È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Produzione di vettori virali adeno-associati in pile cellulari per studi preclinici in modelli animali di grandi dimensioni
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Qui forniamo una procedura dettagliata per la produzione su larga scala di vettori AAV di grado di ricerca utilizzando cellule HEK 293 aderenti coltivate in pile cellulari e purificazione cromatografica di affinità. Questo protocollo produce costantemente >1 x 1013 genomi vettoriali / mL, fornendo quantità vettoriali appropriate per studi su animali di grandi dimensioni.
Abstract
I vettori del virus adeno-associato (AAV) sono tra i vettori di terapia genica clinicamente più avanzati, con tre terapie geniche AAV approvate per l'uomo. Il progresso clinico di nuove applicazioni per AAV comporta la transizione da piccoli modelli animali, come topi, a modelli animali più grandi, tra cui cani, pecore e primati non umani. Uno dei limiti della somministrazione di AAV ad animali più grandi è il requisito di grandi quantità di virus ad alto titolo. Mentre la coltura cellulare in sospensione è un metodo scalabile per la produzione di vettori AAV, pochi laboratori di ricerca hanno l'attrezzatura (ad esempio, bioreattori) o sanno come produrre AAV in questo modo. Inoltre, i titoli AAV sono spesso significativamente più bassi se prodotti in sospensione hek 293 celle rispetto alle celle HEK293 aderenti. Qui è descritto un metodo per produrre grandi quantità di AAV ad alto titolo utilizzando pile di celle. Vengono inoltre descritti un protocollo dettagliato per il titolo AAV e metodi per convalidare la purezza del vettore. Infine, vengono presentati i risultati rappresentativi dell'espressione transgenica mediata da AAV in un modello di pecora. Questo protocollo ottimizzato per la produzione su larga scala di vettori AAV in cellule aderenti consentirà ai laboratori di biologia molecolare di far progredire la sperimentazione delle loro nuove terapie AAV in modelli animali più grandi.
Introduzione
La terapia genica che utilizza vettori di virus adeno-associati (AAV) ha fatto passi da gigante negli ultimi tre decenni1,2. I miglioramenti dimostrati in una vasta gamma di malattie genetiche, tra cui cecità congenita, emofilia e malattie del sistema muscolo-scheletrico e del sistema nervoso centrale, hanno portato la terapia genica AAV in prima linea nella ricerca clinica3,4. Nel 2012, l'Agenzia europea per i medicinali (EMA) ha approvato Glybera, un vettore AAV1 che esprime lipoproteina lipasi (LPL) per il trattamento del deficit di LPL, rendendolo la prima autorizzazione all'immissione in commercio per un trattamento di terapia genica in Europa o negli Stati Uniti5. Da allora, due ulteriori terapie geniche AAV, Luxturna6 e Zolgensma7,hanno ricevuto l'approvazione della FDA e il mercato dovrebbe espandersi rapidamente nei prossimi 5 anni con ben 10-20 terapie geniche previste entro il 20258. I dati clinici disponibili indicano che la terapia genica AAV è una modalità sicura, ben tollerata ed efficace che la rende uno dei vettori virali più promettenti, con oltre 244 studi clinici che coinvolgono AAV registrati con ClinicalTrials.gov. Il crescente interesse per le applicazioni cliniche che coinvolgono vettori AAV richiede metodi di produzione robusti e scalabili per facilitare la valutazione delle terapie AAV in modelli animali di grandi dimensioni, in quanto questo è un passo critico nella pipeline traslazionale9.
Per la produzione di vettori AAV, i due requisiti principali sono il genoma AAV e il capside. Il genoma di wild-type (wt)-AAV è DNA a singolo filamento che è di circa 4,7 kb di lunghezza10. Il genoma wt-AAV comprende ripetizioni terminali invertite (ITR) che si trovano ad entrambe le estremità del genoma, che sono importanti per il packaging, e i geni rep e cap 11. I geni rep e cap, necessari per la replicazione del genoma, l'assemblaggio del capside virale e l'incapsulamento del genoma nel capside virale, vengono rimossi dal genoma virale e forniti in trans per la produzione di vettori AAV12. La rimozione di questi geni dal genoma virale fornisce spazio per i transgeni terapeutici e tutti gli elementi regolatori necessari, tra cui il promotore e il segnale polyA. Gli ITR rimangono nel genoma vettoriale per garantire una corretta replicazione del genoma e l'incapsulamento virale13,14. Per migliorare la cinetica dell'espressione transgenica, i genomi vettoriali AAV possono essere progettati per essere auto-complementari, il che mitiga la necessità di conversione dalla conversione del DNA a singolo filamento a quella a doppio filamento durante la replicazione del genoma AAV, ma riduce la capacità di codifica a ~ 2,4 kb15.
Oltre alla progettazione del genoma AAV, la selezione del sierotipo del capside determina il tropismo tissutale e cellulare del vettore AAV in vivo2. Oltre al tropismo tissutale, diversi sierotipi AAV hanno dimostrato di mostrare diverse cinetiche di espressionegenica 16. Ad esempio, Zincarelli et al.17 hanno classificato diversi sierotipi AAV in sierotipi a bassa espressione (AAV2, 3, 4, 5), sierotipi a espressione moderata (AAV1, 6, 8) e sierotipi ad alta espressione (AAV7 e 9). Hanno anche classificato i sierotipi AAV in espressione ad esordio lento (AAV2, 3, 4, 5) o espressione ad esordio rapido (AAV1, 6, 7, 8 e 9). Questi tropismi divergenti e la cinetica di espressione genica sono dovuti a variazioni aminoacidiche nelle proteine del capside, formazioni di proteine del capside e interazioni con i recettori / co-recettori delle cellule ospiti18. Alcuni capsidi AAV hanno caratteristiche benefiche aggiuntive come la capacità di attraversare la barriera emato-encefalica dopo somministrazione intravascolare (AAV9) o risiedere in cellule muscolari longeve per un'espressione transgenica duratura (AAV6, 6.2FF, 8 e 9)19,20.
Questo documento mira a dettagliare un metodo economico per la produzione di vettori AAV ad alta purezza, ad alto titolo e di grado di ricerca da utilizzare in modelli preclinici di animali di grandi dimensioni. La produzione di AAV utilizzando questo protocollo è ottenuta utilizzando la trasfezione a doppio plasmide in cellule aderenti del rene embrionale umano (HEK) 293 coltivate in pile cellulari. Inoltre, lo studio descrive un protocollo per la purificazione della cromatografia dell'affinità dell'eparina solfato, che può essere utilizzato per i sierotipi AAV che contengono domini leganti l'eparina, tra cui AAV2, 3, 6, 6.2FF, 13 e DJ21,22.
Sono disponibili numerosi sistemi di imballaggio per la produzione di vettori AAV. Tra questi, l'uso di un sistema di co-trasfezione a due plasmidi, in cui i geni Rep e Cap e i geni Ad helper (E1A, E1B55K, E2A, E4orf6 e VA RNA) sono contenuti all'interno di un plasmide (pHelper), presenta alcuni vantaggi pratici rispetto al comune metodo di trasfezione a tre plasmidi (triplo), tra cui costi ridotti per la produzione di plasmidi23,24 . Il plasmide del genoma AAV contenente la cassetta di espressione transgenica (pTransgene), deve essere affiancato da ITR e non deve superare ~ 4,7 kb di lunghezza. Il titolo e la purezza del vettore possono essere influenzati dal transgene a causa dei potenziali effetti citotossici durante la trasfezione. La valutazione della purezza del vettore è descritta nel presente documento. I vettori prodotti con questo metodo, che producono un 1 x 1013 vg / mL per ciascuno, sono stati valutati in topi, criceti e modelli animali ovini.
Tabella 1: Composizione delle soluzioni richieste. Informazioni necessarie, incluse percentuali e volumi, dei componenti necessari per varie soluzioni in tutto il protocollo. Fare clic qui per scaricare questa tabella.
Protocollo
1. Doppia trasfezione plasmidica di cellule HEK293 in pile cellulari
- Scongelare un crio-flaconcino di cellule HEK293 in un bagno di perline fissato a 37 °C.
NOTA: DMEM completo preriscaldato a 37 °C durante lo scongelamento delle celle per garantire che la temperatura fredda non urti le celle durante la placcatura. Assicurarsi che le cellule abbiano un basso numero di passaggi, idealmente inferiore a 20, per garantire una crescita ottimale e un'efficienza di trasfezione. Assicurarsi che le cellule siano certificate per essere prive di micoplasma. - Trasferire il contenuto del crio-flaconcino a goccia in un tubo conico da 15 mL contenente 10 mL di DMEM completo preriscaldato e centrifugare le cellule a 500 x g per 5 minuti.
- Aspirare il fluido e quindi risospendere le celle HEK293 in 20 ml di DMEM completo preriscaldato. Seminare le cellule in una piastra di 15 cm e incubare a 37 °C, con il 5% di CO2.
- Dividere le cellule da una piastra di 15 cm in tre per la semina nella camera di coltura cellulare.
- Una volta che le cellule sono confluenti all'80%, aspirare il fluido e lavare delicatamente la piastra con 3 ml di PBS per non interrompere il monostrato. Quindi, aspirare PBS e aggiungere 3 ml di tripsina.
- Incubare per 2 minuti a 37 °C fino a quando le cellule si sollevano dalla piastra, quindi neutralizzare la tripsina aggiungendo 7 ml di DMEM completo alla piastra.
- Raccogliere tutti i fluidi e le celle in un tubo da 15 ml e pellet le celle centrifugando a 500 x g per 5 minuti.
- Aspirare il surnatante dal tubo da 15 ml e risospescere il pellet cellulare in 3 mL di DMEM completo. Aggiungere 1 mL a ogni piastra da 15 cm contenente 20 mL di DMEM completo; scuotere delicatamente le piastre per distribuire uniformemente le cellule e incubare a 37 °C, con il 5% di CO2.
- Una volta che le celle sono confluenti all'80%, ripetere i passaggi 1.4.1 e 1.4.2. Raccogliere il surnatante in tubi conici da 50 ml e invertire delicatamente il tubo per garantire che le cellule siano omogenee.
- Determinare la densità cellulare mescolando 10 μL dei campioni di cellule con 10 μL di tripano blu e aggiungendo la miscela a un vetrino di conteggio delle cellule per l'analisi nel contatore cellulare.
- Mescolare 1 L di DMEM completo preriscaldato con la sospensione cellulare necessaria per seminare la camera di coltura cellulare (superficie di 6360 cm2) con 1 x 104 celle / cm2. Versare la miscela cellulare nella camera di coltura cellulare e ruotare delicatamente per distribuire uniformemente le cellule su ciascun monostrato (Figura 1) e incubare a 37 °C, con il 5% di CO2.
- Oltre alla camera di coltura cellulare, placcare una piastra di 15 cm con 1 x10 4 celle / cm2 come riferimento per la confluenza.
- Dopo l'incubazione di ~ 65 ore, controllare la piastra di riferimento per la confluenza, idealmente ~ 80% -90% confluente.
NOTA: DMEM completo preriscaldato da aggiungere alla camera di coltura cellulare a 37 °C.

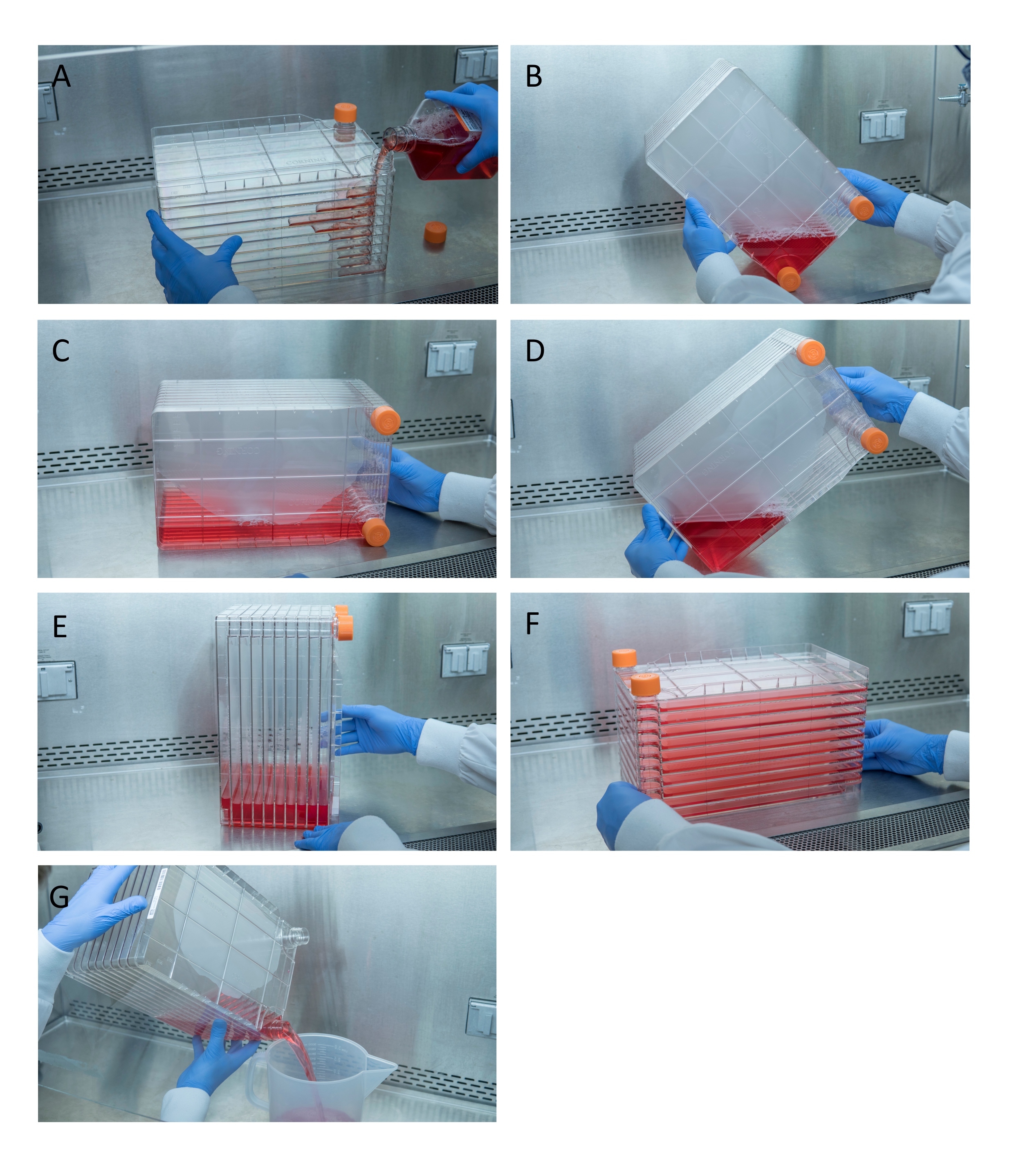
Figura 1: Manovra della pila di cellule per la semina e la trasfezione delle cellule. Per la pila di celle di semina, iniziare rimuovendo uno dei tappi di sfiato e versando 1 L di DMEM completo preriscaldato con la quantità necessaria di celle HEK293 (A). Distribuire uniformemente celle e supporti stringendo entrambi i tappi di sfiato e portare tutti i supporti nell'angolo della pila di celle con uno dei tappi di sfiato e posizionarlo in quell'angolo (B), posizionare lo stack di celle su un lato (C), quindi ruotare lo stack di celle di 90 ° (D) in modo che le porte di sfiato siano verso l'alto (E). Abbassare delicatamente la pila di celle nella sua normale posizione orizzontale e assicurarsi che tutte le camere della pila di celle siano completamente coperte da supporti (F). Durante la trasfezione, svitare entrambi i tappi di sfiato e versare lentamente i vecchi mezzi in un contenitore di rifiuti sterili per un flusso uniforme per non disturbare il monostrato delle cellule (G). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Preparare la miscela di polietileneimmina (PEI)/DNA con un rapporto di concentrazione di 3:1 (p/p).
- Preparare la miscela di DNA in un tubo conico da 50 mL aggiungendo 475 μg di pTrangene e 1425 μg di pHelper a 40 mL di mezzo sierico ridotto per creare un rapporto 3:1 di pHelper:pTrangene.
NOTA: il calcolatore della miscela PEI/DNA può essere trovato utilizzando la Tabella 2. - Aggiungere 5,7 ml di PEI (1 g/L) al mezzo sierico ridotto e alla miscela di DNA a goccia. Quindi, vortice brevemente e incubare per 10 minuti a temperatura ambiente.
NOTA: Quando PEI/DNA incuba a temperatura ambiente, diventerà leggermente nuvoloso.
- Preparare la miscela di DNA in un tubo conico da 50 mL aggiungendo 475 μg di pTrangene e 1425 μg di pHelper a 40 mL di mezzo sierico ridotto per creare un rapporto 3:1 di pHelper:pTrangene.
- Dopo 8 minuti di incubazione PEI/DNA, rimuovere il mezzo dalla camera di coltura cellulare.
NOTA: Assicurarsi di allentare entrambi i cappucci arancioni per mantenere un flusso regolare di fluidi per evitare lo spostamento delle cellule. - Aggiungere PEI/DNA a 1 L di DMEM completo preriscaldato e versare lentamente la miscela nella porta della camera di coltura cellulare. Distribuire il liquido uniformemente su tutte le file (Figura 1) e incubare per 72 h a 37 °C, con il 5% di CO2.
2 Raccolta di AAV e lisi chimica delle cellule HEK293 trasfettate
- Agitare vigorosamente la camera di coltura cellulare per rimuovere le cellule fino a quando il mezzo appare torbido dalle cellule spostate e versare in quattro tubi centrifuga da 500 ml.
- Centrifugare i tubi a 18.000 x g per 30 min a 4 °C per pellettizzare le celle. Versare il surnatante chiarificato in un flacone di copoliestere di polietilene tereftalato (PETG) da 1 L.
NOTA: Se non si ha accesso a una centrifuga ad alta velocità, centrifugare a 12.000 x g per 40 min. Le celle pellettate potrebbero non essere solide a questa velocità e scivoleranno come surnatante versato. - Risospendare i pellet di cella in tubi di centrifuga da 500 mL con 50 mL di tampone di lisi e incubare per 60 minuti a 37 °C.
- Centrifugare i tubi a 18.000 x g per 30 minuti, quindi trasferire il surnatante nella stessa bottiglia di PETG da 1 L. Scartare i detriti della cella pellettati.
NOTA: Purificare immediatamente il surnatante chiarificato e conservare a 4 °C per un massimo di 72 ore. Per la conservazione a lungo termine, conservare a -80 °C. Non conservare a -20 °C.
3 Purificazione vettoriale AAV mediante cromatografia con affinità di eparina
- Rimuovere il lisato grezzo da -80 °C e lasciare scongelare a 4 °C durante la notte. Una volta scongelato, utilizzare un filtro da 0,22 μM per filtrare il lisato grezzo.
- Per passivare il concentratore centrifugo, aggiungere 4 mL di tampone di pretrattamento del filtro a un concentratore centrifugo per ogni colonna di eparina sefarosa utilizzata. Passivare il concentratore centrifugo a temperatura ambiente per 2-8 ore. Impostare la passivazione immediatamente prima delle fasi di purificazione.
- Impostare il tubo e la pompa (Figura 2).
- Posizionare il tubo in una pompa peristaltica e far funzionare 20 mL di 1 M NaOH. Quindi, eseguire 50 ml di acqua di grado molecolare e quindi eseguire 50 ml di DMEM basale.
- Attaccare 5 mL di colonna di eparina sefarosio al tubo ed eseguire 25 mL di DMEM basale per rimuovere il conservante.
- Eseguire 0,2 μM del lisato grezzo filtrato attraverso la colonna ad una portata di 1-2 gocce/s.

Figura 2: Configurazione della pompa peristaltica per la purificazione AAV. Eseguire il tubo dal lisato grezzo, attraverso la pompa peristaltica e nella colonna della matrice di eparina. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: assicurarsi di non introdurre bolle o lasciare asciugare la colonna, in quanto ciò comprometterà la colonna e impedirà l'eluizione di AAV. Scartare la colonna se si asciuga e utilizzare una nuova colonna per il resto del lisato grezzo.
- Caricare tutto il lisato grezzo sulla colonna di eparina e utilizzare le seguenti soluzioni per lavare la colonna.
- Lavare con 50 mL di 1x Hank's Balanced Salt Solutions (HBSS) senza Mg2+ e Ca2+.
- Lavare con 15 mL di N-lauroylsarcosine allo 0,5% in HBSS senza Mg2+ e Ca2+.
- Lavare con 50 mL di HBSS senza Mg2+ e Ca2+.
- Lavare con 50 mL di HBSS con Mg2+ e Ca2+
- Lavare con 50 mL di 200 mM NaCl/HBSS con Mg2+ e Ca2+.
- Elute 5 x 5 mL (25 mL totali) con 300 mM di NaCl/HBSS con Mg2+ e Ca2+ ed etichettare le eluizioni come E1-E5 (ogni eluizione è di 5 mL).
- Concentrazione del virus mediante un concentratore centrifugo
- Ruotare il concentratore centrifugo contenente il tampone di pretrattamento a 900 x g per 2 min. Scartare il flusso attraverso.
- Lavare il filtro concentratore centrifugo con 4 mL di HBSS con Mg2+ e Ca2+ e centrifugare a 1000 x g per 2 min; scartare il flusso attraverso.
- Aggiungere l'eluizione E2 al concentratore centrifugo. Girare a 1000 x g per 5 minuti ed eliminare il flusso attraverso.
- Terminare l'aggiunta di E2 e quindi aggiungere E3 al concentratore centrifugo e girare a 1000 x g per 5 minuti fino a quando il virus concentrato è di circa 1 ml.
NOTA: evitare di centrifugare il vettore in modo tale che il volume sia inferiore al livello del filtro. Non concentrare E1, E4 o E5 nel concentratore centrifugo, poiché contengono pochissimo o nessun vettore e contengono contaminanti. - Rimuovere il virus concentrato dal concentratore centrifugo utilizzando una punta filtrata p200 e posizionarlo in un tubo centrifugo sterile da 1,5 ml.
- Risciacquare il concentratore centrifugo con 200 μL di HBSS con Mg2+ e Ca2+ per rimuovere eventuali AAV rimanenti dal filtro. Pipettare su e giù vigorosamente più volte (per ~ 30 s) per rimuovere qualsiasi virus aderente alla membrana e posizionarlo nel tubo della centrifuga da 1,5 ml con il resto del virus. Mescolare bene il tubo.
- Aliquota 5 μL per estrazioni di DNA e immagazzinare il vettore purificato a -80 °C.
- Lavare la colonna utilizzando 25 mL di 2 M NaCl. Inoltre, utilizzare 25 mL di Triton X-100 allo 0,1%, preriscaldato a 37 °C per lavare la colonna. Quindi, lavare la colonna utilizzando 50 ml di dH2O sterile e quindi lavare con 25 mL di etanolo al 20%.
- Assicurarsi che la membrana della colonna sia completamente satura di etanolo al 20%, poiché questa è la soluzione di stoccaggio. Sigillare la colonna con tappi in dotazione e conservare a 4 °C.
- Conservare il tubo in 1 M NaOH.
NOTA: Se pulite correttamente, le colonne di eparina sefarosio possono essere riutilizzate fino a cinque volte.
4 Estrazione del DNA genomico AAV
- Preparare la miscela di reazione menzionata nella Tabella 3 in un tubo PCR per il trattamento con DNasi.
| Componente | Volume |
| Vettore AAV purificato | 5 μL |
| 10x Buffer DNasi | 2 μL |
| DNasi | 1 μL |
| ddH2O | 12 μL |
| Volume finale | 20 μL |
Tabella 3: Formula di miscelazione master per il trattamento DNasi. Componenti e volumi raccomandati necessari per il trattamento con DNasi dei vettori virali AAV durante l'estrazione del DNA.
- Ruotare il tubo PCR per mescolare e pulsare il tubo PCR per far girare verso il basso il contenuto.
- Utilizzando un termociclatore, incubare a 37 °C per 20 minuti seguiti da 75 °C per 15 minuti per riscaldare inattivare la DNasi.
- Aggiungere 5 μL di proteinasi K.
- Utilizzando un termociclatore, incubare a 50 °C per 60 minuti e poi a 95 °C per 30 minuti per riscaldare la proteinasi K inattivata.
- Utilizzare un kit di pulizia del DNA per rimuovere potenziali contaminanti.
NOTA: Questa fase è stata eseguita utilizzando un kit di pulizia del sangue e dei tessuti disponibile in commercio(Tabella dei materiali).- Aggiungere 200 μL di tampone AL (Kit di pulizia del sangue e dei tessuti, Tabella dei materiali)al tubo PCR contenente il vettore trattato con DNasi/Proteinasi K.
- Ruotare il tubo PCR e incubare a 56 °C per 10 minuti in un termociclatore.
- Pipettare il liquido dal tubo PCR in una colonna di spin sterile seduta in un tubo di raccolta.
- Aggiungere 200 μL di etanolo al 100% alla colonna e mescolare accuratamente mediante vortice.
- Centrifugare a 6.000 x g per 1 minuto ed eliminare il flusso.
- Aggiungere 500 μL di tampone AW1 (Kit di pulizia del sangue e dei tessuti, Tabella dei materiali)alla colonna di rotazione.
- Centrifugare a 6.000 x g per 1 minuto ed eliminare il flusso.
- Aggiungere 500 μL di tampone AW2 (Kit di pulizia del sangue e dei tessuti, Tabella dei materiali)alla colonna di rotazione.
- Centrifugare a 15.000 x g per 3 minuti ed eliminare il flusso.
- Posizionare la colonna di spin in un tubo centrifugo sterile da 1,5 mL e aggiungere 200 μL di tampone AE (Kit di pulizia del sangue e dei tessuti, Tabella dei materiali)direttamente alla membrana della colonna di spin.
- Incubare a temperatura ambiente per 1 min.
- Centrifugare a 6.000 x gper 1 minuto per eluire il DNA.
- Conservare il DNA a -20 °C.
5 Titolazione di genomi vettoriali AAV mediante reazione a catena quantitativa della polimerasi e sonda Simian Virus 40 (SV40)
NOTA: Eseguire tutto il lavoro qPCR in una cappa PCR utilizzando punte di pipetta filtrate per evitare la contaminazione esterna del DNA. Se il genoma AAV non codifica una sequenza di poliA SV40, utilizzare una sonda contro l'ITR descritto altrove25. Assicurarsi che il DNA plasmidico selezionato come standard contenga la sequenza SV40 polyA.
- Preparazione standard delle scorte
- Diluire lo standard del DNA plasmidico di riserva (plasmide pTransgene contenente sequenza SV40 polyA) ad una concentrazione finale di 10 μg/μL e conservare a -20 °C in aliquote da 6 μL.
- Determinare il numero di copie presente nello standard del DNA plasmidico utilizzando il seguente calcolatore online26.
NOTA: Utilizzare un DNA plasmidico utilizzato per lo standard prodotto da un fornitore commerciale per garantire la qualità e la corretta concentrazione. Preparare una grande quantità di standard (ad esempio, 10 ml) per condurre studi ponte durante la transizione a uno standard appena preparato.
- Preparare la seguente miscela di reagenti menzionata nella Tabella 4 sia per i campioni che per lo standard in un tubo di centrifuga da 1,5 mL.
NOTA: Preparare un eccesso sufficiente di miscela master. Vedere la Tabella 5 per le sequenze di primer/sonda.
| Componente | Volume |
| Mix master qPCR universale (2X) | 10 μL |
| Acqua di grado molecolare | 4,5 μL |
| 40x SV40 polyA primer/sonda | 0,5 μL |
| Volume finale | 15 μL |
Tabella 4: miscela master qPCR per titolazione AAV. Componenti e volumi raccomandati necessari per la qPCR del DNA estratto da vettori virali AAV.
| Componente | Sequenza |
| Primer in avanti | 5'-AGCAATAGCATCACAAATTTCACAA-3' |
| Primer inverso | 5'-CCAGACATGATAAGATACATTGATGAGTT-3' |
| Sonda | /56-FAM/AGCATTTTT/Zen/TTCACTGCATTCTAGTTGTGGTTTGTC/3IABkFQ |
Tabella 5: Sequenze di primer contro la sequenza di DNA poliA SV40. Sequenze dei primer e della sonda utilizzati per la titolazione qPCR, che si legano a specifiche aree di vettori virali AAV che contengono la sequenza SV40 polyA.
- Pipettare il master mescolare su e giù per mescolare.
- Impostare la piastra di diluizione.
- Utilizzare una piastra trasparente a 96 pozzetti per preparare diluizioni standard e campione, aggiungere 45 μL di acqua di grado molecolare a ciascun pozzo in ogni altra colonna a partire dalla colonna 1 (colonne 1, 3, 5, 7, 9 e 11).
- Aggiungere 5 μL dello standard al pozzo A1 e pipetta per mescolare.
- Utilizzare una nuova punta della pipetta filtrata per creare una diluizione di 1/10 dal pozzo A1 a B1.
- Continuare una serie di diluizioni 10 volte lungo la colonna fino a raggiungere G1.
- Non aggiungere a H1, in quanto ciò fungerà da controllo negativo.
- Applicare il primo campione (S1) aggiungendolo al pozzo A3, formando una diluizione 1/10. Pipettare questa miscela e trasferire 5 μLto bene B3. Scartare la punta della pipetta dopo questo trasferimento.
- Con una nuova punta della pipetta, mescolare la soluzione in un pozzo B3 e formare una diluizione di 1/100. Trasferire 5 μLof questa miscela al pozzo C3 ed eliminare la punta dopo il trasferimento.
- Con una nuova punta della pipetta, pipettare su e giù la soluzione nel pozzo C3 per effettuare una diluizione 1/1000. Scartare la punta.
- Continuare a diluire i campioni senza aggiungere campioni alle colonne 2, 4, 6, 8, 10 o 12.
- Una volta diluiti tutti i campioni, mescolare il contenuto nei pozzetti della colonna 1 e quindi trasferire 20 μL alla colonna 2.
- Ripetere questa operazione per le colonne 3, 5, 7, 9 e 11 per creare repliche di ogni diluizione standard e campione. Fare riferimento alla Figura 3 per il layout delle piastre.
NOTA: quando si segue l'impostazione della piastra nella Figura 3,i campioni diluiti nelle righe G e H avranno solo 1/10 e 1/100 diluizioni.

Figura 3: Layout delle piastre per la titolazione QPCR AAV. Il blu indica il posizionamento della diluizione seriale dello standard; verde indica il posizionamento del controllo negativo; viola indica il posizionamento della diluizione dei campioni. Ogni standard, negativo o campione viene aggiunto in replica. È stato aggiunto un esempio per la concentrazione dello standard per mostrare la serie di diluizione dello standard e il posizionamento delle diluizioni del campione è stato aggiunto ai rispettivi pozzetti. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Titolazione mediante qPCR basata sul rilevamento di poliA SV40
- Aggiungere 15 μL di miscela master qPCR a ciascun pozzetto di una piastra qPCR a 96 pozzetti semi-bordata bianca.
- Trasferire 5 μL di ciascun campione dalla piastra trasparente a 96 pozzetti alla piastra qPCR a 96 pozzetti semi-bordata bianca.
- Utilizzare una pipetta multicanale per garantire un'adeguata miscelazione della miscela e del campione master qPCR.
- Sigillare la piastra con un film sigillante e centrifugare la piastra qPCR a 1500 x g per 30 s.
- Eseguire la reazione qPCR su uno strumento di amplificazione e rilevamento PCR in tempo reale basato su piastra, utilizzando le condizioni suggerite nella Tabella 6.
| Sezione | Cicli | Ore | Temperatura | Descrizione |
| Pre-incubazione | 1x | 5 minuti | 95 °C | Denaturazione del DNA. |
| Amplificazione | 38 volte | 15 s | 95 °C | Amplificazione del DNA. Le impostazioni possono essere modificate se si utilizzano primer alternativi con temperature di ricottura diverse. |
| 60 anni | 60 °C | |||
| Raffreddamento | 1x | 60 anni | 40 °C | Raffreddamento a piastre. Fine della corsa. |
Tabella 6: Protocollo termociclatore per la titolazione qPCR basata su sonda di idrolisi. Protocollo termociclatore raccomandato per l'uso della titolazione qPCR basata su sonda di vettori AAV purificati estratti dal DNA.
NOTA: per il foglio di lavoro di titolazione AAV qPCR vedere tabella 7.
- Analisi dei dati per determinare i numeri di copia del genoma AAV.
- Compilare le celle di dati del foglio di calcolo (Tabella 7A) con i valori di concentrazione ottenuti dall'esecuzione qPCR per diluizioni sia standard che campione.
- Utilizzare i valori di concentrazione della tabella 7A per produrre una curva standard (Tabella 7B).
NOTA: La curva standard verrà mostrata come un logaritmo naturale (y = a ln(x) + b) insieme all'efficienza R2. Una curva standard deve avere un'efficienza vicina al 100 % e R2 vicina a 1,0 (≥0,99). - Compila l'efficienza della pendenza compilando questo calcolatore online27.
NOTA: un'efficienza compresa tra il 90% e il 110% è accettabile. Se l'efficienza di qPCR è al di fuori di questo intervallo, eseguire nuovamente il qPCR. - Utilizzare i valori di concentrazione della Tabella 7A per calcolare la media delle diluizioni di ciascun campione e determinare la deviazione standard di ciascun campione (Tabella 7C).
NOTA: escludere le diluizioni da campioni che si trovano a più di una deviazione standard dalla media delle diluizioni del campione. - Usando la concentrazione media di ogni diluizione, moltiplicare per il fattore di diluizione, quindi dividere per cinque per ottenere i genomi vettoriali (vg)/μL di ciascun campione (Tabella 7C).
- Calcolare il vg/mL di ciascun campione moltiplicando la media delle concentrazioni di ciascun campione per 80.000 (Tabella 7C).
- Mediare il vg/mL di ciascuna diluizione per produrre il vg/mL finale di ciascun campione (Tabella 7C).
NOTA: l'utente deve dividere la concentrazione media di ciascuna diluizione per un fattore cinque per tenere conto dei 5 μL caricati in ciascun pozzo affinché la qPCR venga eseguita per produrre la concentrazione in vg/μL. Il fattore di 80.000 rappresenta la transizione dal valore medio di concentrazione di ciascun campione a vg/mL. In primo luogo, la media del valore di concentrazione di ciascun campione deve essere moltiplicata per 2 per tenere conto dei genomi a singolo filamento, poiché il set di primer-sonda quantifica solo il DNA a senso positivo e a singolo filamento (ssDNA) e il genoma AAV esiste in un rapporto approssimativo 1: 1 tra ssDNA positivo e negativo25,28. La media del valore di concentrazione di ciascun campione deve essere moltiplicata x40 per tenere conto della diluizione del campione da 5 μL di vettore purificato (punto 4.1) a 200 μL di DNA estratto (sezione 4.6.12). Infine, la media del valore di concentrazione di ciascun campione deve essere moltiplicata x1000 per convertire da vg/μL a vg/mL.
6 Valutazione della qualità e della purezza del vettore
- Controllo Qualità - Western Blot
- Preparare un gel SDS PAGE al 12%.
- Eseguire l'elettroforesi su gel di poliacrilammide.
NOTA: Caricare 6 x 1010 vg di campioni per pozzetto. - Trasferire le proteine alla membrana di polivinilidene difluoruro (PVDF).
- Membrana in PVDF bloccante
- Rimuovere la membrana dall'apparecchiatura di trasferimento e risciacquare in 0,1% PBST per rimuovere l'acrilammide sciolta.
- Porre la membrana in soluzione bloccante per almeno 1 ora a temperatura ambiente o durante la notte a 4 °C.
NOTA: il tampone di blocco può essere ulteriormente integrato con siero di capra al 2%.
- Incubazione con anticorpo primario
- Decantare il tampone bloccante e aggiungere l'anticorpo primario, un anticorpo monoclonale anti-AAV di topo a una diluizione di 1:200.
- Incubare durante la notte a 4 °C.
- Decantare l'anticorpo primario e lavare cinque volte con 0,1% PBST per 5 minuti a temperatura ambiente con agitazione.
- Incubazione con anticorpo secondario
- Decantare la soluzione di lavaggio e aggiungere l'anticorpo secondario coniugato HRP, diluito a 1:7500 in tampone bloccante, e incubare per 1 ora a temperatura ambiente con agitazione.
- Decantare l'anticorpo secondario e lavare cinque volte con PBST allo 0,1% per 5 minuti a temperatura ambiente con agitazione.
- Eseguire un lavaggio finale con PBS a temperatura ambiente con agitazione.
- Rilevare le proteine utilizzando il substrato chemiluminescente avanzato (ECL).
- Immagine del gel per visualizzare le proteine virali (subunità VP1, VP2 e VP3) (Figura 4).

Figura 4: Western blot che mostra le proteine del capside AAV. Corsia A; Scala MW, Corsia B; AAV6.2FF-hIgG01, Corsia C; AAV6.2FF-hIgG02, Corsia D; AAV6.2FF-hIgG03 e Corsia E; AAV6.2FF-hIgG04. 6 x 1010 vg di ogni AAV6.2FF-hIgG è stato caricato nelle rispettive corsie. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Controllo della purezza - SDS PAGE e Coomassie Stain
- Preparare il gel SDS-PAGE e i campioni come descritto nei passaggi 6.1.1 e 6.1.2.
- Fissare il gel in soluzione fissante per 1 ora o durante la notte con una leggera agitazione. Cambiare la soluzione di fissaggio una volta durante la prima ora.
- Macchiare il gel in soluzione colorante per 2-4 ore con una delicata agitazione.
- Dedurre il gel con una soluzione di destaining. Ricostituire la soluzione di detaining più volte fino a quando lo sfondo del gel è completamente destained (4-24 h).
- Conservare il gel decorticato in una soluzione di conservazione.
- Immagina il gel per visualizzare tutte le proteine colorate dalla soluzione di colorazione Coomassie.

Figura 5: Gel macchiato di coomassie. Corsia A; Scala MW, Corsia B; AAV6.2FF-hIgG01, Corsia C; AAV6.2FF-hIgG02, Corsia D; AAV6.2FF-hIgG03, Corsia E; AAV6.2FF-hIgG04, Corsia F; AAV6.2FF-hIgG05 e Lane G; AAV6.2FF-hIgG06. 6 x 1010 vg di ogni AAV6.2FF-hIgG è stato caricato nelle rispettive corsie. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Saggio alternativo di controllo della purezza - HEK293 rilevamento della proteina delle cellule ospiti ELISA
- Eseguire il rilevamento della proteina della cellula ospite HEK293 tramite ELISA secondo le istruzioni del produttore.
NOTA: utilizzare diluizioni di 5 x 10-2 e 1 x 10-3 per campioni rAAV purificati. Una volta aggiunto TMB al pozzo, incubare lontano dalla luce. La regressione lineare non può essere utilizzata per analizzare i risultati. - Eseguire un'analisi punto-punto, una spline cubica o un metodo di adattamento logistico a quattro parametri per interpolare le concentrazioni di incognite e moltiplicare per il fattore di diluizione per determinare la concentrazione del campione originale.
- Eseguire il rilevamento della proteina della cellula ospite HEK293 tramite ELISA secondo le istruzioni del produttore.
Risultati
La traduzione da modelli di piccoli roditori a modelli animali più grandi e l'eventuale applicazione clinica rappresentano una sfida significativa a causa della grande quantità di AAV necessaria per trasdurre animali più grandi e ottenere effetti terapeutici. Per confrontare l'efficienza di trasduzione del capside AAV6.2FF progettato razionalmente, precedentemente dimostrato un aumento di 101 volte dell'efficienza di trasduzione nelle cellule muscolari murine rispetto ad AAV63,topi, criceti e a...
Discussione
La produzione di vettori AAV ricombinanti (rAAV) descritti in questo documento utilizza materiali, reagenti e attrezzature comuni presenti nella maggior parte dei laboratori e delle strutture di ricerca di biologia molecolare. Questo documento consente al lettore di produrre rAAV di alta qualità in vitro e in vivo. Soprattutto, questo protocollo per la produzione di rAAV, rispetto ai protocolli più noiosi che coinvolgono la purificazione del cloruro di cesio, è efficiente ed evita l'uso dell'ultracen...
Divulgazioni
Sarah K. Wootton è un inventore di un brevetto statunitense US10806802B2 per il capside AAV6.2FF.
Riconoscimenti
Amira D. Rghei, Brenna A. Y. Stevens, Sylvia P. Thomas e Jacob G. E. Yates hanno ricevuto stipendi per studenti dell'Ontario Veterinary College e borse di studio per laureati dell'Ontario. Amira D. Rghei è stata la destinataria di un Mitacs Accelerate Studentship. Questo lavoro è stato finanziato dal Canadian Institutes for Health Research (CIHR) Project Grant (# 66009) e da una sovvenzione Collaborative Health Research Projects (NSERC partnered) (# 433339) a SKW.
Materiali
| Name | Company | Catalog Number | Comments |
| 0.22 μm filter | Millipore Sigma | S2GPU05RE | |
| 0.25% Trypsin | Fisher Scientific | SM2001C | |
| 1-Butanol | Thermo Fisher Scientific | A399-4 | CAUTION. Use under a laminar flow hood. Wear gloves |
| 10 chamber cellstack | Corning | 3271 | |
| 1L PETG bottle | Thermo Fisher Scientific | 2019-1000 | |
| 30% Acrylamide/Bis Solution | Bio-Rad | 1610158 | |
| 96-well skirted plate | FroggaBio | FS-96 | |
| Adhesive plate seals | Thermo Fisher Scientific | 08-408-240 | |
| Ammonium persulfate (APS) | Bio-Rad | 161-0700 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Blood and Tissue Clean up Kit | Qiagen | 69506 | Use for DNA clean up in section 4.6 of protocol |
| Bromophenol blue | Fisher Scientific | B392-5 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Cell Culture Dishes | Greiner bio-one | 7000232 | 15 cm plates |
| Culture Conical Tube | Thermo Fisher Scientific | 339650 | 15 mL conical tube |
| Culture Conical Tube | Fisher Scientific | 14955240 | 50 mL conical tube |
| Dulbecco's Modified Eagle Medium (DMEM) with 1000 mg/L D-glucose, L-glutamine | Cytiva Life Sciences | SH30022.01 | |
| ECL Western Blotting Substrate | Thermo Fisher Scientific | 32209 | |
| Ethanol | Greenfield | P016EA95 | Dilute ethyl alcohol(95% vol) to 20% for section 3.7.4 and 70% for section 6.1.1.1 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | SH30396.03 | |
| Glacial acetic acid | Fisher Scientific | A38-500 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | Fisher Scientific | BP381-500 | |
| HBSS with Mg2+ and Ca2+ | Thermo Fisher Scientific | SH302268.02 | |
| HBSS without Mg2+ and Ca2+ | Thermo Fisher Scientific | SH30588.02 | |
| HEK293 cells | American Tissue Culture Collection | CRL-1573 | Upon receipt, thaw the cells and culture as described in manufacturer’s protocol. Once cells have been minimally passaged and are growing well, freeze a subfraction for future in aliquots and store in liquid nitrogen. Always use cells below passage number 30. Once cultured cells have been passaged more than 30 times, it is recommended to restart a culture from the stored aliquots |
| HEK293 host cell protein ELISA kit | Cygnus Technologies | F650S | Follow manufacturer’s instructions |
| Heparin sulfate column | Cytiva Life Sciences | 17040703 | |
| Kimwipe | Thermo Fisher Scientific | KC34120 | |
| L-glutamine (200 mM) | Thermo Fisher Scientific | SH30034.02 | |
| Large Volume Centrifuge Tube Support Cushion | Corning | CLS431124 | Support cushion must be used with large volume centrifuge tubes uless the centrifuge rotor has the approriate V-bottom cushions |
| Large Volume Centrifuge Tubes | Corning | CLS431123-6EA | 500 mL centrifuge tubes |
| MgCl2 | Thermo Fisher Scientific | 7791-18-6 | |
| Microcentrifuge tube | Fisher Scientific | 05-408-129 | 1.5 mL microcentrifuge tube, sterilize prior to use |
| Molecular Grade Water | Cytiva Life Sciences | SH30538.03 | |
| N-Lauroylsarcosine sodium salt | Sigma Aldrich | L5125 | CAUTION. Wear gloves |
| NaCl | Thermo Fisher Scientific | BP35810 | |
| Optimem, reduced serum medium | Thermo Fisher Scientific | 31985070 | |
| Pasteur pipets | Fisher Scientific | 13-678-20D | Sterilize prior to use |
| PBS (10x) | Thermo Fisher Scientific | 70011044 | Dilute to 1x for use on cells |
| Penicillin-Streptomycin Solution | Cytiva Life Sciences | SV30010 | |
| pHelper plasmid | De novo design or obtained from plasmid repository | NA | |
| Pipet basin | Thermo Fisher Scientific | 13-681-502 | Purchase sterile pipet basins |
| Polyethylene glycol tert-octylphenyl ether (Triton X-100) | Thermo Fisher Scientific | 9002-93-1 | CAUTION. Wear gloves |
| Polyethylenimine (PEI) | Polyscience | 24765-1 | Follow manufacturer’s instructions to produce a 1L solution. 0.22μm filter and store at 4°C |
| Polypropylene semi-skirted PCR Plate | FroggaBio | WS-96 | |
| Polysorbate 20 (Tween 20) | Thermo Fisher Scientific | BP337-100 | CAUTION. Wear gloves |
| polyvinylidene difluoride (PVDF) membrane | Cytiva Life Sciences | 10600023 | Use forceps to manipulate. Wear gloves. |
| Primary antibody | Progen | 65158 | |
| Protein Ladder | FroggaBio | PM008-0500 | |
| Proteinase K | Thermo Fisher Scientific | AM2546 | |
| pTrangene plasmid | De novo design or obtained from plasmid repository | NA | Must contain SV40 polyA in genome to be compatible with AAV titration in section 5.0 |
| Pump tubing | Cole-Parmer | RK-96440-14 | Optimize length of tubing and containment of virus in fractions E1-E5 |
| RQ1 Dnase 10 Reaction Buffer | Promega | M6101 | Use at 10x concentration in protocol from section 4.0 |
| RQ1 Rnase-free Dnase | Promega | M6101 | |
| Sample dilutent | Cygnus Technologies | I700 | Must be purchased separately for use with HEK293 host cell protein ELISA kit |
| Secondary antibody, HRP | Thermo Fisher Scientific | G-21040 | |
| Skim milk powder | Oxoid | LP0033B | |
| Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 28312 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Sodium hydroxide (NaOH) | Thermo Fisher Scientific | SS266-4 | |
| SV40 polyA primer probe | IDT | Use sequence in Table X for quote from IDT for synthesis | |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | 15524010 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Tris Base | Fisher Scientific | BP152-5 | |
| Trypan blue | Bio-Rad | 1450021 | |
| Ultra-Filter | Millipore Sigma | UFC810024 | Ultra-4 Centrifugal 10K device must be used, as it has a 10000 molecular weight cutoff |
| Universal Nuclease for cell lysis | Thermo Fisher Scientific | 88702 | |
| Universal qPCR master mix | NEB | M3003L | |
| Whatman Paper | Millipore Sigma | WHA1001325 | |
| β-mercaptoethanol | Fisher Scientific | 21985023 | CAUTION. Use under a laminar flow hood. Wear gloves |
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. |
Riferimenti
- Hastie, E., Samulski, R. J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success-a personal perspective. Human Gene Therapy. 26 (5), 257-265 (2015).
- Wang, D., Tai, P. W. L., Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. 18 (5), 358-378 (2019).
- Nathwani, A. C., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England Journal of Medicine. 371 (21), 1994-2004 (2014).
- Kuzmin, D. A., et al. The clinical landscape for AAV gene therapies. Nature Reviews Drug Discovery. 20 (3), 173-174 (2021).
- Ylä-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (10), 1831-1832 (2012).
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020)
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (2020)
- Statement from FDA Commissioner Scott Gottlieb, MD and Peter Marks, MD Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. FDA Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (2020)
- Asokan, A., Schaffer, D. V., Samulski, R. J. The AAV vector toolkit: poised at the clinical crossroads. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (4), 699-708 (2012).
- Rose, J. A., Hoggan, M. D., Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proceedings of the National Academy of Sciences of the United States of America. 56 (1), 86-92 (1966).
- Lusby, E., Fife, K. H., Berns, K. I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. Journal of Virology. 34 (2), 402-409 (1980).
- Masat, E., Pavani, G., Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discovery Medicine. 15 (85), 379-389 (2013).
- Ling, C. Enhanced Transgene Expression from Recombinant Single-Stranded D-Sequence-Substituted Adeno-Associated Virus Vectors in Human Cell Lines In Vitro and in Murine Hepatocytes In Vivo. Journal of Virology. 89 (2), 952-961 (2014).
- Cathomen, T., Stracker, T. H., Gilbert, L. B., Weitzman, M. D. A genetic screen identifies a cellular regulator of adeno-associated virus. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 14991-14996 (2001).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Aschauer, D. F., Kreuz, S., Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLOS One. 8 (9), 76310 (2013).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Pillay, S., et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. Journal of Virology. 91 (18), (2017).
- Merkel, S. F. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. Journal of Neurochemistry. 140 (2), 216-230 (2017).
- van Lieshout, L. P., et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Molecular Therapy. Methods & Clinical Development. 9, 323-329 (2018).
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M., Samulski, R. J. single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. Journal of Virology. 80 (22), 11393-11397 (2006).
- Liu, J., Moon, Y. -. A. Simple purification of adeno-associated virus-DJ for liver-specific gene expression. Yonsei Medical Journal. 57 (3), 790-794 (2016).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adeno-associated virus vectors. Human Gene Therapy. 9 (18), 2745-2760 (1998).
- Kimura, T., et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Scientific Reports. 9 (1), 13601 (2019).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2012).
- . Paramyxoviridae: The viruses and their replication. Fields Virology Available from: https://www.scholars.northwestern.edu/en/publications/paramyxoviridae-the-viruses-and-their-replication (1996)
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Kaludov, N., Brown, K. E., Walters, R. W., Zabner, J., Chiorini, J. A. Adeno-associated virus serotype 4 (AAV4) and AAV5 Both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of Virology. 75 (15), 6884-6893 (2001).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Human Gene Therapy Methods. 26 (6), 228-242 (2015).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Backovic, A., et al. Capsid protein expression and adeno-associated virus like particles assembly in Saccharomyces cerevisiae. Microbial Cell Factories. 11, 124 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati