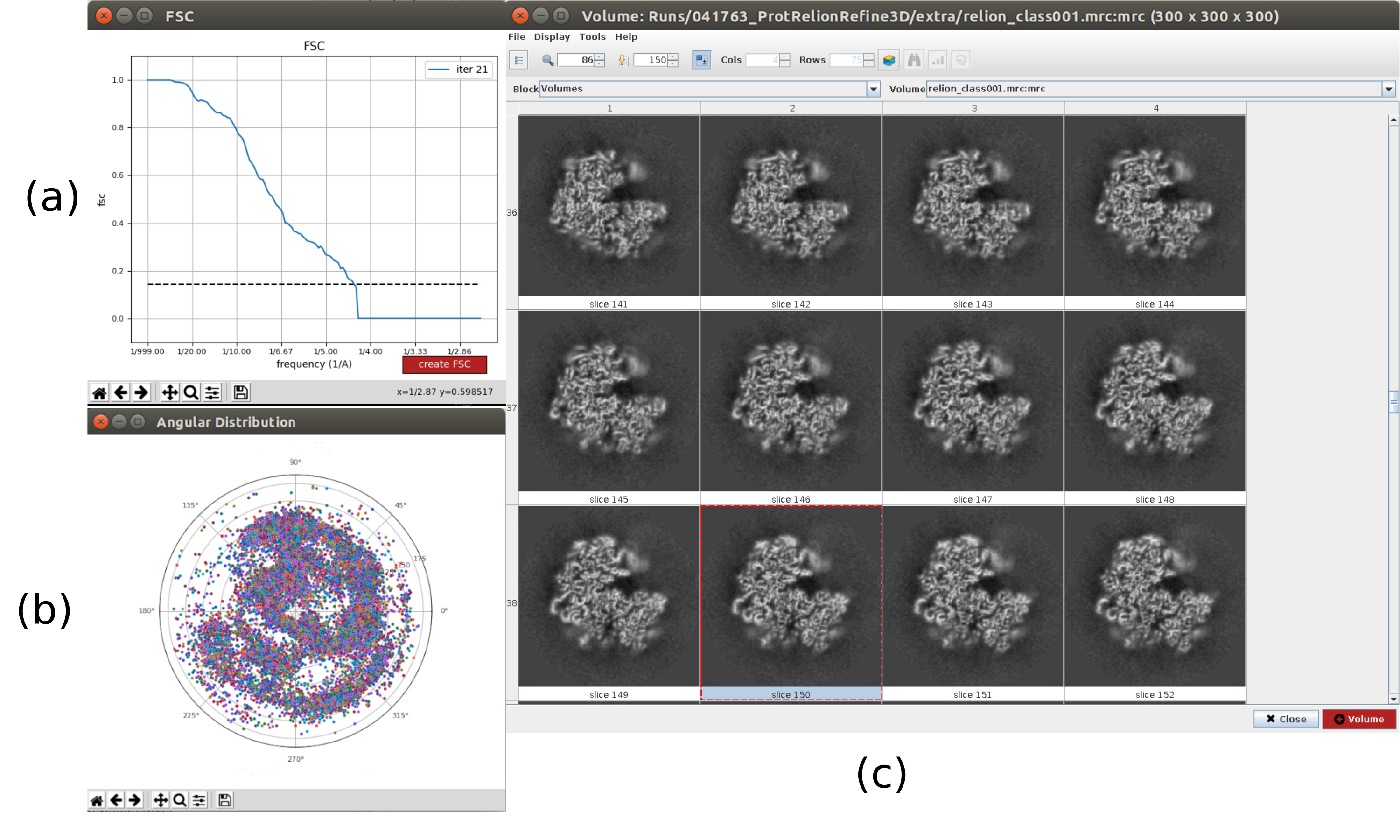
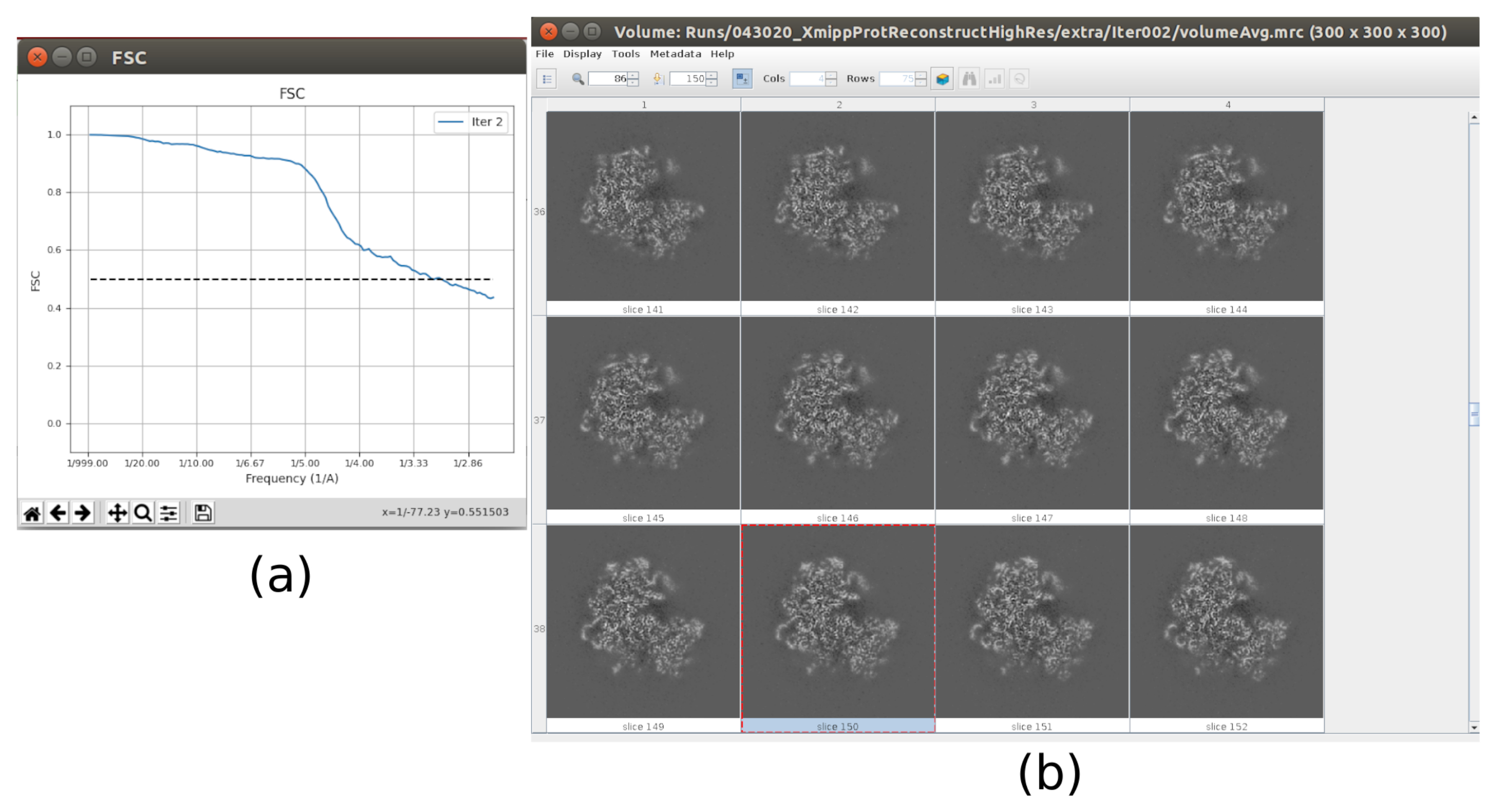
참고: UCSF Chimera26 을 사용할 수 있는 경우 창 상단의 마지막 아이콘을 사용하여 획득한 볼륨의 3D 시각화를 만듭니다. - 릴리온을 열고 실행 - 3D 자동 정제10을 통해 파티클의 첫 번째 3D 각도 할당을 합니다. 입력 입자로 5.5의 출력을 선택하고 파티클 마스크 직경을 402Å로 설정합니다. 참조 3D 맵 탭에서 이전 단계에서 얻은 입력 볼륨, c1로 대칭, 초기 로우패스 필터를 30Å로 선택합니다(보충 도 14).
- 결과 분석을 클릭합니다. 새 창에서 파이널을 볼륨으로 선택하고 표시 볼륨을 클릭합니다. 또한 결과 창의 디스플레이 해상도 플롯과 디스플레이 각 분포의 각 범위: 2D 플롯(그림 7)을 클릭하여 푸리에 쉘 상관관계(FSC)를 확인합니다. 재구성된 볼륨에는 훨씬 더 많은 세부 정보(구조의 바깥부분에 일부 흐린 영역)가 포함되어 있으며 FSC는 4.5Å 주위의 0.143 임계값을 교차합니다. 각 범위는 전체 3D 구를 다룹니다.
7.3D 분류: 형태 적 상태 발견
- 합의 방식을 사용하여 다른 형태 상태가 데이터에 있는 경우 발견할 수 있습니다. 열기 리온 - 3D 분류10 (보충 도 15). 입력 입자 는 6.10에서 얻은 입자 를 사용하고 파티클 마스크 직경 을 402Å로 설정합니다. 참조 3D 맵 탭에서 6.10단계 이후에 얻은 입력 볼륨 으로 사용하고 대칭을 c1로 설정하고 초기 로우패스 필터 를 15Å로 설정합니다. 마지막으로 최적화 탭에서 클래스 수를 3개로 설정합니다. 실행합니다.
- 결과 분석 을 클릭하여 결과를 확인하고 Scipion에서 분류 표시를 선택합니다. 생성된 세 가지 클래스와 몇 가지 흥미로운 조치가 표시됩니다. 처음 두 클래스에는 비슷한 수의 할당된 이미지(크기 열)가 있어야 하며 매우 유사해야 하며, 세 번째 클래스는 이미지 수가 적고 모양이 더 흐려집니다. 또한, rln정확도 회전 및 rln정확도번역은 처음 두 클래스에 대해 명확하게 더 나은 것이어야 합니다. 두 개의 최고의 클래스를 선택하고 클래스 버튼을 클릭하여 클래스가 포함된 하위 집합을 생성합니다.
- 7.1 및 7.2 단계를 반복하여 두 번째 좋은 클래스 그룹을 생성합니다. 둘 다 컨센서스 도구의 입력이 될 것입니다.
- xmipp3 열기 및 실행 - 합의 클래스 3D및 입력 클래스로 이전 단계에서 생성 된 두 개의 하위 집합을 선택합니다.
- 결과 분석을 클릭합니다. 클래스 간의 일치 입자의 수가 제시된다: 첫 번째 값은 서브 세트 1의 첫 번째 클래스와 서브 세트 2의 첫 번째 클래스에서 일치 입자의 수이며, 두 번째 값은 하위 집합 1및 하위 집합 2의 첫 번째 클래스에서 일치 입자의 수입니다. 파티클이 하나 또는 두 개의 클래스에 무작위로 할당되어 있는지 확인하므로 3D 분류 방법이 변형 변화를 찾을 수 없습니다. 이 결과를 감안할 때 전체 파티클 집합이 처리를 계속하는 데 사용됩니다.
8.3D 세련미: 동질모의 각할당 을 정제
- 다시 말하지만, 이 단계에서 합의 접근 방식을 적용합니다. 첫째, pwem을 열고 실행 - 6.9의 출력으로 항목의 전체 세트와 하위 집합, 예에 임의의 하위 집합을 확인하고, 5000 요소의 수. 이를 통해 다음 단계에서 사용되는 메서드를 학습하기 위해 이전 정렬이 있는 이미지의 하위 집합이 만들어집니다.
- open xmipp3 - 딥 정렬, 입력 이미지를 5.5에서 얻은 양량의 출력으로 설정하고, 6.10 이후에 얻은 양체 , 입력 훈련 세트 는 이전 단계에서 생성된 것으로 설정되고, 목표 해상도 를 10Å로 설정하고, 나머지 매개 변수를 기본 값으로 유지한다(보충 도 16). 실행을 클릭 합니다.
- 결과를 분석하려면 검색된 각도 분포를 확인하며, 경로가 누락되지 않고 각 커버리지가 6.10(그림 8)에 비해 약간 향상됩니다.
- xmipp3 열기 및 실행 - 각도를 비교 하고 입력 입자 1 의 출력6.9 및 입력 입자 2 의 출력으로 선택하여 8.2의 출력을 선택하여 대칭 그룹이 c1인지 확인합니다. 이 방법은 xmipp3 - 깊은 정렬 및 리온 - 3D 자동 정제 사이의 계약을 계산합니다.
- 결과 분석을 클릭하면 이동과 각도의 차이가 있는 파티클 목록이 표시됩니다. 창 의 상단 부분에있는 막대 아이콘을 클릭, 계산 된 변수의 플롯을 만들 수 있도록 다른 창이 열립니다 열립니다. _xmipp_angleDiff 선택하고 플롯 을 클릭하여 파티클당 각 차이의 표현을 확인합니다. _xmipp_shiftDiff 똑같이 하십시오. 이 수치에서, 입자의 절반정도에서 두 방법이 모두 동의한다(도 9). 각도 차이가 10º보다 낮은 파티클을 선택하고 새 하위 집합을 만듭니다.
- 지금, 오픈 xmipp3 - highres27 할당 된 각도의 로컬 개선을 할 수 있습니다. 먼저, 이전 단계에서 얻은 이미지를 풀 사이즈 이미지로 선택하고 초기 볼륨으로 출력6.9, 파티클 반경을 150픽셀로 설정하고 대칭 그룹을 c1로 설정합니다. 각도 할당 탭에서 이미지 정렬을 로컬로 설정, 반복 수를 1로 설정하고 Max를 설정합니다. 5Å/px로 목표 해상도 (보충 그림 17). 실행합니다.
- 요약 탭에서 출력 볼륨이 300x300x300 픽셀보다 작고 픽셀 크기가 약간 높은지 확인합니다.
- 결과를 분석하려면 얻은 결과를 확인합니다. FSC를 보려면 디스플레이 해상도 플롯을 클릭하고 디스플레이 볼륨에서: 재구성된 볼륨(추가 그림 18)을 확인합니다. 4-3.5Å에 가까운 좋은 해상도 볼륨이 얻어진다.
- 출력 파티클 표시를 클릭하고 파티클 목록이 있는 창에서 막대 아이콘을 클릭합니다. 새 창에서 100개의 빈이 있는 히스토그램으로 유형을 선택하고 _xmipp_cost 레이블을 선택하고 마지막으로 플롯(추가 그림 19)을 누릅니다. 이렇게 하면 비용 레이블의 히스토그램이 제시되며, 여기에는 파티클의 상관관계가 선택된 투영 방향과 포함됩니다. 이 경우, 단모달 밀도 함수가 얻어지며, 이는 입자 집합에 다른 집단을 갖지 않는다는 신호이다. 따라서 그들 모두는 정제를 계속하는 데 사용됩니다
참고: 다중 모달 밀도 함수를 볼 경우 더 높은 최대값에 속하는 파티클 집합을 선택하여 워크플로우만 계속해야 합니다. - 열고 다시 xmipp3 실행 - 이전 실행에서 계속 높은? 예, 8.5 이후 얻은 전체 크기 이미지 로 설정하고 Xmipp Highres의 이전 실행과 함께 이전 실행을 선택합니다. 각 할당 탭에서 이미지 정렬 을 로컬로 설정하고 1회 반복과 2.6Å/px를 대상 해상도(전체 해상도)로 설정합니다.
- 이제 출력에는 전체 해상도(크기 300x300x300 픽셀)의 볼륨이 포함되어야 합니다. 결과를 분석 하려면 얻은 볼륨과 FSC를 다시 확인하며, 이제 약 3Å(그림 10)에서 고해상도 볼륨이어야 합니다.
9. 평가 및 후처리
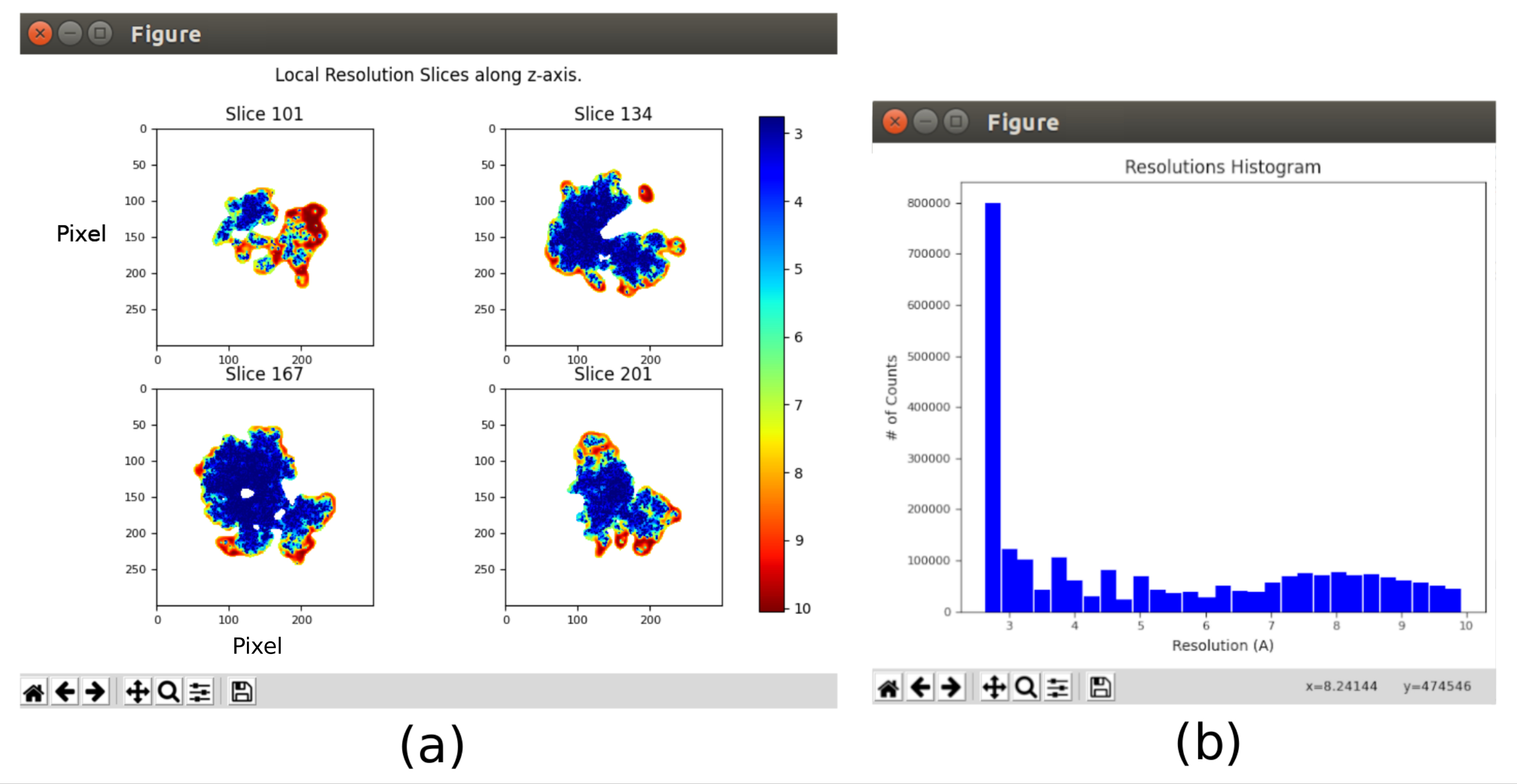
- 오픈 xmipp3 - 로컬 MonoRes28. 이 메서드는 로컬로 해상도를 계산합니다. 입력 볼륨으로 설정 8.10 후 얻은 볼륨, 설정 절반 볼륨을 사용 하시겠습니까? 실행합니다.
- 결과 분석을 클릭하고 해상도 히스토그램 표시를 선택하고 컬러 슬라이스 표시(그림 11)를 선택합니다. 볼륨의 다른 부분의 해상도가 표시됩니다. 구조의 중앙 부분의 복셀의 대부분은 3Å 주위에 해상도를 제시해야하며, 최악의 해상도는 외부 부분에서 달성된다. 또한 복셀 당 해상도의 히스토그램은 (아래도) 3Å 주위에 피크와 함께 표시됩니다.
- xmipp3를 열고 실행 - localdeblur 선명하게 29를 선명하게 적용합니다. 8.10에서 얻은 입력 맵으로 선택하고 해상도 맵으로 모노Re를 사용하여 이전 단계에서 얻은 맵으로 선택합니다.
- 결과 분석 분석을 클릭하여 얻은 볼륨을 확인합니다. 알고리즘의 마지막 반복에 해당하는 마지막 반복을 엽니다. UCSF Chimera26과 같은 다른 도구로 볼륨을 열어 볼륨의 특징을 3D로 더 잘 볼 수 있도록 하는 것이 좋습니다(그림 12).
- 마지막으로 xmipp3에 포함된 유효성 검사 도구를 엽니다- 파티클 수와 함께 해상도가 어떻게 변경되는지 보여 주는 overfitting30의 유효성을 검사합니다. 그것을 열고 8.5 단계에서 얻은 입자를 입력 입자로 포함하고, 해상도에 대한 경계 노이즈 계산을 설정하려면 초기 3D 참조 볼륨을 8.10의 출력으로 설정합니다. 고급 옵션에서는 파티클 수를 "500 1000 1500 2000 2000 3000 5000 1000 15000 2000"(보충 그림 20)으로 설정합니다. 실행합니다.
- 결과를 분석하려면 클릭합니다. 재구성에 사용되는 입자수가 증가함에 따라 녹색 선에서 해상도의 진화와 함께 두 플롯(그림 13)이 나타납니다. 빨간색 선은 정렬 된 가우시안 소음의 재구성으로 달성 된 해상도를 나타냅니다. 해상도는 입자의 수와 노이즈로부터의 입자에 비해 재구성의 큰 차이로 향상되며, 이는 좋은 구조 정보를 갖는 입자를 갖는 지표이다.
- 이전 결과에서, 처리후 부피에서 모델의 피팅이 수행될 수 있으며, 이는 거대 분자의 생물학적 구조를 발견할 수 있게 하였다.
결과
우리는 플라스모듐 팔시파룸 80S 리보솜 (EMPIAR 항목 : 10028의 데이터 세트를 사용했습니다. EMDB 항목: 2660) 테스트를 실시하고, 이전 섹션에서 제시된 시피온 프로토콜을 통해, 이 특정 예에서 거대 분자의 고해상도 3D 재구성 부피를 달성하였으며, 시편의 임의의 방향에서 2D 프로젝션을 포함하는 매우 시끄러운 이미지로 구성된 현미경으로 수집된 정보로 시작하여 달성되었다.
전체 프로토콜을 실행한 후 얻은 주요 결과는 도 10, 도 11 및 도 12로 표시됩니다. 도 10은 후처리 전에 얻어진 3D 볼륨을 나타낸다. 도 10a에서 3Å의 FSC는 나이퀴스트 한계에 매우 가깝다는 것을 알 수 있습니다(픽셀 크기가 1.34Å인 데이터, 나이퀴스트 제한은 2.6Å). 그림 10b는 높은 수준의 세부 사항과 잘 정의된 구조로 재구성된 3D 볼륨의 일부 조각을 보여줍니다. 도 11에서 얻어진 3D 부피의 분해능을 로컬로 분석한 후의 결과가 제시된다. 구조의 복셀의 대부분은 구조의 중앙 부분에 주로 위치한 3 Å 이하의 해상도를 달성하는 것을 볼 수 있습니다. 그러나 외부 부품은 그림 10b의 조각에서 해당 영역에 나타나는 흐림과 일치하는 더 나쁜 해상도를 보여줍니다. 도 12는 볼륨의 더 높은 주파수를 강조 표시할 수 있는 후 동일한 3D 맵을 나타내며, 자세한 내용을 공개하고 표현이 개선되어 특히 도 12c의 3D 프레젠테이션에서 볼 수 있습니다.
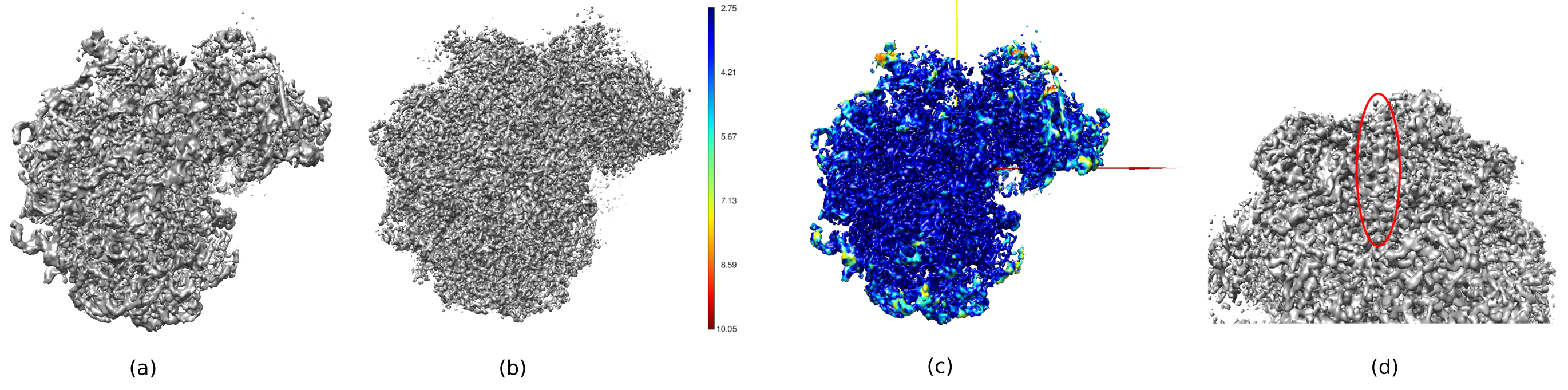
도 14에서, Chimera26은 획득된 볼륨(도 14a), 사후 처리(도 14b), 및 해상도 맵(도 14c)의 3D 표현을 로컬 해상도의 색상 코드로 채색하는 데 사용하였다. 이것은 얻어진 구조에 대한 더 많은 정보를 제공 할 수 있습니다. 이 도구는 구조의 전체 3D 컨텍스트에서 매우 작은 세부 사항을 볼 수 있기 때문에 얻어진 볼륨의 품질에 대한 통찰력을 얻는 데 매우 유용합니다. 달성된 해상도가 충분할 때, 구조의 일부 생화학적 부분도 발견될 수 있습니다(예: 도 14d의 알파-헬릭). 이 그림에서는 도 14c의 진한 파란색 영역으로 볼 수 있는 3D 구조의 모든 중앙 부분에서 달성된 고해상도를 강조해야 한다.
이전 의 모든 결과는 전체 프로토콜의 좋은 성능 덕분에 달성되었지만 그렇지 않을 수 있습니다. 잘못된 행동을 식별하는 방법에는 여러 가지가 있습니다. 가장 일반적인 경우, 이것은 얻은 구조가 낮은 해상도를 가지고 더 나은 것으로 진화 할 수없는 경우에 발생합니다. 이 것의 한 가지 예는 그림 15에 제시됩니다. 흐린 부피(도 15c)는 FSC 곡선(도 15a)과 로컬 추정의 히스토그램(도 15b)에서 볼 수 있는 낮은 FSC를 생성합니다. 이 예제는 잘못된 입력 데이터가 있는 3D 정제 방법을 사용하여 생성되었으며, 이는 충족되지 않는 입자의 입력 집합에 일부 특정 특성을 예상했기 때문이다. 볼 수 있듯이, 다른 방법이 데이터를 수신하고 제대로 준비하는 방법을 알고 항상 매우 중요합니다. 일반적으로 그림 15 의 출력과 같은 출력을 얻을 때 처리 워크플로 또는 기본 데이터에 문제가 있을 수 있습니다.
프로토콜이 제대로 진화하는지 아닌지 알아서 분석할 수 있는 워크플로를 따라 몇 가지 검사점이 있습니다. 예를 들어, 따기 직후, 앞에서 설명한 몇 가지 방법은 파티클의 순위를 지정하고 각 메서드에 대해 점수를 줄 수 있습니다. 잘못된 파티클이 있는 경우 이러한 방법을 사용하면 이러한 방법을 사용하여 파티클을 식별하고 제거할 수 있습니다. 또한, 2D 분류는 입자의 나쁜 세트를 갖는 좋은 지표가 될 수 있습니다. 도 16 은 이러한 나쁜 집합의 예를 나타낸다. 도 16a에서는 구조의 일부 세부 사항을 포함하는 좋은 클래스가 표시되고 , 도 16b 는 시끄러운 또는 중심이 없는 나쁜 클래스를 나타내며, 이 마지막 경우에는 따기가 부정확하고 두 개의 입자가 함께 나타나는 것을 볼 수 있습니다. 또 다른 검사점은 초기 볼륨 추정, 도 17 은 좋은 (그림 17a) 및 나쁜 (그림 17b) 초기 추정의 예를 보여줍니다. 잘못된 추정은 메서드에 대한 잘못된 설정을 사용하여 작성되었습니다. 분석되는 데이터에 따라 모든 매개 변수를 적절하게 선택하여 모든 설정을 신중하게 수행해야 한다는 점을 고려해야 합니다. 최소한의 구조 정보가 있는 맵이 없는 경우 다음과 같은 구체화는 양호한 재건을 얻을 수 없습니다.
영화가 구조적 정보를 보존하지 않는 나쁜 획득이 문제가 발생하면 좋은 입자를 추출하고 성공적인 처리를 하는 것은 불가능합니다. 이 경우 고해상도 3D 재구성을 위해 더 많은 영화를 수집해야 합니다. 그러나 그렇지 않은 경우 처리 워크플로를 따라 문제를 관리하는 몇 가지 방법이 있습니다. 피킹이 충분하지 않은 경우, 피킹을 반복하거나, 다른 방법을 사용하거나, 수동으로 더 많은 입자를 선택하여 학습하는 방법을 시도하는 등 이를 고치는 여러 가지 방법이 있습니다. 2D 분류 중에 몇 가지 클래스만 좋은 경우 선택 프로세스를 반복하는 것도 고려해 보십시오. 초기 볼륨 추정에서 일부 메서드가 부정확한 결과를 준 경우 여러 가지 방법을 사용하려고 합니다. 3D 세련미도 마찬가지입니다. 이 추론에 이어 이 원고에는 여러 가지 합의 도구가 제공되었으며, 이는 문제를 방지하고 정확한 데이터로 처리를 계속하는 데 매우 유용할 수 있습니다. 여러 가지 방법 간의 합의를 통해 잘못된 데이터의 지표인 선택, 분류, 정렬 등 어려운 데이터를 폐기할 수 있습니다. 그러나 생성된 출력에 여러 가지 메서드가 동의할 수 있는 경우 이러한 데이터에는 처리를 계속할 중요한 정보가 포함되어 있을 수 있습니다.
독자는 더 많은 데이터 집합을 다운로드하고 이 원고에 제시된 권장 사항에 따라 데이터를 처리하고 Scipion을 사용하여 처리 패키지를 결합한 유사한 워크플로우를 만드는 것이 좋습니다. 데이터 집합을 처리하는 것은 Cryo-EM의 최첨단에서 사용할 수 있는 처리 도구의 능력을 배우고, 처리 중에 나타나는 가능한 단점을 극복하고, 각 특정 테스트 사례에서 사용 가능한 방법의 성능을 향상시키는 가장 좋은 규칙을 아는 가장 좋은 방법입니다.

그림 1. 동영상 정렬 결과. (a) 생성된 모든 현미경 그래프의 목록과 추가 정보: 전력 스펙트럼 밀도, 극좌의 추정 정렬궤적, 카르테시안 좌표에서 동일, 생성된 현미경그래프의 파일이름. (b) 카르테시안 좌표로 표현된 정렬 궤적. (c) 생성된 현미경 그래프. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2. Ctffind 결과를 가진 CTF 추정. 결과를 가진 메인 창에는 데이터에서 오는 PSD와 함께 추정된 PSD(모서리)가 있는 그림및 여러 디포커스 파라임이 포함됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 3. Xmipp와 수동 따기 창. (a) 처리할 현미경 사진 목록과 다른 매개 변수가 있는 주 창입니다. (b) 마이크로그래프 영역 내에서 수동으로 입자를 골라. (c) 및 (d) 자동으로 선택된 입자를 선택하여 Xmipp 자동 피킹 방법에 대한 교육 입자 집합을 생성합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 4. Xmipp 결과와 깊은 합의 따기. 매개 변수 zScoreDeepLearning 입자의 양호에 무게를 제공하고 나쁜 입자를 발견하는 열쇠입니다. (a) 가장 낮은 zScores 값은 아티팩트와 연결됩니다. (b) 가장 높은 zScores는 거대 분자를 포함하는 입자와 연관된다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 5. Cryosparc 결과와 2D 분류. 생성된 클래스(동일한 방향에서 나오는 파티클의 하위 집합의 평균)가 표시됩니다. 빨간색으로 선택된 몇 가지 좋은 클래스(일부 세부 수준) 및 일부 잘못된 클래스는 선택되지 않은 클래스(시끄러운 클래스 및 중심되지 않은 클래스)입니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 6. 3D 초기 볼륨과 무리 합의 결과. 합의 도구 xmipp3를 실행한 후 얻은 3D 초기 부피의 보기 - Xmipp 및 Relion의 이전 3D 초기 볼륨 추정을 사용하여 군단 컨센서스. (a) 볼륨은 조각으로 표시됩니다. (b) 볼륨의 3D 시각화. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 7. 리온 결과3D 초기 볼륨의 정제. (a) FSC 커브를 얻어4.5Å로 임계값을 가로지르며 약 4.5Å로 교차한다. (b) 3D 구의 상층뷰로 도시된 각 커버리지. 이 경우 대칭이 없기 때문에 할당된 파티클이 전체 구를 덮어야 합니다. (c) 슬라이스로 표현된 정제 된 볼륨. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 8. Xmipp 결과와 딥 러닝을 기반으로 하는 3D 정렬. xmipp3에 의해 생성 된 결과 - 3D 정렬을위한 깊은 정렬 방법. (a) 변환 행렬의 형태로 모든 파티클에 대한 각 할당입니다. (b) 각 커버리지. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 9. 3D 정렬 합의 결과. (a) 이동 및 각도 매개 변수의 획득 된 차이를 가진 입자 목록. (b) 파티클당 각도 차이의 플롯입니다. (c) 파티클당 시프트 차이의 플롯. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 10. 3D 개선 결과의 최종 반복. (a) FSC 곡선. (b) 슬라이스로 전체 해상도로 볼륨을 획득했습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 11. Xmipp 결과를 가진 로컬 해상도 분석. 방법 xmipp3 의 결과 - 로컬 모노리스. (a) 색상 코드에 표시된 대로 복셀당 해상도 값으로 착색된 일부 대표적인 슬라이스입니다. (b) 로컬 해상도 히스토그램. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 12. Xmipp 결과 선명. xmipp3 의 결과 - 로컬데블러 선명하게 하는 방법. (a) 반복당 획득된 볼륨 목록입니다. (b) 슬라이스로 표시된 마지막 반복 후에 얻은 3D 볼륨입니다. (c) 최종 볼륨의 3D 표현. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 13. Xmipp 결과에서 도구의 피팅 유효성을 검사합니다. xmipp3 의 결과 - 유효성 검사 오버 피팅. 녹색 선은 데이터에서 재구성하는 데 해당하며 노이즈에서 빨간색 선이 표시됩니다. (a) 입자 수의 로가릿을 곱한 해상도의 역. (b) 파티클 수를 가진 해상도. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 14. 획득된 볼륨의 몇 가지 3D 표현. (a) 미리 처리된 볼륨. (b) 처리 후 볼륨. (c) 로컬 해상도, 진한 파란색 복셀은 해상도가 높은(2.75Å)이고 어두운 빨간색 복셀은 해상도가 낮은 복셀(10.05Å)입니다. (d) 알파-나선(빨간색 타원형)을 볼 수 있는 후 처리된 볼륨을 확대합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 15. 나쁜 3D 재구성의 예입니다. (a) FSC 곡선은 급격한 하락과 낮은 해상도로 임계값을 넘습니다. (b) 로컬 해상도 히스토그램. (c) 슬라이스별 3D 볼륨. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 16. 2D 클래스의 예입니다. (a) 일부 수준의 세부 사항을 보여주는 좋은 클래스입니다. (b) 소음과 아티팩트가 포함된 불량 클래스(Xmipp로 얻은 상부, CryoSparc로 낮음). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 17. 서로 다른 특성을 가진 3D 초기 볼륨의 예입니다. (a) 거대 분자의 형상을 관찰할 수 있는 좋은 초기 부피. (b) 얻어진 형상이 예상된 형상과 완전히 다른 불량 초기 볼륨. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
보충 도 1. 시피온 프로젝트 만들기. 이전 프로젝트를 선택할 수 있는 Scipion에서 표시한 창또는 해당 프로젝트의 이름과 위치를 제공하는 새 프로젝트를 만들 수 있습니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 2. 가져오기 동영상 메서드입니다. pwem - 가져오기 영화가 열려있을 때 Scipion에 의해 표시되는 창. 여기서는 Scipion에서 영화를 처리할 수 있도록 주요 획득 매개 변수를 포함해야 합니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 3. 동영상 정렬 메서드입니다. xmipp3 - 광학 정렬 이 사용되는 경우 Scipion에 의해 표시되는 창. 입력 영화, 정렬로 간주되는 프레임 범위 및 영화를 처리하는 다른 매개 변수가 채워져야 합니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 4. Ctffind와 CTF 추정 방법. 프로그램 Ctffind를 실행하는 데 필요한 모든 필드와 시피온의 양식. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 5. 시피온의 마법사. 사용자가 양식의 일부 매개 변수를 채우는 데 도움이 되는 마법사입니다. 이 경우 마법사는 ctffind 메서드인 grigoriefflab 에서 해상도 필드를 완료하는 것입니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 6. CtF 미세 한 방법 Xmipp. xmipp3 의 형태 - 이전에 추정 된 CTF의 구체화를 만들기 위해 모든 매개 변수와 ctf 추정. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 7. 마이크로 그래프 전처리 방법. xmipp3 의 형태 - 그들 위에 일부 작업을 수행 할 수있는 사전 처리 현미경 사진. 이 예제에서는 불량 픽셀 제거 및 다운샘플 현미경 그래프가 유용한 샘플입니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 8. Cryolo와 함께 선택 방법. 미리 학습된 네트워크를 사용하여 Cryolo 따기 메서드를 실행하는 양식입니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 9. Xmipp와 합의 따기 방법. xmipp3 의 형태 - 다른 따기 방법으로 얻은 좌표의 여러 세트를 통해 미리 훈련 된 네트워크를 사용하여 좌표의 합의를 계산하는 딥 학습에 기초한 깊은 합의 따기. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 10. 추출 입자 메서드. xmipp3 의 입력 및 사전 처리 탭 - 추출 입자. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 11.3D Xmipp와 초기 볼륨 방법. 메서드 xmipp3 의 형태 - 초기 3D 맵을 얻기 위해 중요한 재구성. 입력 및 기준 탭이 표시됩니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 12. 볼륨 메서드의 크기를 조정합니다. 볼륨의 자르기 또는 크기를 조정하는 양식입니다. 이 예제에서는 xmipp3 이후에 전체 크기 볼륨을 생성하는 데 이 메서드 를 사용합니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 13.3D 리온 결과 초기 볼륨. 슬라이스에 의한 리 온 - 3D 초기 모델 방법을 이용한 얻어진 3D 초기 볼륨의 뷰입니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 14. 리온으로 초기 볼륨의 정제. 방법 리온 의 형태 - 3D 자동 정제. 이 예제에서는 합의 후 예상된 초기 볼륨을 구체화하는 데 사용되었습니다. 입력 및 참조 3D 맵 탭이 표시됩니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 15.3D 분류 방법. 리온 의 형태 - 3D 분류. 탭 입력, 참조 3D 맵 및 최적화가 표시됩니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
딥 러닝 방법에 기반한 보충 도 16.3D 정렬. 메서드 xmipp3 - 깊은 정렬에 대한 양식이 열렸습니다. 여기서는 교육 세트로 네트워크를 학습한 다음 해당 네트워크가 파티클당 각도 할당을 예측할 필요가 있습니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 17.3D 세련 미 방법. xmipp3 의 형태 - 하이레스 방법. 탭 입력 및 각도 할당이 표시됩니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 18. 3D 개선 결과의 첫 번째 반복. (a) FSC 곡선. (b) 조각으로 표현된 볼륨(전체 해상도보다 작은 크기). 이 파일을 다운로드하려면 여기를 클릭하십시오.
보충 도 19. 3D 미세 조정 상관 관계 분석의 첫 번째 반복. 파티클 목록이 있는 창 상단의 막대 아이콘을 클릭하여 새 창이 나타납니다. 플롯 열 창에서 원하는 추정 매개 변수의 히스토그램을 만들 수 있습니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
추가 그림 20. 유효성 검사 부적합 도구입니다. xmipp3 의 형태 - 과적합 방법을 검증합니다. 이 파일을 다운로드하려면 여기를 클릭하십시오.
토론
현재, 저온-EM은 생물학적 샘플의 3D 구조를 드러내는 핵심 도구입니다. 현미경으로 좋은 데이터를 수집하면 사용 가능한 처리 도구를 사용하면 연구 하에 거대 분자의 3D 재구성을 얻을 수 있습니다. Cryo-EM 데이터 처리는 거대 분자의 기능 적 행동을 이해하는 열쇠이며 약물 발견에서도 중요한 거의 원자 해상도를 달성 할 수 있습니다.
Scipion은 통합 방식으로 가장 관련성이 뛰어난 이미지 처리 패키지를 결합한 전체 워크플로우를 만들 수 있는 소프트웨어로 전체 이미지 처리 워크플로우의 추적성과 재현성을 지원합니다. Scipion은 처리를 수행하는 매우 완벽한 도구 세트를 제공합니다. 그러나 고해상도 재구성을 얻는 것은 획득한 데이터의 품질과 이러한 데이터가 처리되는 방식에 완전히 달려 있습니다.
고해상도 3D 재구성을 위해 첫 번째 요구 사항은 구조적 정보를 고해상도로 보존하는 현미경으로부터 좋은 영화를 얻는 것입니다. 그렇지 않은 경우 워크플로는 데이터에서 고화질 정보를 추출할 수 없습니다. 그런 다음 성공적인 처리 워크플로우가 구조에 실제로 해당하는 입자를 추출하고 3D 공간에서 이러한 파티클의 방향을 찾을 수 있어야 합니다. 워크플로의 단계가 실패하면 재구성된 볼륨의 품질이 저하됩니다. Scipion은 모든 처리 단계에서 다른 패키지를 사용할 수 있으므로 데이터를 처리하는 가장 적절한 방법을 찾는 데 도움이 됩니다. 또한 다양한 방법의 예상 출력에서 합의를 찾아 정확도를 높이는 컨센서스 도구를 사용할 수 있는 패키지가 많기 때문에 사용할 수 있습니다. 또한 대표 결과 섹션에서 여러 유효성 검사 도구와 워크플로의 모든 단계에서 정확하고 부정확한 결과를 식별하고 잠재적인 문제를 감지하는 방법 및 이를 해결하는 방법에 대해 자세히 설명했습니다. 프로토콜을 따라 프로토콜이 제대로 실행중인지 아닌지 깨닫는 데 도움이 되는 몇 가지 검사점이 있습니다. 가장 관련성이 높은 것 중 일부는 피킹, 2D 분류, 초기 볼륨 추정 및 3D 정렬입니다. 입력을 확인하거나, 다른 방법으로 단계를 반복하거나, 컨센서스를 사용하면 사용자가 문제가 나타날 때 솔루션을 찾는 데 사용할 수 있는 옵션입니다.
Cryo-EM 필드의 패키지 통합에 대한 이전의 접근 방식과 관련하여 Appion31 은 다른 소프트웨어 패키지를 실제 통합할 수 있는 유일한 방법이다. 그러나, Appion은 단단히 레기논32, 전자 현미경에서 이미지의 자동화 된 수집을위한 시스템과 연결되어 있습니다. Scipion의 주요 차이점은 데이터 모델과 스토리지가 덜 결합된다는 것입니다. 이러한 방식으로 Scipion에서 새 프로토콜을 만들려면 파이썬 스크립트만 개발해야 합니다. 그러나 Appion에서 개발자는 스크립트를 작성하고 기본 데이터베이스를 변경해야 합니다. 요약하자면, Scipion은 유지 보수 및 확장성을 단순화하기 위해 개발되었습니다.
우리는이 원고에 Plasmodium falciparum 80S 리보솜의 실제 사례 데이터 세트를 사용하여 Cryo-EM 처리를위한 완전한 워크 플로우를 제시했습니다 (EMPIAR 항목 : 10028, EMDB 항목 : 2660). 여기서 다루어지고 논의된 단계는 영화 정렬, CTF 추정, 입자 따기, 2D 분류, 초기 지도 추정, 3D 분류, 3D 정제, 평가 및 사후 처리로 요약할 수 있습니다. 다른 패키지가 사용되었으며 이러한 여러 단계에서 합의 도구가 적용되었습니다. 최종 3D 재구성 볼륨은 3Å의 해상도를 달성했으며, 처리 후 부피에서 일부 보조 구조는 알파 헬릭과 같이 구별될 수 있으며, 이는 원자가 공간에서 배열되는 방식을 설명하는 데 도움이 됩니다.
이 원고에 제시된 워크플로우는 Scipion을 사용하여 다양한 Cryo-EM 패키지를 간단하고 통합적인 방식으로 결합하여 처리를 단순화하고 동시에 보다 신뢰할 수 있는 결과를 얻을 수 있는 방법을 보여줍니다.
미래에새로운 방법과 패키지의 개발은 성장을 계속하고 쉽게 그들 모두를 통합하는 Scipion 같은 소프트웨어는 연구원에 대한 더 중요 할 것이다. 합의 접근법은 다른 기초가 있는 많은 방법이 제공될 때, Cryo-EM의 재건 과정에 관련된 모든 매개 변수에 대한 보다 정확한 추정을 얻는 데 도움이 될 때 더욱 관련성이 높을 것입니다. 추적 및 재현성은 연구 프로세스의 핵심이며 완전한 워크플로우 실행을 위한 공통프레임워크를 가지고 있기 때문에 Scipion을 통해 쉽게 달성할 수 있습니다.
공개
저자는 공개 할 것이 없습니다.
감사의 말
저자는 에서 경제적 지원을 인정하고 싶습니다: 보조금을 통해 과학 혁신의 스페인 부: PID2019-104757RB-I00/AEI/10.13039/501100011033, 그랜트를 통해 "코무니다드 Autónoma 드 마드리드" 그랜트를 통해: S2017/BMD-3817, 인스티투토 드 살루드 카를로스 III, PT17/0009/0010 (ISCIII-SGEFI/ERDF), 유럽 연합(EU) 및 호라이즌 2020 보조금: 교육 - ULTRA (INFRADEV-03-2016-2017, 제안: 731005), EOSC 생활 (INFRAOSC-0418, 2418 824087), iNEXT - 디스커버리 (제안 : 871037), 및 HighResCells (ERC - 2018 - SyG, 제안 : 810057). 이러한 결과를 낳은 프로젝트는 "라 카이사"재단 (ID 100010434)의 펠로우십의 지원을 받았다. 펠로우십 코드는 LCF/BQ/DI18/11660021. 이 프로젝트는 마리 Skłodowska-Curie 보조금 계약 No. 713673 따라 유럽 연합의 호라이즌 2020 연구 및 혁신 프로그램에서 자금을 받았습니다. 저자는 랜드마크 ESFRI 프로젝트인 지도의 지원과 자원의 사용을 인정합니다.
자료
| Name | Company | Catalog Number | Comments |
| no material is used in this article | - | - | - |
참고문헌
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 13 (1), 24-27 (2016).
- Kühlbrandt, W. The Resolution Revolution. Science. 343 (6178), 1443-1444 (2014).
- . 1.15 A structure of human apoferritin obtained from Titan Mono- BCOR microscope Available from: https://www.rcsb.org/structure/7A6A (2021)
- Arnold, S. A., et al. Miniaturizing EM Sample Preparation: Opportunities, Challenges, and "Visual Proteomics". PROTEOMICS. 18 (5-6), 1700176 (2018).
- Faruqi, A. R., McMullan, G. Direct imaging detectors for electron microscopy. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 878, 180-190 (2018).
- Vilas, J. L., et al. Advances in image processing for single-particle analysis by electron cryomicroscopy and challenges ahead. Current Opinion in Structural Biology. 52, 127-145 (2018).
- Martinez, M., et al. Integration of Cryo-EM Model Building Software in Scipion. Journal of Chemical Information and Modeling. 60, 2533-2540 (2020).
- de la Rosa-Trevín, J. M., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195, 93-99 (2016).
- de la Rosa-Trevín, J. M., et al. Xmipp 3.0: an improved software suite for image processing in electron microscopy. Journal of Structural Biology. 184, 321-328 (2013).
- Scheres, S. H. W. . Methods in Enzymology. The Resolution Revolution: Recent Advances In cryoEM. , 125-157 (2016).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods. 14, 290-296 (2017).
- Ludtke, S. J. 3-D structures of macromolecules using single-particle analysis in EMAN. Methods in Molecular Biology. 673, 157-173 (2010).
- Shaikh, T. R., et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 3, 1941-1974 (2008).
- Wagner, T., et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications Biology. 2, (2019).
- Mindell, J. A., Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology. 142, 334-347 (2003).
- Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography. 67, 235-242 (2011).
- Liebschner, D., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D. 75, 861-887 (2019).
- Wong, W., et al. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 3, 03080 (2014).
- Abrishami, V., et al. Alignment of direct detection device micrographs using a robust Optical Flow approach. Journal of Structural Biology. 189, 163-176 (2015).
- Sorzano, C. O. S., Jonic, S., Nunez Ramirez, R., Boisset, N., Carazo, J. M. Fast, robust and accurate determination of transmission electron microscopy contrast transfer function. Journal of Structural Biology. 160, 249-262 (2007).
- Abrishami, V., et al. A pattern matching approach to the automatic selection of particles from low-contrast electron micrographs. Bioinformatics. 29, 2460-2468 (2013).
- Sanchez-Garcia, R., Segura, J., Maluenda, D., Carazo, J. M., Sorzano, C. O. S. Deep Consensus, a deep learning-based approach for particle pruning in cryo-electron microscopy. IUCrJ. 5, 854-865 (2018).
- Sorzano, C. O. S., et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. Journal of Structural Biology. 171, 197-206 (2010).
- Sorzano, C. O. S., et al. A statistical approach to the initial volume problem in Single Particle Analysis by Electron Microscopy. Journal of Structural Biology. 189, 213-219 (2015).
- Sorzano, C. O. S., et al. Swarm optimization as a consensus technique for Electron Microscopy Initial Volume. Applied Analysis and Optimization. 2, 299-313 (2018).
- Pettersen, E. F., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 25, 1605-1612 (2004).
- Sorzano, C. O. S., et al. A new algorithm for high-resolution reconstruction of single particles by electron microscopy. Journal of Structural Biology. 204, 329-337 (2018).
- Vilas, J. L., et al. MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure. 26, 337-344 (2018).
- Ramirez-Aportela, E., et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics. 36, 765-772 (2020).
- Heymann, J. B. Validation of 3D EM Reconstructions: The Phantom in the Noise. AIMS Biophys. 2, 21-35 (2015).
- Lander, G. C., et al. Appion: An integrated, database-drive pipeline to facilitate EM image processing. Journal of Structural Biology. 166, 95-102 (2009).
- Suloway, C., et al. Automated molecular microscopy: The new Leginon system. Journal of Structural Biology. 151, 41-60 (2005).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유