
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Herstellung von Adeno-assoziierten Virusvektoren in Zellstapeln für präklinische Studien in Großtiermodellen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Hier bieten wir ein detailliertes Verfahren für die großtechnische Produktion von AAV-Vektoren in Forschungsqualität unter Verwendung von adhärenten HEK 293-Zellen, die in Zellstapeln gezüchtet wurden, und affinitätschromatographischer Aufreinigung. Dieses Protokoll liefert konsistent >1 x 1013 Vektorgenomen/ml und liefert Vektormengen, die für Großtierstudien geeignet sind.
Zusammenfassung
Adeno-assoziierte Virus (AAV) -Vektoren gehören zu den klinisch fortschrittlichsten Gentherapievektoren, wobei drei AAV-Gentherapien für den Menschen zugelassen sind. Die klinische Weiterentwicklung neuartiger Anwendungen für AAV beinhaltet den Übergang von Kleintiermodellen wie Mäusen zu größeren Tiermodellen, einschließlich Hunden, Schafen und nichtmenschlichen Primaten. Eine der Einschränkungen bei der Verabreichung von AAV an größere Tiere ist der Bedarf an großen Mengen an Hochtiterviren. Während die Suspensionszellkultur eine skalierbare Methode für die AAV-Vektorproduktion ist, verfügen nur wenige Forschungslabore über die Ausrüstung (z. B. Bioreaktoren) oder wissen, wie man AAV auf diese Weise herstellt. Darüber hinaus sind AAV-Titer oft signifikant niedriger, wenn sie in HEK 293-Suspensionszellen im Vergleich zu adhärenten HEK293-Zellen hergestellt werden. Hier beschrieben ist ein Verfahren zur Herstellung großer Mengen von High-Titer-AAV unter Verwendung von Zellstapeln. Ein detailliertes Protokoll zur Titerierung von AAV sowie Methoden zur Validierung der Vektorreinheit werden ebenfalls beschrieben. Abschließend werden repräsentative Ergebnisse der AAV-vermittelten Transgenexpression in einem Schafmodell vorgestellt. Dieses optimierte Protokoll für die Großproduktion von AAV-Vektoren in adhärenten Zellen wird es molekularbiologischen Labors ermöglichen, die Erprobung ihrer neuartigen AAV-Therapien in größeren Tiermodellen voranzutreiben.
Einleitung
Die Gentherapie unter Verwendung von Adeno-assoziierten Virus (AAV) -Vektoren hat in den letzten drei Jahrzehnten große Fortschritte gemacht1,2. Nachgewiesene Verbesserungen bei einer Vielzahl von genetischen Erkrankungen, einschließlich angeborener Blindheit, Hämophilie und Erkrankungen des Bewegungsapparates und des zentralen Nervensystems, haben die AAV-Gentherapie an die Spitze der klinischen Forschung gebracht3,4. Im Jahr 2012 genehmigte die Europäische Arzneimittel-Agentur (EMA) Glybera, einen AAV1-Vektor, der Lipoproteinlipase (LPL) zur Behandlung von LPL-Mangel exprimiert, und ist damit die erste Marktzulassung für eine Gentherapie in Europa oder den Vereinigten Staaten5. Seitdem haben zwei weitere AAV-Gentherapien, Luxturna6 und Zolgensma7,die FDA-Zulassung erhalten, und es wird erwartet, dass der Markt in den nächsten 5 Jahren mit bis zu 10-20 Gentherapien, die bis 2025 erwartet werden, schnell expandierenwird 8. Verfügbare klinische Daten deuten darauf hin, dass die AAV-Gentherapie eine sichere, gut verträgliche und wirksame Modalität ist, die sie zu einem der vielversprechendsten viralen Vektoren macht, mit über 244 klinischen Studien mit AAV, die bei ClinicalTrials.gov registriert wurden. Das zunehmende Interesse an klinischen Anwendungen mit AAV-Vektoren erfordert robuste und skalierbare Produktionsmethoden, um die Bewertung von AAV-Therapien in Großtiermodellen zu erleichtern, da dies ein kritischer Schritt in der translationalen Pipelineist 9.
Für die AAV-Vektorproduktion sind die beiden Hauptanforderungen das AAV-Genom und das Kapsid. Das Genom des Wildtyps (wt)-AAV ist eine einzelsträngige DNA mit einer Länge von etwa 4,7 kb10. Das wt-AAV-Genom umfasst invertierte terminale Wiederholungen (ITRs) an beiden Enden des Genoms, die für die Verpackung wichtig sind, sowie die Rep- und Cap-Gene 11. Die Rep- und Cap-Gene, die für die Genomreplikation, die Assemblierung des viralen Kapsids und die Verkapselung des Genoms in das virale Kapsid notwendig sind, werden aus dem viralen Genom entfernt und in trans für die AAV-Vektorproduktionbereitgestellt 12. Die Entfernung dieser Gene aus dem viralen Genom bietet Raum für therapeutische Transgene und alle notwendigen regulatorischen Elemente, einschließlich des Promotors und des PolyA-Signals. Die ITRs verbleiben im Vektorgenom, um eine ordnungsgemäße Genomreplikation und Virusverkapselung zu gewährleisten13,14. Um die Kinetik der Transgenexpression zu verbessern, können AAV-Vektorgenome so konstruiert werden, dass sie selbstkomplementär sind, was die Notwendigkeit einer Umwandlung von einzelsträngiger zu doppelsträngiger DNA-Umwandlung während der AAV-Genomreplikation verringert, aber die Kodierungskapazität auf ~ 2,4 kb15reduziert.
Über das AAV-Genomdesign hinaus bestimmt die Auswahl des Kapsidserotyps den Gewebe- und Zelltropismus des AAV-Vektors in vivo2. Zusätzlich zum Gewebetropismus wurde gezeigt, dass verschiedene AAV-Serotypen unterschiedliche Genexpressionskinetik aufweisen16. Zum Beispiel klassifizierten Zincarelli et al.17 verschiedene AAV-Serotypen in Serotypen mit niedriger Expression (AAV2, 3, 4, 5), Serotypen mit moderater Expression (AAV1, 6, 8) und Serotypen mit hoher Expression (AAV7 und 9). Sie kategorisierten auch AAV-Serotypen in langsam einsetzende Expression (AAV2, 3, 4, 5) oder schnell einsetzende Expression (AAV1, 6, 7, 8 und 9). Diese divergierenden Tropismen und Genexpressionskinetik sind auf Aminosäurevariationen in den Kapsidproteinen, Kapsidproteinbildungen und Wechselwirkungen mit Wirtszellrezeptoren / Co-Rezeptoren zurückzuführen18. Einige AAV-Kapside haben zusätzliche vorteilhafte Eigenschaften wie die Fähigkeit, die Blut-Hirn-Schranke nach intravaskulärer Verabreichung (AAV9) zu überwinden oder sich in langlebigen Muskelzellen für eine dauerhafte Transgenexpression zu befinden (AAV6, 6.2FF, 8 und 9)19,20.
Dieser Artikel zielt darauf ab, eine kostengünstige Methode zur Herstellung von hochreinen, hochtiterhaltigen AAV-Vektoren in Forschungsqualität für den Einsatz in präklinischen Großtiermodellen zu beschreiben. Die Produktion von AAV unter Verwendung dieses Protokolls wird durch Dual-Plasmid-Transfektion in adhärente menschliche embryonale Nieren (HEK) 293 Zellen erreicht, die in Zellstapeln gezüchtet werden. Darüber hinaus beschreibt die Studie ein Protokoll zur Aufreinigung der Heparinsulfataffinitätschromatographie, das für AAV-Serotypen verwendet werden kann, die Heparin-bindende Domänen enthalten, einschließlich AAV2, 3, 6, 6.2FF, 13 und DJ21,22.
Für die Herstellung von AAV-Vektoren stehen eine Reihe von Verpackungssystemen zur Verfügung. Unter diesen hat die Verwendung eines Zwei-Plasmid-Co-Transfektionssystems, bei dem die Rep- und Cap-Gene sowie die Ad-Helfergene (E1A, E1B55K, E2A, E4orf6 und VA-RNA) in einem Plasmid (pHelper) enthalten sind, einige praktische Vorteile gegenüber der üblichen Drei-Plasmid-(Dreifach-) Transfektionsmethode, einschließlich reduzierter Kosten für die Plasmidproduktion23,24 . Das AAV-Genomplasmid, das die Transgenexpressionskassette (pTransgene) enthält, muss von ITRs flankiert werden und darf eine Länge von ~4,7 kb nicht überschreiten. Vektortiter und Reinheit können durch das Transgen aufgrund möglicher zytotoxischer Wirkungen während der Transfektion beeinflusst werden. Die Beurteilung der Vektorreinheit wird hierin beschrieben. Mit dieser Methode hergestellte Vektoren, die jeweils 1 x10 13 vg/ml ergeben, wurden in Mäusen, Hamstern und Schafmodellen bewertet.
Tabelle 1: Zusammensetzung der erforderlichen Lösungen. Notwendige Informationen, einschließlich Prozentsätze und Volumen, von Komponenten, die für verschiedene Lösungen im gesamten Protokoll benötigt werden. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Protokoll
1. Doppelplasmidtransfektion von HEK293-Zellen in Zellstapeln
- Tauen Sie eine Kryo-Durchstechflasche mit HEK293-Zellen in einem Perlenbad bei 37 °C auf.
HINWEIS: Komplettes DMEM während des Auftauens der Zellen auf 37 °C vorwärmen, um sicherzustellen, dass die kalte Temperatur die Zellen beim Beschichten nicht schockt. Stellen Sie sicher, dass die Zellen eine niedrige Passagenzahl haben, idealerweise weniger als 20, um eine optimale Wachstums- und Transfektionseffizienz zu gewährleisten. Stellen Sie sicher, dass die Zellen als mykoplasmenfrei zertifiziert sind. - Der Inhalt der Kryo-Durchstechflasche wird tropfenweise in ein konisches 15-ml-Röhrchen mit 10 ml vorgewärmtem vollständigem DMEM überführt und die Zellen bei 500 x g für 5 min zentrifugiert.
- Aspirieren Sie die Medien und resuspenieren Sie dann die HEK293-Zellen in 20 ml vorgewärmtem vollständigem DMEM. Säen Sie die Zellen in eine 15 cm große Platte und inkubieren Sie bei 37 °C mit 5%CO2.
- Teilen Sie die Zellen von einer 15 cm großen Platte in drei auf, um sie in der Zellkulturkammer zu säen.
- Sobald die Zellen zu 80% konfluent sind, saugen Sie das Medium ab und waschen Sie die Platte vorsichtig mit 3 ml PBS, um die Monoschicht nicht zu stören. Dann aspirieren Sie PBS und fügen Sie 3 ml Trypsin hinzu.
- 2 min bei 37 °C inkubieren, bis sich die Zellen von der Platte abheben, und dann Trypsin neutralisieren, indem man 7 ml vollständiges DMEM auf die Platte gibt.
- Sammeln Sie alle Medien und Zellen in einem 15-ml-Röhrchen und pelletieren Sie die Zellen durch Zentrifugieren bei 500 x g für 5 min.
- Den Überstand aus dem 15-ml-Röhrchen absaugen und das Zellpellet in 3 ml vollständigem DMEM resuspendieren. Zu jeder 15-cm-Platte, die 20 ml vollständiges DMEM enthält, 1 ml hinzufügen; Die Platten sanft schaukeln, um die Zellen gleichmäßig zu verteilen, und bei 37 °C mit 5%CO2inkubieren.
- Sobald die Zellen zu 80% konfluent sind, wiederholen Sie die Schritte 1.4.1 und 1.4.2. Sammeln Sie den Überstand in 50 ml konischen Rohren und kehren Sie das Rohr vorsichtig um, um sicherzustellen, dass die Zellen homogen sind.
- Bestimmen Sie die Zelldichte, indem Sie 10 μL der Zellproben mit 10 μL Trypanblau mischen und die Mischung zu einem Zellzählobjektträger zur Analyse im Zellzähler geben.
- Mischen Sie 1 l vorgewärmtes komplettes DMEM mit der benötigten Zellsuspension, um die Zellkulturkammer (Oberfläche von 6360 cm2)mit 1 x 104 Zellen/cm2 zusäen. Gießen Sie die Zellmischung in die Zellkulturkammer und drehen Sie sie vorsichtig, um die Zellen gleichmäßig über jede Monoschicht zu verteilen (Abbildung 1) und inkubieren Sie bei 37 ° C mit 5%CO2.
- Platten Sie neben der Zellkulturkammer eine 15 cm große Platte mit 1 x 104 Zellen/cm2 als Referenz für die Konfluenz.
- Überprüfen Sie nach ~ 65-h-Inkubation die Referenzplatte auf Konfluenz - idealerweise ~ 80% -90% Konfluent.
HINWEIS: Komplettes DMEM zur Zugabe in die Zellkulturkammer bei 37 °C vorwärmen.

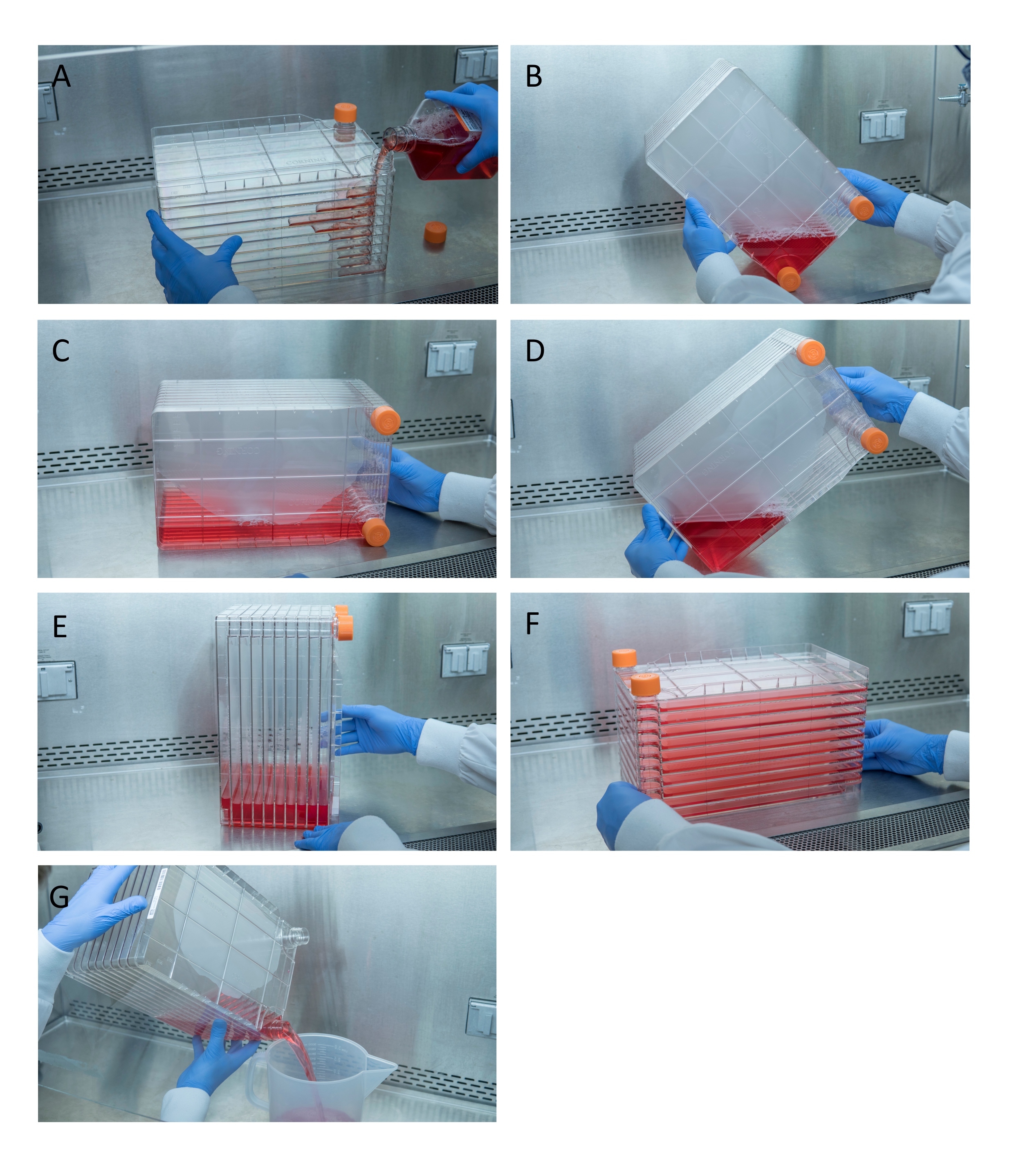
Abbildung 1: Manövrieren des Zellstapels zur Zellaussaat und Transfektion. Für die Aussaat des Zellstapels entfernen Sie zunächst eine der Entlüftungskappen und gießen Sie 1 l vorgewärmtes komplettes DMEM mit der erforderlichen Menge an HEK293-Zellen (A). Verteilen Sie Zellen und Medien gleichmäßig, indem Sie beide Belüftungskappen anziehen und alle Medien mit einer der Entlüftungskappen in die Ecke des Zellenstapels bringen und in diese Ecke (B) legen, den Zellenstapel auf die Seite legen (C) und dann den Zellenstapel um 90 ° (D) drehen, so dass die Entlüftungsanschlüsse oben sind (E). Senken Sie den Zellstapel vorsichtig auf seine normale horizontale Position und stellen Sie sicher, dass alle Kammern des Zellstapels vollständig mit Medien bedeckt sind (F). Schrauben Sie bei der Transfektion beide Entlüftungskappen ab und gießen Sie das alte Medium langsam in einen sterilen Abfallbehälter für einen gleichmäßigen Fluss, um die Monoschicht der Zellen nicht zu stören (G). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Polyethylenimin (PEI)/DNA-Gemisch in einem Konzentrationsverhältnis von 3:1 (w/w) herstellen.
- Bereiten Sie die DNA-Mischung in einem 50 ml konischen Röhrchen vor, indem Sie 475 μg pTrangen und 1425 μg pHelper zu 40 ml reduziertem Serummedium hinzufügen, um ein 3: 1-Verhältnis von pHelper: pTrangen zu erzeugen.
HINWEIS: Der PEI/DNA-Mischungsrechner kann anhand von Tabelle 2ermittelt werden. - 5,7 ml PEI (1 g/L) in das reduzierte Serummedium und die DNA-Mischung tropfenweise geben. Dann kurz wirbeln und 10 min bei Raumtemperatur inkubieren.
HINWEIS: Wenn PEI/DNA bei Raumtemperatur inkubiert, wird es leicht trüb.
- Bereiten Sie die DNA-Mischung in einem 50 ml konischen Röhrchen vor, indem Sie 475 μg pTrangen und 1425 μg pHelper zu 40 ml reduziertem Serummedium hinzufügen, um ein 3: 1-Verhältnis von pHelper: pTrangen zu erzeugen.
- Nach 8 min PEI/DNA-Inkubation werden die Medien aus der Zellkulturkammer entfernt.
HINWEIS: Stellen Sie sicher, dass Sie beide orangefarbenen Kappen lösen, um einen reibungslosen Medienfluss aufrechtzuerhalten und ein Entfernen der Zellen zu vermeiden. - Fügen Sie PEI / DNA zu 1 l vorgewärmtem vollständigem DMEM hinzu und gießen Sie die Mischung langsam in den Zellkulturkammeranschluss. Die Flüssigkeit gleichmäßig auf alle Reihen verteilen (Abbildung 1) und 72 h bei 37 °C mit 5%CO2inkubieren.
2 Ernte von AAV und chemische Lyse der transfizierten HEK293-Zellen
- Schütteln Sie die Zellkulturkammer kräftig, um die Zellen zu entfernen, bis das Medium von den entfernten Zellen trüb erscheint, und gießen Sie es in vier 500-ml-Zentrifugenröhrchen.
- Zentrifugieren Sie die Röhrchen bei 18.000 x g für 30 min bei 4 °C, um die Zellen zu pelletieren. Gießen Sie den geklärten Überstand in eine 1-Liter-Flasche aus Polyethylenterephthalat-Copolyester (PETG).
HINWEIS: Wenn man keinen Zugang zu einer Hochgeschwindigkeitszentrifuge hat, zentrifugieren Sie bei 12.000 x g für 40 min. Die pelletierten Zellen sind bei dieser Geschwindigkeit möglicherweise nicht fest und gleiten als Ausgießen des Überstands. - Die Zellpellets in 500 mL Zentrifugenröhrchen mit 50 mL Lysepuffer resuspenieren und 60 min bei 37 °C inkubieren.
- Zentrifugieren Sie die Röhrchen bei 18.000 x g für 30 min und geben Sie dann den Überstand in dieselbe 1 L PETG-Flasche. Entsorgen Sie die pelletierten Zellreste.
HINWEIS: Den geklärten Überstand sofort reinigen und bei 4 °C bis zu 72 h lagern. Bei längerer Lagerung bei -80 °C lagern. Nicht bei -20 °C lagern.
3 AAV-Vektorreinigung mittels Heparin-Affinitätschromatographie
- Das Rohlysat von -80 °C entfernen und über Nacht bei 4 °C auftauen lassen. Verwenden Sie nach dem Auftauen einen 0,22 μM-Filter, um das Rohlysat zu filtern.
- Um den Zentrifugalkonzentrator zu passivieren, geben Sie 4 ml Filtervorbehandlungspuffer zu einem Zentrifugalkonzentrator für jede verwendete Heparin-Sepharosesäule hinzu. Passivieren Sie den Zentrifugalkonzentrator bei Raumtemperatur für 2-8 h. Richten Sie die Passivierung unmittelbar vor den Reinigungsschritten ein.
- Richten Sie den Schlauch und die Pumpe ein (Abbildung 2).
- Legen Sie den Schlauch in eine Peristaltikpumpe und lassen Sie 20 ml 1 M NaOH laufen. Als nächstes laufen Sie 50 ml molekulares Wasser und dann 50 ml basales DMEM.
- Befestigen Sie 5 ml Heparin-Sepharose-Säule an Schläuchen und führen Sie 25 ml basales DMEM, um das Konservierungsmittel zu entfernen.
- 0,2 μM des gefilterten Rohlysats werden mit einer Durchflussrate von 1-2 Tropfen/s durch die Säule geführt.

Abbildung 2: Einrichtung für peristaltische Pumpe zur AAV-Reinigung. Führen Sie den Schlauch vom Rohlysat durch die Peristaltikpumpe und in die Heparinmatrixsäule. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
HINWEIS: Stellen Sie sicher, dass Sie keine Blasen einführen oder die Säule trocken laufen lassen, da dies die Säule gefährdet und die Elution von AAV verhindert. Verwerfen Sie die Säule, wenn sie trocken ist, und verwenden Sie eine neue Säule für den Rest von Rohlysat.
- Laden Sie das gesamte Rohlysat auf die Heparinsäule und verwenden Sie die folgenden Lösungen, um die Säule zu waschen.
- Waschen Sie mit 50 ml 1x Hank's Balanced Salt Solutions (HBSS) ohne Mg2+ und Ca2+.
- Waschen Sie mit 15 ml 0,5 % N-Lauroylsarcosin in HBSS ohneMg2+ und Ca2+.
- Waschen Sie mit 50 ml HBSS ohne Mg2+ und Ca2+.
- Waschen mit 50 ml HBSS mit Mg2+ und Ca2+
- Waschen Sie mit 50 ml 200 mM NaCl/HBSS mit Mg2+ und Ca2+.
- Eluieren Sie 5 x 5 ml (25 ml insgesamt) mit 300 mM NaCl /HBSS mit Mg2+ und Ca2+ und kennzeichnen Sie die Elutionen als E1-E5 (jede Elution ist von 5 ml).
- Konzentration des Virus mit einem Zentrifugalkonzentrator
- Den Zentrifugalkonzentrator, der den Vorbehandlungspuffer enthält, 2 min lang mit 900 x g drehen. Verwerfen Sie den Durchfluss.
- Waschen Sie den Zentrifugalkonzentratorfilter mit 4 ml HBSS mit Mg2+ und Ca2+ und zentrifugieren Sie bei 1000 x g für 2 min; Verwerfen Sie den Durchfluss.
- Die Elution E2 in den Zentrifugalkonzentrator geben. 5 min bei 1000 x g drehen und den Durchfluss verwerfen.
- Beenden Sie die Zugabe von E2 und dann E3 in den Zentrifugalkonzentrator und drehen Sie bei 1000 x g für 5 min, bis das konzentrierte Virus ungefähr 1 ml beträgt.
HINWEIS: Vermeiden Sie es, den Vektor so zu zentrifugieren, dass das Volumen unter dem Niveau des Filters liegt. Konzentrieren Sie E1, E4 oder E5 nicht im Zentrifugalkonzentrator, da sie sehr wenig bis gar keinen Vektor enthalten und Verunreinigungen enthalten. - Entfernen Sie das konzentrierte Virus mit einer p200-filtrierten Spitze aus dem Zentrifugenkonzentrator und legen Sie es in ein steriles 1,5-ml-Zentrifugenröhrchen.
- Spülen Sie den Zentrifugalkonzentrator mit 200 μL HBSS mit Mg2+ und Ca2+ ab, um verbleibende AAV aus dem Filter zu entfernen. Mehrmals kräftig auf und ab pipettieren (für ~ 30 s), um alle an der Membran haftenden Viren zu entfernen, und mit dem Rest des Virus in das 1,5 ml Zentrifugenröhrchen geben. Mischen Sie die Tube gut.
- Aliquot 5 μL für DNA-Extraktionen und lagern den gereinigten Vektor bei -80 °C.
- Waschen Sie die Säule mit 25 ml 2 M NaCl. Verwenden Sie außerdem 25 ml 0,1% Triton X-100, die auf 37 ° C vorgewärmt sind, um die Säule zu waschen. Als nächstes waschen Sie die Säule mit 50 ml sterilemdH2O und waschen Sie dann mit 25 ml 20% igem Ethanol.
- Stellen Sie sicher, dass die Säulenmembran vollständig mit 20% Ethanol gesättigt ist, da dies die Speicherlösung ist. Die Säule mit mitgelieferten Stopfen abdichten und bei 4 °C lagern.
- Lagern Sie den Schlauch in 1 M NaOH.
HINWEIS: Bei richtiger Reinigung können Heparin-Sepharose-Säulen bis zu fünf Mal wiederverwendet werden.
4 AAV genomische DNA-Extraktion
- Bereiten Sie die in Tabelle 3 erwähnte Reaktionsmischung in einem PCR-Röhrchen für die DNase-Behandlung vor.
| Bestandteil | Volumen |
| Gereinigter AAV-Vektor | 5 μL |
| 10x DNase Puffer | 2 μL |
| DNase | 1 μL |
| ddH2O | 12 μL |
| Letzter Band | 20 μL |
Tabelle 3: Master-Mix-Formel für die DNase-Behandlung. Empfohlene Komponenten und Volumina, die für die DNase-Behandlung von AAV-Virusvektoren während der DNA-Extraktion erforderlich sind.
- Wirbeln Sie die PCR-Röhre, um die PCR-Röhre zu mischen und zu pulsieren, um den Inhalt herunterzufahren.
- Mit einem Thermocycler bei 37 °C für 20 min inkubieren, gefolgt von 75 °C für 15 min, um die DNase zu erhitzen.
- Fügen Sie 5 μL Proteinase K hinzu.
- Mit einem Thermocycler bei 50 °C für 60 min und dann bei 95 °C für 30 min inkubieren, um Proteinase K zu inaktivieren.
- Verwenden Sie ein DNA-Reinigungskit, um potenzielle Verunreinigungen zu entfernen.
HINWEIS: Dieser Schritt wurde mit einem handelsüblichen Blut- und Gewebereinigungskit (Materialtabelle) durchgeführt.- 200 μL AL-Puffer (Blood and tissue clean up kit, Table of Materials)in das PCR-Röhrchen geben, das den mit DNase/Proteinase K behandelten Vektor enthält.
- Das PCR-Röhrchen vorwirbeln und bei 56 °C für 10 min in einem Thermocycler inkubieren.
- Die Flüssigkeit aus dem PCR-Röhrchen wird in eine sterile Spinsäule pipettiert, die in einem Auffangröhrchen sitzt.
- Geben Sie 200 μL 100% Ethanol in die Säule und mischen Sie gründlich durch Wirbeln.
- Zentrifugiere bei 6.000 x g für 1 min und entsorge den Durchfluss.
- Fügen Sie 500 μL Puffer AW1 (Blood and tissue clean up kit, Table of Materials) zur Spin-Spalte hinzu.
- Zentrifugiere bei 6.000 x g für 1 min und entsorge den Durchfluss.
- Fügen Sie 500 μL Puffer AW2 (Blood and tissue clean up kit, Table of Materials) zur Spin-Spalte hinzu.
- Zentrifugiere bei 15.000 x g für 3 min und entsorge den Durchfluss.
- Legen Sie die Spinsäule in ein steriles 1,5 ml Zentrifugenröhrchen und geben Sie 200 μL Puffer AE (Blood and tissue clean up kit, Table of Materials)direkt in die Spinsäulenmembran.
- Bei Raumtemperatur 1 Min. inkubieren.
- Zentrifugiere bei 6.000 x gfür 1 min, um die DNA zu eluieren.
- Lagern Sie die DNA bei -20 °C.
5 Titration von AAV-Vektorgenomen mittels quantitativer Polymerase-Kettenreaktion und einer Simian Virus 40 (SV40) Sonde
HINWEIS: Führen Sie alle qPCR-Arbeiten in einer PCR-Haube mit gefilterten Pipettenspitzen durch, um eine externe DNA-Kontamination zu vermeiden. Wenn das AAV-Genom keine SV40-PolyA-Sequenz kodiert, verwenden Sie eine Sonde gegen die an anderer Stelle beschriebene ITR25. Stellen Sie sicher, dass die standardmäßig ausgewählte Plasmid-DNA die SV40-PolyA-Sequenz enthält.
- Standardaufbereitung
- Verdünnen Sie den Stammplasmid-DNA-Standard (pTransgene Plasmid mit SV40 polyA-Sequenz) auf eine Endkonzentration von 10 μg/μL und lagern Sie bei -20 °C in 6 μL Aliquots.
- Bestimmen Sie die im Plasmid-DNA-Standard vorhandene Kopienzahl mit dem folgendenOnline-Rechner 26.
HINWEIS: Verwenden Sie eine Plasmid-DNA, die für den von einem kommerziellen Anbieter hergestellten Standard verwendet wird, um Qualität und korrekte Konzentration sicherzustellen. Bereiten Sie eine große Menge an Standards (z. B. 10 ml) vor, um beim Übergang zu einem neu vorbereiteten Standard Brückenstudien durchzuführen.
- Die folgende in Tabelle 4 genannte Reagenzmischung sowohl für die Proben als auch für die Norm ist in einem 1,5-ml-Zentrifugenröhrchen herzustellen.
HINWEIS: Bereiten Sie eine ausreichende Überschreitung der Mastermischung vor. Siehe Tabelle 5 für Primer/Sonden-Sequenzen.
| Bestandteil | Volumen |
| Universeller qPCR Master Mix (2X) | 10 μL |
| Wasser in molekularer Qualität | 4,5 μL |
| 40x SV40 PolyA Primer/Sonde | 0,5 μL |
| Letzter Band | 15 μL |
Tabelle 4: qPCR-Master-Mix für die AAV-Titration. Empfohlene Komponenten und Volumen, die für die qPCR von DNA erforderlich sind, die aus AAV-Virusvektoren extrahiert wurde.
| Bestandteil | Reihenfolge |
| Vorwärts-Primer | 5'-AGCAATAGCATCACAAATTTCACAA-3' |
| Reverse Primer | 5'-CCAGACATGATAAGATACATTGATGAGTT-3' |
| Sonde | /56-FAM/AGCATTTTT/Zen/TTCACTGCATTCTAGTTGTGGTTTGTC/3IABkFQ |
Tabelle 5: Primersequenzen gegen die SV40 PolyA DNA-Sequenz. Sequenzen der Primer und sonden, die für die qPCR-Titration verwendet werden und an bestimmte Bereiche von AAV-Virusvektoren binden, die die SV40-PolyA-Sequenz enthalten.
- Pipettieren Sie den Master-Mix nach oben und unten, um ihn zu mischen.
- Stellen Sie die Verdünnungsplatte auf.
- Verwenden Sie eine klare 96-Well-Platte, um Standard- und Probenverdünnungen vorzubereiten, und geben Sie 45 μL molekulares Wasser in jede Vertiefung in jede zweite Spalte, beginnend mit Spalte 1 (Spalten 1, 3, 5, 7, 9 und 11).
- Fügen Sie 5 μL des Standards in die Vertiefung A1 hinzu und pipettieren Sie zum Mischen.
- Verwenden Sie eine neue gefilterte Pipettenspitze, um eine 1/10-Verdünnung von Vertiefung A1 nach B1 zu erzeugen.
- Setzen Sie eine Reihe von 10-fachen Verdünnungen in der Säule fort, bis G1 erreicht ist.
- Fügen Sie H1 nicht hinzu, da dies als Negativkontrolle wirkt.
- Tragen Sie die erste Probe (S1) auf, indem Sie sie in die Vertiefung A3 geben und eine 1/10-Verdünnung bilden. Diese Mischung pipettieren und 5 μL in die Vertiefung B3 überführen. Entsorgen Sie die Pipettenspitze nach diesem Transfer.
- Mit einer neuen Pipettenspitze die Lösung in Vertiefung B3 mischen und eine 1/100 Verdünnung bilden. 5 μLof diese Mischung in die Vertiefung C3 geben und die Spitze nach dem Transfer verwerfen.
- Mit einer neuen Pipettenspitze die Lösung in Vertiefung C3 auf und ab pipettieren, um eine Verdünnung von 1/1000 herzustellen. Verwerfen Sie die Spitze.
- Verdünnen Sie die Proben weiter, ohne Proben zu den Spalten 2, 4, 6, 8, 10 oder 12 hinzuzufügen.
- Sobald alle Proben verdünnt sind, mischen Sie den Inhalt in Vertiefungen von Spalte 1 und übertragen Sie dann 20 μL in Spalte 2.
- Wiederholen Sie dies für die Spalten 3, 5, 7, 9 und 11, um Replikate jeder Standard- und Probenverdünnung zu erstellen. Siehe Abbildung 3 für das Plattenlayout.
ANMERKUNG: Bei folgendem Plattenaufbau in Abbildung 3weisen proben, die in den Reihen G und H verdünnt sind, nur 1/10 und 1/100 Verdünnungen auf.

Abbildung 3: Plattenlayout für die qPCR-AAV-Titration. Blau zeigt die Platzierung der seriellen Verdünnung des Standards an; grün zeigt die Platzierung der Negativkontrolle an; lila zeigt die Platzierung der Verdünnung der Proben an. Jede Standard-, Negativ- oder Probe wird im Replikat hinzugefügt. Ein Beispiel für die Konzentration des Standards wurde hinzugefügt, um die Verdünnungsreihe des Standards zu zeigen, und die Platzierung von Probenverdünnungen wurde zu ihren jeweiligen Vertiefungen hinzugefügt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Titration durch SV40 polyA detektionsbasierte qPCR
- Fügen Sie 15 μL qPCR-Mastermischung zu jeder Vertiefung einer weiß-halbschürzten 96-Well-qPCR-Platte hinzu.
- Übertragen Sie 5 μL jeder Probe von der klaren 96-Well-Platte auf die weiß-halbsampige 96-Well-qPCR-Platte.
- Verwenden Sie eine Mehrkanalpipette, um eine ausreichende Mischung des qPCR-Master-Mixes und der Probe sicherzustellen.
- Verschließen Sie die Platte mit einer Siegelfolie und zentrifugieren Sie die qPCR-Platte bei 1500 x g für 30 s.
- Führen Sie die qPCR-Reaktion auf einem plattenbasierten Real-Time-PCR-Amplifikations- und Detektionsinstrument unter Verwendung der in Tabelle 6 vorgeschlagenen Bedingungen durch.
| Abschnitt | Zyklen | Zeit | Temperatur | Beschreibung |
| Vorinkubation | 1x | 5 Minuten | 95 °C | DNA-Denaturierung. |
| Verstärkung | 38-fach | 15 Sek. | 95 °C | Amplifikation der DNA. Die Einstellungen können geändert werden, wenn alternative Primer mit unterschiedlichen Glühtemperaturen verwendet werden. |
| 60 Sek. | 60 °C | |||
| Kühlung | 1x | 60 Sek. | 40 °C | Plattenkühlung. Ende des Laufs. |
Tabelle 6: Thermocycler-Protokoll für die hydrolysesondenbasierte qPCR-Titration. Empfohlenes Thermocycler-Protokoll für die Verwendung der sondenbasierten qPCR-Titration von DNA-extrahierten gereinigten AAV-Vektoren.
HINWEIS: Für das Arbeitsblatt zur qPCR-AAV-Titration siehe Tabelle 7.
- Datenanalyse zur Bestimmung der AAV-Genomkopienzahlen.
- Füllen Sie die Tabellenkalkulationsdatenzellen (Tabelle 7A) mit den Konzentrationswerten aus, die aus dem qPCR-Lauf sowohl für Standard- als auch für Probenverdünnungen erhalten wurden.
- Verwenden Sie Konzentrationswerte aus Tabelle 7A, um eine Standardkurve zu erstellen (Tabelle 7B).
HINWEIS: Die Standardkurve wird als natürlicher Logarithmus (y = a ln(x) + b) zusammen mitR2-Effizienz dargestellt. Eine Normkurve muss einen Wirkungsgrad nahe 100 % undR2 nahe 1,0 (≥0,99) aufweisen. - Füllen Sie die Pisteneffizienz aus, indem Sie diesen Online-Rechner27ausfüllen.
HINWEIS: Ein Wirkungsgrad zwischen 90% -110% ist akzeptabel. Wenn die Effizienz von qPCR außerhalb dieses Bereichs liegt, führen Sie die qPCR erneut aus. - Verwenden Sie Konzentrationswerte aus Tabelle 7A, um die Verdünnungen jeder Probe zu mittelen und die Standardabweichung jeder Probe zu bestimmen (Tabelle 7C).
ANMERKUNG: Schließen Sie Verdünnungen von Proben aus, die mehr als eine Standardabweichung vom Durchschnitt der Probenverdünnungen entfernt sind. - Unter Verwendung der mittleren Konzentration jeder Verdünnung mit dem Verdünnungsfaktor multiplizieren und dann durch fünf teilen, um die Vektorgenome (vg) / μL jeder Probe zu erhalten (Tabelle 7C).
- Berechnen Sie den vg/ml jeder Probe, indem Sie den Mittelwert der Konzentrationen jeder Probe mit 80.000 multiplizieren (Tabelle 7C).
- Mittelwert der durchschnittlichen/ml jeder Verdünnung, um den endgültigen vg/ml jeder Probe zu erhalten (Tabelle 7C).
HINWEIS: Der Anwender muss die mittlere Konzentration jeder Verdünnung durch den Faktor fünf teilen, um die 5 μL zu berücksichtigen, die in jede Vertiefung für den qPCR-Lauf geladen werden, um die Konzentration in vg/μL zu erzeugen. Der Faktor von 80.000 berücksichtigt den Übergang vom mittleren Konzentrationswert jeder Probe zu vg/ml. Erstens muss der Mittelwert des Konzentrationswerts jeder Probe mit 2 multipliziert werden, um die einzelsträngigen Genome zu berücksichtigen, da der Primer-Sonden-Satz nur positiv-sinnige, einzelsträngige DNA (ssDNA) quantifiziert und das AAV-Genom in einem ungefähren Verhältnis von 1: 1 zwischen positiver und negativer ssDNAexistiert 25,28. Der Mittelwert des Konzentrationswerts jeder Probe ist x40 zu multiplizieren, um die Verdünnung der Probe von 5 μL des gereinigten Vektors (Abschnitt 4.1) auf 200 μL extrahierter DNA (Abschnitt 4.6.12) zu berücksichtigen. Schließlich muss der Mittelwert des Konzentrationswerts jeder Probe mit x1000 multipliziert werden, um von vg/μL in vg/mL umzurechnen.
6 Beurteilung der Vektorqualität und -reinheit
- Qualitätskontrolle - Western Blot
- Bereiten Sie ein 12% SDS PAGE Gel vor.
- Führen Sie polyacrylamid-Gelelektrophorese durch.
HINWEIS: Laden Sie 6 x 1010 Durchschnitt der Proben pro Bohrloch. - Übertragen Sie die Proteine auf die Polyvinylidendifluorid (PVDF) -Membran.
- Blockierende PVDF-Membran
- Entfernen Sie die Membran aus der Transfervorrichtung und spülen Sie 0,1% PBST ein, um loses Acrylamid zu entfernen.
- Legen Sie die Membran in Blocklösung für mindestens 1 h bei Raumtemperatur oder über Nacht bei 4 °C.
HINWEIS: Blocking Buffer kann mit 2% Ziegenserum ergänzt werden.
- Inkubation mit primärem Antikörper
- Dekantieren Sie den Blockierungspuffer und fügen Sie den primären Antikörper, einen monoklonalen Anti-AAV-Maus-Antikörper, in einer Verdünnung von 1:200 hinzu.
- Über Nacht bei 4 °C inkubieren.
- Dekantieren Sie den primären Antikörper und waschen Sie ihn fünfmal mit 0,1% PBST für 5 min bei Raumtemperatur unter Bewegung.
- Inkubation mit sekundären Antikörpern
- Dekantieren Sie die Waschlösung und fügen Sie HRP-konjugierten sekundären Antikörper hinzu, verdünnt bei einem 1:7500 in Blocking-Puffer, und inkubieren Sie für 1 h bei Raumtemperatur unter Rühren.
- Dekantieren Sie den Sekundärantikörper und waschen Sie ihn fünfmal mit 0,1% PBST für 5 min bei Raumtemperatur unter Rühren.
- Führen Sie eine abschließende Wäsche mit PBS bei Raumtemperatur unter Rühren durch.
- Nachweis der Proteine unter Verwendung von ECL-Substrat (Enhanced Chemiluminescent).
- Stellen Sie sich das Gel vor, um die viralen Proteine (VP1-, VP2- und VP3-Untereinheiten) zu visualisieren (Abbildung 4).

Abbildung 4: Western Blot zeigt AAV-Kapsidproteine. Spur A; MW-Leiter, Spur B; AAV6.2FF-hIgG01, Spur C; AAV6.2FF-hIgG02, Spur D; AAV6.2FF-hIgG03 und Spur E; AAV6.2FF-hIgG04. 6 x 1010 vg jedes AAV6.2FF-hIgG wurde in die jeweiligen Lanes geladen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Reinheitskontrolle - SDS PAGE und Coomassie Stain
- Bereiten Sie SDS-PAGE Gel und Proben wie in Schritt 6.1.1 und 6.1.2 beschrieben vor.
- Fixieren Sie das Gel in Fixierlösung für 1 h oder über Nacht mit sanfter Bewegung. Wechseln Sie die Fixierlösung einmal in der ersten Stunde.
- Färben Sie das Gel in Färbelösung für 2-4 h mit sanfter Bewegung.
- Entfärben Sie das Gel mit einer Entfärbungslösung. Füllen Sie die Entfärbungslösung mehrmals auf, bis der Hintergrund des Gels vollständig entfleckt ist (4-24 h).
- Bewahren Sie das entfärbte Gel in einer Lagerlösung auf.
- Stellen Sie sich das Gel vor, um alle Proteine zu visualisieren, die von der Coomassie-Färbelösung gefärbt wurden.

Abbildung 5: Coomassie-gefärbtes Gel. Spur A; MW-Leiter, Spur B; AAV6.2FF-hIgG01, Spur C; AAV6.2FF-hIgG02, Spur D; AAV6.2FF-hIgG03, Spur E; AAV6.2FF-hIgG04, Spur F; AAV6.2FF-hIgG05 und Spur G; AAV6.2FF-hIgG06. 6 x 1010 vg jedes AAV6.2FF-hIgG wurde in die jeweiligen Lanes geladen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Alternativer Reinheitskontrolltest - HEK293 Wirtszellprotein-Nachweis ELISA
- Führen Sie den HEK293-Wirtszellproteinnachweis über ELISA gemäß den Anweisungen des Herstellers durch.
HINWEIS: Verwenden Sie Verdünnungen von 5 x 10-2 und 1 x 10-3 für gereinigte rAAV-Proben. Sobald TMB zum Brunnen hinzugefügt wurde, inkubieren Sie vor Licht. Die lineare Regression kann nicht zur Analyse der Ergebnisse verwendet werden. - Führen Sie eine Punkt-zu-Punkt-Analyse, einen kubischen Spline oder eine logistische Anpassungsmethode mit vier Parametern durch, um Konzentrationen von Unbekannten zu interpolieren und mit dem Verdünnungsfaktor zu multiplizieren, um die ursprüngliche Probenkonzentration zu bestimmen.
- Führen Sie den HEK293-Wirtszellproteinnachweis über ELISA gemäß den Anweisungen des Herstellers durch.
Ergebnisse
Die Translation von kleinen Nagetiermodellen in größere Tiermodelle und die letztendliche klinische Anwendung stellt aufgrund der großen Menge an AAV, die erforderlich ist, um größere Tiere zu transduzieren und therapeutische Effekte zu erzielen, eine erhebliche Herausforderung dar. Um die Transduktionseffizienz des rational entwickelten AAV6.2FF-Kapsids zu vergleichen, das zuvor eine 101-fache Steigerung der Transduktionseffizienz in murinen Muskelzellen im Vergleich zu AAV63zeigte, wurde M?...
Diskussion
Die Herstellung von rekombinanten AAV (rAAV) -Vektoren, die in diesem Artikel beschrieben werden, verwendet gängige Materialien, Reagenzien und Geräte, die in den meisten molekularbiologischen Forschungslabors und -einrichtungen zu finden sind. Dieses Papier ermöglicht die Herstellung hochwertiger in vitro und in vivo rAAV durch das Lesegerät. Vor allem dieses Protokoll für die rAAV-Produktion ist im Vergleich zu langwierigeren Protokollen mit Cäsiumchloridreinigung effizient und vermeidet den Ein...
Offenlegungen
Sarah K. Wootton ist Erfinderin eines US-Patents US10806802B2 für das AAV6.2FF-Kapsid.
Danksagungen
Amira D. Rghei, Brenna A. Y. Stevens, Sylvia P. Thomas und Jacob G. E. Yates waren Empfänger von Ontario Veterinary College Student Stipends sowie Ontario Graduate Scholarships. Amira D. Rghei erhielt ein Mitacs Accelerate Studentship. Diese Arbeit wurde durch den Canadian Institutes for Health Research (CIHR) Project Grant (#66009) und einen Collaborative Health Research Projects (NSERC Partnered) Grant (#433339) an SKW finanziert.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.22 μm filter | Millipore Sigma | S2GPU05RE | |
| 0.25% Trypsin | Fisher Scientific | SM2001C | |
| 1-Butanol | Thermo Fisher Scientific | A399-4 | CAUTION. Use under a laminar flow hood. Wear gloves |
| 10 chamber cellstack | Corning | 3271 | |
| 1L PETG bottle | Thermo Fisher Scientific | 2019-1000 | |
| 30% Acrylamide/Bis Solution | Bio-Rad | 1610158 | |
| 96-well skirted plate | FroggaBio | FS-96 | |
| Adhesive plate seals | Thermo Fisher Scientific | 08-408-240 | |
| Ammonium persulfate (APS) | Bio-Rad | 161-0700 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Blood and Tissue Clean up Kit | Qiagen | 69506 | Use for DNA clean up in section 4.6 of protocol |
| Bromophenol blue | Fisher Scientific | B392-5 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Cell Culture Dishes | Greiner bio-one | 7000232 | 15 cm plates |
| Culture Conical Tube | Thermo Fisher Scientific | 339650 | 15 mL conical tube |
| Culture Conical Tube | Fisher Scientific | 14955240 | 50 mL conical tube |
| Dulbecco's Modified Eagle Medium (DMEM) with 1000 mg/L D-glucose, L-glutamine | Cytiva Life Sciences | SH30022.01 | |
| ECL Western Blotting Substrate | Thermo Fisher Scientific | 32209 | |
| Ethanol | Greenfield | P016EA95 | Dilute ethyl alcohol(95% vol) to 20% for section 3.7.4 and 70% for section 6.1.1.1 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | SH30396.03 | |
| Glacial acetic acid | Fisher Scientific | A38-500 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | Fisher Scientific | BP381-500 | |
| HBSS with Mg2+ and Ca2+ | Thermo Fisher Scientific | SH302268.02 | |
| HBSS without Mg2+ and Ca2+ | Thermo Fisher Scientific | SH30588.02 | |
| HEK293 cells | American Tissue Culture Collection | CRL-1573 | Upon receipt, thaw the cells and culture as described in manufacturer’s protocol. Once cells have been minimally passaged and are growing well, freeze a subfraction for future in aliquots and store in liquid nitrogen. Always use cells below passage number 30. Once cultured cells have been passaged more than 30 times, it is recommended to restart a culture from the stored aliquots |
| HEK293 host cell protein ELISA kit | Cygnus Technologies | F650S | Follow manufacturer’s instructions |
| Heparin sulfate column | Cytiva Life Sciences | 17040703 | |
| Kimwipe | Thermo Fisher Scientific | KC34120 | |
| L-glutamine (200 mM) | Thermo Fisher Scientific | SH30034.02 | |
| Large Volume Centrifuge Tube Support Cushion | Corning | CLS431124 | Support cushion must be used with large volume centrifuge tubes uless the centrifuge rotor has the approriate V-bottom cushions |
| Large Volume Centrifuge Tubes | Corning | CLS431123-6EA | 500 mL centrifuge tubes |
| MgCl2 | Thermo Fisher Scientific | 7791-18-6 | |
| Microcentrifuge tube | Fisher Scientific | 05-408-129 | 1.5 mL microcentrifuge tube, sterilize prior to use |
| Molecular Grade Water | Cytiva Life Sciences | SH30538.03 | |
| N-Lauroylsarcosine sodium salt | Sigma Aldrich | L5125 | CAUTION. Wear gloves |
| NaCl | Thermo Fisher Scientific | BP35810 | |
| Optimem, reduced serum medium | Thermo Fisher Scientific | 31985070 | |
| Pasteur pipets | Fisher Scientific | 13-678-20D | Sterilize prior to use |
| PBS (10x) | Thermo Fisher Scientific | 70011044 | Dilute to 1x for use on cells |
| Penicillin-Streptomycin Solution | Cytiva Life Sciences | SV30010 | |
| pHelper plasmid | De novo design or obtained from plasmid repository | NA | |
| Pipet basin | Thermo Fisher Scientific | 13-681-502 | Purchase sterile pipet basins |
| Polyethylene glycol tert-octylphenyl ether (Triton X-100) | Thermo Fisher Scientific | 9002-93-1 | CAUTION. Wear gloves |
| Polyethylenimine (PEI) | Polyscience | 24765-1 | Follow manufacturer’s instructions to produce a 1L solution. 0.22μm filter and store at 4°C |
| Polypropylene semi-skirted PCR Plate | FroggaBio | WS-96 | |
| Polysorbate 20 (Tween 20) | Thermo Fisher Scientific | BP337-100 | CAUTION. Wear gloves |
| polyvinylidene difluoride (PVDF) membrane | Cytiva Life Sciences | 10600023 | Use forceps to manipulate. Wear gloves. |
| Primary antibody | Progen | 65158 | |
| Protein Ladder | FroggaBio | PM008-0500 | |
| Proteinase K | Thermo Fisher Scientific | AM2546 | |
| pTrangene plasmid | De novo design or obtained from plasmid repository | NA | Must contain SV40 polyA in genome to be compatible with AAV titration in section 5.0 |
| Pump tubing | Cole-Parmer | RK-96440-14 | Optimize length of tubing and containment of virus in fractions E1-E5 |
| RQ1 Dnase 10 Reaction Buffer | Promega | M6101 | Use at 10x concentration in protocol from section 4.0 |
| RQ1 Rnase-free Dnase | Promega | M6101 | |
| Sample dilutent | Cygnus Technologies | I700 | Must be purchased separately for use with HEK293 host cell protein ELISA kit |
| Secondary antibody, HRP | Thermo Fisher Scientific | G-21040 | |
| Skim milk powder | Oxoid | LP0033B | |
| Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 28312 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Sodium hydroxide (NaOH) | Thermo Fisher Scientific | SS266-4 | |
| SV40 polyA primer probe | IDT | Use sequence in Table X for quote from IDT for synthesis | |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | 15524010 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Tris Base | Fisher Scientific | BP152-5 | |
| Trypan blue | Bio-Rad | 1450021 | |
| Ultra-Filter | Millipore Sigma | UFC810024 | Ultra-4 Centrifugal 10K device must be used, as it has a 10000 molecular weight cutoff |
| Universal Nuclease for cell lysis | Thermo Fisher Scientific | 88702 | |
| Universal qPCR master mix | NEB | M3003L | |
| Whatman Paper | Millipore Sigma | WHA1001325 | |
| β-mercaptoethanol | Fisher Scientific | 21985023 | CAUTION. Use under a laminar flow hood. Wear gloves |
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. |
Referenzen
- Hastie, E., Samulski, R. J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success-a personal perspective. Human Gene Therapy. 26 (5), 257-265 (2015).
- Wang, D., Tai, P. W. L., Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. 18 (5), 358-378 (2019).
- Nathwani, A. C., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England Journal of Medicine. 371 (21), 1994-2004 (2014).
- Kuzmin, D. A., et al. The clinical landscape for AAV gene therapies. Nature Reviews Drug Discovery. 20 (3), 173-174 (2021).
- Ylä-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (10), 1831-1832 (2012).
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020)
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (2020)
- Statement from FDA Commissioner Scott Gottlieb, MD and Peter Marks, MD Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. FDA Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (2020)
- Asokan, A., Schaffer, D. V., Samulski, R. J. The AAV vector toolkit: poised at the clinical crossroads. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (4), 699-708 (2012).
- Rose, J. A., Hoggan, M. D., Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proceedings of the National Academy of Sciences of the United States of America. 56 (1), 86-92 (1966).
- Lusby, E., Fife, K. H., Berns, K. I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. Journal of Virology. 34 (2), 402-409 (1980).
- Masat, E., Pavani, G., Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discovery Medicine. 15 (85), 379-389 (2013).
- Ling, C. Enhanced Transgene Expression from Recombinant Single-Stranded D-Sequence-Substituted Adeno-Associated Virus Vectors in Human Cell Lines In Vitro and in Murine Hepatocytes In Vivo. Journal of Virology. 89 (2), 952-961 (2014).
- Cathomen, T., Stracker, T. H., Gilbert, L. B., Weitzman, M. D. A genetic screen identifies a cellular regulator of adeno-associated virus. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 14991-14996 (2001).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Aschauer, D. F., Kreuz, S., Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLOS One. 8 (9), 76310 (2013).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Pillay, S., et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. Journal of Virology. 91 (18), (2017).
- Merkel, S. F. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. Journal of Neurochemistry. 140 (2), 216-230 (2017).
- van Lieshout, L. P., et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Molecular Therapy. Methods & Clinical Development. 9, 323-329 (2018).
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M., Samulski, R. J. single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. Journal of Virology. 80 (22), 11393-11397 (2006).
- Liu, J., Moon, Y. -. A. Simple purification of adeno-associated virus-DJ for liver-specific gene expression. Yonsei Medical Journal. 57 (3), 790-794 (2016).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adeno-associated virus vectors. Human Gene Therapy. 9 (18), 2745-2760 (1998).
- Kimura, T., et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Scientific Reports. 9 (1), 13601 (2019).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2012).
- . Paramyxoviridae: The viruses and their replication. Fields Virology Available from: https://www.scholars.northwestern.edu/en/publications/paramyxoviridae-the-viruses-and-their-replication (1996)
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Kaludov, N., Brown, K. E., Walters, R. W., Zabner, J., Chiorini, J. A. Adeno-associated virus serotype 4 (AAV4) and AAV5 Both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of Virology. 75 (15), 6884-6893 (2001).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Human Gene Therapy Methods. 26 (6), 228-242 (2015).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Backovic, A., et al. Capsid protein expression and adeno-associated virus like particles assembly in Saccharomyces cerevisiae. Microbial Cell Factories. 11, 124 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten