
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Produção de vetores de vírus associados a Adeno em pilhas de células para estudos pré-clínicos em grandes modelos animais
Neste Artigo
Resumo
Aqui fornecemos um procedimento detalhado para a produção em larga escala de vetores AAV de grau de pesquisa usando células hek 293 aderentes cultivadas em pilhas de células e purificação de cromatografia de afinidade. Este protocolo produz consistentemente >1 x 1013 genomas vetoriais/mL, fornecendo quantidades vetoriais apropriadas para grandes estudos em animais.
Resumo
Os vetores de vírus associados ao Adeno (AAV) estão entre os vetores de terapia genética mais clinicamente avançados, com três terapias genéticas AAV aprovadas para humanos. O avanço clínico de novas aplicações para AAV envolve a transição de pequenos modelos animais, como ratos, para modelos animais maiores, incluindo cães, ovelhas e primatas não humanos. Uma das limitações da administração de AAV para animais maiores é a exigência de grandes quantidades de vírus de alta titulação. Embora a cultura celular de suspensão seja um método escalável para a produção de vetores AAV, poucos laboratórios de pesquisa têm o equipamento (por exemplo, bioreatores) ou sabem como produzir AAV desta maneira. Além disso, os títulos AAV são muitas vezes significativamente mais baixos quando produzidos em células HEK 293 de suspensão em comparação com as células HEK293 aderentes. Descrito aqui é um método para produzir grandes quantidades de AAV de alta titer usando pilhas de células. Um protocolo detalhado para titering AAV, bem como métodos para validar a pureza vetorial também são descritos. Finalmente, são apresentados resultados representativos da expressão transgênica mediada pela AAV em um modelo de ovinos. Este protocolo otimizado para a produção em larga escala de vetores AAV em células aderentes permitirá que laboratórios de biologia molecular avancem os testes de suas novas terapias AAV em modelos animais maiores.
Introdução
A terapia genética utilizando vetores de vírus associados ao adeno (AAV) fez grandes avanços nas últimas três décadas1,2. Melhorias demonstradas em uma variedade diversificada de doenças genéticas, incluindo cegueira congênita, hemofilia e doenças do sistema nervoso musculoesquelético e central, trouxeram a terapia genética AAV à vanguarda da pesquisa clínica3,4. Em 2012, a Agência Europeia de Medicamentos (EMA) aprovou a Glybera, um vetor AAV1 que expressa lipoproteína lipase (LPL) para o tratamento da deficiência de LPL, tornando-se a primeira autorização de comercialização para um tratamento de terapia genética na Europa ou nos Estados Unidos5. Desde então, duas terapias genéticas AAV adicionais, Luxturna6 e Zolgensma7,receberam aprovação da FDA, e espera-se que o mercado se expanda rapidamente nos próximos 5 anos, com até 10-20 terapias genéticas esperadas até 20258. Os dados clínicos disponíveis indicam que a terapia genética AAV é uma modalidade segura, bem tolerada e eficaz, tornando-a um dos vetores virais mais promissores, com mais de 244 ensaios clínicos envolvendo AAV registrados com ClinicalTrials.gov. O crescente interesse por aplicações clínicas envolvendo vetores AAV requer métodos de produção robustos e escaláveis para facilitar a avaliação das terapias AAV em grandes modelos animais, pois este é um passo crítico no pipeline translacional9.
Para a produção de vetores AAV, os dois principais requisitos são o genoma AAV e o capsídeo. O genoma do tipo selvagem (wt)-AAV é um DNA de um único fio que tem aproximadamente 4,7 kb de comprimento10. O genoma wt-AAV compreende repetições terminais invertidas (ITRs) encontradas em ambas as extremidades do genoma, que são importantes para a embalagem, e os genes rep e cap 11. Os genes de representação e tampa, necessários para a replicação do genoma, montagem do capsídeo viral e encapsulamento do genoma no capsídeo viral, são removidos do genoma viral e fornecidos em trans para a produção de vetores AAV12. A remoção desses genes do genoma viral abre espaço para transgênicos terapêuticos e todos os elementos regulatórios necessários, incluindo o promotor e o sinal poliA. Os ITRs permanecem no genoma vetorial para garantir a replicação adequada do genoma e o encapsulamento viral13,14. Para melhorar a cinética da expressão transgênica, os genomas vetores AAV podem ser projetados para serem autocons complementares, o que mitiga a necessidade de conversão de conversão de DNA de uma só vez para dupla mente durante a replicação do genoma AAV, mas reduz a capacidade de codificação para ~2,4 kb15.
Além do desenho do genoma AAV, a seleção do sorotipo capsid determina o tropismo tecidual e celular do vetor AAV in vivo2. Além do tropismo tecidual, diferentes sorotipos AAV têm sido mostrados para exibir cinética de expressão genética diferente16. Por exemplo, Zincarelli et al.17 classificaram diferentes sorotipos AAV em sorotipos de baixa expressão (AAV2, 3, 4, 5), sorotipos de expressão moderada (AAV1, 6, 8) e sorotipos de alta expressão (AAV7 e 9). Eles também categorizaram os sorotipos AAV em expressão de início lento (AAV2, 3, 4, 5) ou expressão de início rápido (AAV1, 6, 7, 8 e 9). Esses tropismos divergentes e cinéticas de expressão genética são devido a variações de aminoácidos nas proteínas capsóides, formações de proteína capsíide e interações com receptores/co-receptores de células hospedeiras18. Alguns capsídeos AAV têm características benéficas adicionais, como a capacidade de atravessar a barreira hemoencefálica após a administração intravascular (AAV9) ou residir em células musculares de longa duração para expressão transgênica durável (AAV6, 6.2FF, 8 e 9)19,20.
Este artigo tem como objetivo detalhar um método econômico para produzir vetores AAV de alta pureza, alto título e grau de pesquisa para uso em modelos animais grandes pré-clínicos. A produção de AAV usando este protocolo é alcançada usando transfecção dual-plasmid em rim embrionário humano aderente (HEK)293 células cultivadas em pilhas de células. Além disso, o estudo descreve um protocolo para purificação cromatografia de afinidade de sulfato de heparina, que pode ser usado para sorotipos AAV que contêm domínios de ligação de heparina, incluindo AAV2, 3, 6, 6.2FF, 13 e DJ21,22.
Uma série de sistemas de embalagem estão disponíveis para a produção de vetores AAV. Entre eles, o uso de um sistema de co-transfection de dois plasmídeos, nos quais os genes Rep e Cap e os genes auxiliares de anúncios (E1A, E1B55K, E2A, E4orf6 e VA RNA) estão contidos dentro de um plasmídeo (pHelper), tem algumas vantagens práticas sobre o método comum de transfecção de três plasmídeos (triplos), incluindo custo reduzido para produção plasmida23,24 . O plasmídeo genoma AAV contendo o de expressão transgene (pTransgene), deve ser ladeado por ITRs, e não deve exceder ~4,7 kb de comprimento. O título vetorial e a pureza podem ser afetados pelo transgene devido a potenciais efeitos citotóxicos durante a transfecção. A avaliação da pureza vetorial é descrita aqui. Os vetores produzidos utilizando este método, que produzem um 1 x 1013 vg/mL para cada um, foram avaliados em camundongos, hamsters e modelos de animais ovinos.
Tabela 1: Composição das soluções necessárias. Informações necessárias, incluindo percentuais e volumes, de componentes necessários para diversas soluções ao longo do protocolo. Clique aqui para baixar esta Tabela.
Protocolo
1. Transfecção dupla de plasmídeos de células HEK293 em pilhas de células
- Descongele um crio-frasco de células HEK293 em um banho de contas a 37 °C.
NOTA: DMEM completo pré-aquecido a 37 °C enquanto as células estão descongelando para garantir que a temperatura fria não choque as células ao emplacarem. Certifique-se de que as células tenham um número de passagem baixo, idealmente inferior a 20, para garantir o crescimento ideal e eficiência de transfecção. Certifique-se de que as células são certificadas para serem livres de mycoplasma. - Transfira o conteúdo do crio-vial dropwise em um tubo cônico de 15 mL contendo 10 mL de DMEM completo pré-aquecido e centrifugar as células a 500 x g por 5 min.
- Aspire a mídia e, em seguida, resuspenque as células HEK293 em 20 mL de DMEM completo pré-aquecido. Semente as células em uma placa de 15 cm e incubar a 37 °C, com 5% de CO2.
- Divida as células de uma placa de 15 cm em três para semeadura na câmara de cultura celular.
- Uma vez que as células sejam 80% confluentes, aspire a mídia e lave suavemente a placa com 3 mL de PBS para não interromper a monocamada. Em seguida, aspire PBS e adicione 3 mL de trippsina.
- Incubar por 2 min a 37 °C até que as células levantem da placa e, em seguida, neutralizar a trippsina adicionando 7 mL de DMEM completo à placa.
- Colete todas as mídias e células em um tubo de 15 mL e pelota as células por centrifugação a 500 x g por 5 min.
- Aspire o supernatante do tubo de 15 mL e resuspense a pelota celular em 3 mL de DMEM completo. Adicione 1 mL a cada placa de 15 cm contendo 20 mL de DMEM completo; balance suavemente as placas para distribuir as células uniformemente, e incubar a 37 °C, com 5% de CO2.
- Uma vez que as células são 80% confluentes, repita as etapas 1.4.1 e 1.4.2. Colete o supernatante em tubos cônicos de 50 mL e inverta suavemente o tubo para garantir que as células sejam homogêneas.
- Determine a densidade celular misturando 10 μL das amostras de células com 10 μL de azul trypan e adicionando a mistura a um slide de contagem celular para análise no contador celular.
- Misture 1 L de DMEM completo pré-aquecido com a suspensão celular necessária para semear a câmara de cultura celular (área de superfície de 6360 cm2) com 1 x 104 células/cm2. Despeje a mistura celular na câmara de cultura celular e gire suavemente para distribuir uniformemente as células em cada monocamada (Figura 1) e incubar a 37 °C, com 5% de CO2.
- Além da câmara de cultura celular, emplaque uma placa de 15 cm com 1 x 104 células/cm2 como referência para confluência.
- Após a incubação ~65 h, verifique a placa de referência para confluência idealmente ~80%-90% confluente.
NOTA: DMEM completo pré-aquecido para adicionar à câmara de cultura celular a 37 °C.

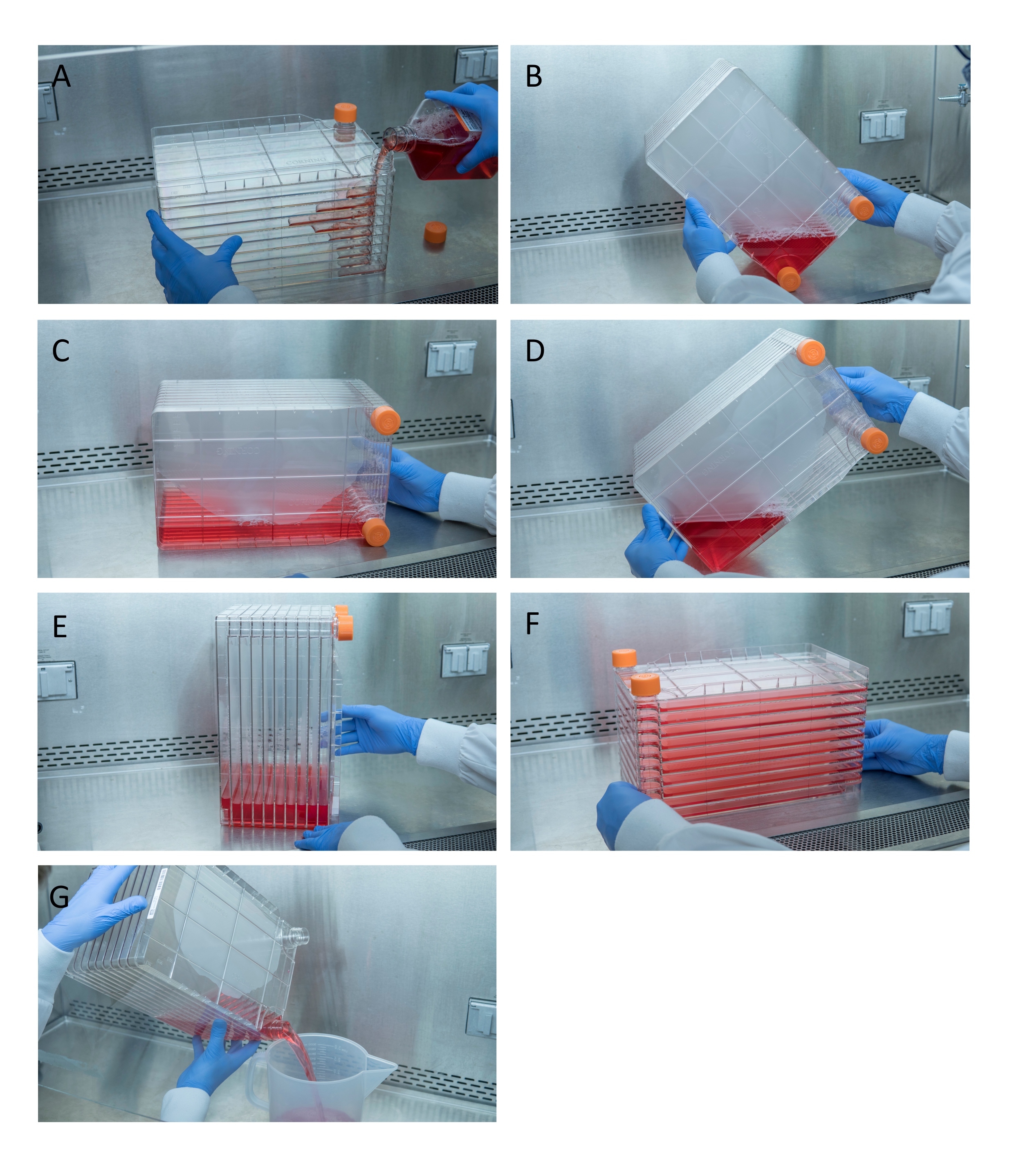
Figura 1: Manobra da pilha de células para semeadura celular e transfecção. Para a pilha de células de semeadura, comece removendo uma das tampas de ventilação e derramando em 1 L de DMEM completo pré-aquecido com quantidade necessária de células HEK293(A). Distribua uniformemente células e mídias apertando as duas tampas de ventilação e leve toda a mídia para o canto da pilha de células com uma das tampas de ventilação e coloque-a nesse canto(B),coloque a pilha de células de lado (C),e depois gire a pilha de células 90°(D)para que as portas de ventilação estejam para cima(E). Abaixe suavemente a pilha de células para sua posição horizontal normal e certifique-se de que todas as câmaras da pilha de células estejam completamente cobertas de mídia(F). Ao transfetar, desaparafusar as duas tampas de ventilação e lentamente despejar a mídia antiga em um recipiente de resíduos estéreis para mesmo fluir para não perturbar a monocamada das células(G). Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Prepare a mistura de polietileneimina (PEI)/DNA a uma razão de concentração de 3:1 (w/w).
- Prepare a mistura de DNA em um tubo cônico de 50 mL adicionando 475 μg de pTrangene e 1425 μg de pHelper a 40 mL de meio de soro reduzido para criar uma razão de 3:1 de pHelper:pTrangene.
NOTA: A calculadora de mistura PEI/DNA pode ser encontrada usando a Tabela 2. - Adicione 5,7 mL de PEI (1 g/L) ao meio de soro reduzido e à mistura de DNA dropwise. Em seguida, vórtice brevemente e incubar por 10 minutos à temperatura ambiente.
NOTA: À medida que o PEI/DNA incuba à temperatura ambiente, ele ficará ligeiramente nublado.
- Prepare a mistura de DNA em um tubo cônico de 50 mL adicionando 475 μg de pTrangene e 1425 μg de pHelper a 40 mL de meio de soro reduzido para criar uma razão de 3:1 de pHelper:pTrangene.
- Após 8 minutos de incubação pei/DNA, remova a mídia da câmara de cultura celular.
NOTA: Certifique-se de afrouxar ambas as tampas laranjas para manter um fluxo suave de mídia para evitar o desalojamento das células. - Adicione PEI/DNA a 1 L de DMEM completo pré-aquecido e despeje lentamente a mistura na porta da câmara de cultura celular. Distribua o líquido uniformemente para todas as linhas(Figura 1) e incubar por 72h a 37 °C, com 5% de CO2.
2 Colheita de AAV e lise química das células HEK293 transfectadas
- Agite a câmara de cultura celular vigorosamente para desalojar as células até que a mídia pareça nublada a partir de células desalojadas e despeje em quatro tubos de centrífuga de 500 mL.
- Centrifugar os tubos a 18.000 x g por 30 min a 4 °C para pelotar as células. Despeje o supernatante esclarecido em 1 L garrafa de copolídalato de polietileno (PETG).
NOTA: Se não tiver acesso a uma centrífuga de alta velocidade, centrífuga a 12.000 x g por 40 min. As células pelleted podem não ser sólidas a esta velocidade e deslizarão como derramando supernasce. - Resuspense as pelotas de célula em tubos de centrífugas de 500 mL com 50 mL de tampão de lise e incubar por 60 min a 37 °C.
- Centrifugar os tubos a 18.000 x g por 30 min, e depois transferir o supernante para a mesma garrafa 1 L PETG. Descarte os detritos celulares pelleted.
NOTA: Purifique imediatamente o sobrenante esclarecido e armazene a 4 °C por até 72 h. Para armazenamento a longo prazo, armazene a -80 °C. Não armazene a -20 °C.
3 Purificação vetorial AAV usando cromatografia de afinidade de heparina
- Retire o lise bruto de -80 °C e deixe a 4 °C durante a noite para descongelar. Uma vez descongelado, use um filtro de 0,22 μM para filtrar o lise bruto.
- Para passivar o concentrador centrífugo, adicione 4 mL de filtro pré-tratamento tampão a um concentrador centrífugo para cada coluna de sepharose de heparina que está sendo usada. Passivate o concentrador centrífuga à temperatura ambiente por 2-8 h. Configure a passivação imediatamente antes das etapas de purificação.
- Configure a tubulação e a bomba(Figura 2).
- Coloque a tubulação em uma bomba peristáltica e execute 20 mL de 1 M NaOH. Em seguida, execute 50 mL de água de grau molecular, e depois execute 50 mL de DMEM basal.
- Conecte a coluna de sepharose heparina de 5 mL à tubulação e execute 25 mL de DMEM basal para remover o conservante.
- Execute 0,2 μM do lise bruto filtrado através da coluna a uma taxa de fluxo de 1-2 gotas/s.

Figura 2: Configuração para bomba peristáltica para purificação de AAV. Passe a tubulação do lise bruto, através da bomba peristáltica, e para a coluna matriz de heparina. Clique aqui para ver uma versão maior desta figura.
{kind=link}
NOTA: Certifique-se de não introduzir bolhas ou permitir que a coluna seque, pois isso comprometerá a coluna e impedirá a eluição do AAV. Descarte a coluna se ela estiver seca e use uma nova coluna para o restante do lise bruto.
- Carregue todo o lise bruto na coluna heparina e use as seguintes soluções para lavar a coluna.
- Lave usando 50 mL de 1x Hank's Balanced Salt Solutions (HBSS) sem Mg2+ e Ca2+.
- Lave usando 15 mL de 0,5 % N-Lauroylsarcosine em HBSS sem Mg2+ e Ca2+.
- Lave usando 50 mL de HBSS sem Mg2+ e Ca2+.
- Lave usando 50 mL de HBSS com Mg2+ e Ca2+
- Lave usando 50 mL de 200 mM NaCl/HBSS com Mg2+ e Ca2+.
- Elute 5 x 5 mL (25 mL total) com 300 mM de NaCl/HBSS com Mg2+ e Ca2+ e rotular as eluções como E1-E5 (cada eluição é de 5 mL).
- Concentrando o vírus usando um concentrador centrífuga
- Gire o concentrador centrífugo contendo o tampão de pré-tratamento a 900 x g por 2 min. Descarte o fluxo.
- Lave o filtro concentrador centrífugo com 4 mL de HBSS com Mg2+ e Ca2+ e centrífuga a 1000 x g por 2 min; descartar o fluxo-through.
- Adicione a elução E2 ao concentrador centrífuga. Gire a 1000 x g por 5 min e descarte o fluxo.
- Finalize a adição de E2 e, em seguida, adicione E3 ao concentrador centrífuga e gire a 1000 x g por 5 min até que o vírus concentrado seja de aproximadamente 1 mL.
NOTA: Evite centrifugar o vetor de tal forma que o volume esteja abaixo do nível do filtro. Não concentre E1, E4 ou E5 no concentrador centrífugo, pois eles contêm muito pouco ou nenhum vetor e contêm contaminantes. - Remova o vírus concentrado do concentrador centrífuga usando uma ponta filtrada p200 e coloque-o em um tubo de centrífuga estéril de 1,5 mL.
- Enxágüe o concentrador centrífuga com 200 μL de HBSS com Mg2+ e Ca2+ para desalojar qualquer AAV restante do filtro. Pipeta para cima e para baixo vigorosamente várias vezes (para ~30 s) para desalojar qualquer vírus aderido à membrana e colocar no tubo centrífuga de 1,5 mL com o restante do vírus. Misture bem o tubo.
- Alíquota 5 μL para extrações de DNA e armazene o vetor purificado a -80 °C.
- Lave a coluna usando 25 mL de 2 M NaCl. Além disso, use 25 mL de 0,1% Triton X-100, pré-aquecido a 37 °C para lavar a coluna. Em seguida, lave a coluna usando 50 mL de dH2O estéril e depois lave usando 25 mL de 20% de etanol.
- Certifique-se de que a membrana da coluna está totalmente saturada em 20% de etanol, pois esta é a solução de armazenamento. Sele a coluna com plugues fornecidos e armazene a 4 °C.
- Guarde a tubulação em 1 M NaOH.
NOTA: Se limpas corretamente, as colunas de heparina sepharose podem ser reutilizadas até cinco vezes.
4 Extração genômica de DNA AAV
- Prepare a mistura de reação mencionada na Tabela 3 em um tubo PCR para tratamento DNase.
| Componente | Volume |
| Vetor AAV purificado | 5 μL |
| 10x DNase Buffer | 2 μL |
| DNase | 1 μL |
| ddH2O | 12 μL |
| Final Volume | 20 μL |
Tabela 3: Fórmula de mistura mestre de tratamento DNase. Componentes e volumes recomendados necessários para o tratamento DNase de vetores virais AAV durante a extração de DNA.
- Vórtice do tubo PCR para misturar e pulsar o tubo PCR para girar o conteúdo.
- Usando um termociclador, incubar a 37 °C por 20 min seguido de 75 °C por 15 min para aquecer a inativação do DNase.
- Adicione 5 μL de Proteinase K.
- Usando um termociclador, incubar a 50 °C por 60 min e depois a 95 °C por 30 min para aquecer a inativação Proteinase K.
- Use um kit de limpeza de DNA para remover potenciais contaminantes.
NOTA: Esta etapa foi realizada utilizando-se um kit de limpeza de sangue e tecido comercialmente disponível(Tabela de Materiais).- Adicione 200 μL de tampão de AL (kit de limpeza de sangue e tecido, Tabela de Materiais) ao tubo PCR contendo o vetor tratado DNase/Proteinase K.
- Vórtice do tubo PCR e incubar a 56 °C por 10 min em um termociclador.
- Pipeta o líquido do tubo PCR em uma coluna de giro estéril sentada em um tubo de coleta.
- Adicione 200 μL de 100% de etanol à coluna e misture bem por vórtice.
- Centrifugar a 6.000 x g por 1 min e descartar o fluxo através.
- Adicione 500 μL de tampão AW1 (kit de limpeza de sangue e tecido, Tabela de Materiais) à coluna de giro.
- Centrifugar a 6.000 x g por 1 min e descartar o fluxo através.
- Adicione 500 μL de tampão AW2 (kit de limpeza de sangue e tecido, Tabela de Materiais) à coluna de giro.
- Centrifugar a 15.000 x g por 3 min e descartar o fluxo através.
- Coloque a coluna de giro em um tubo de centrífuga de 1,5 mL estéril e adicione 200 μL de AE tampão (kit de limpeza de sangue e tecido, Tabela de Materiais) diretamente à membrana da coluna de spin.
- Incubar em temperatura ambiente por 1 min.
- Centrifugar a 6.000 x gpor 1 min para eluto o DNA.
- Armazene o DNA a -20 °C.
5 Titulação de genomas vetores AAV usando reação quantitativa em cadeia de polimerase e uma sonda Sian Virus 40 (SV40)
NOTA: Realize todo o trabalho qPCR em um capô PCR usando pontas de pipeta filtradas para evitar contaminação externa do DNA. Se o genoma AAV não codificar uma sequência de poliA SV40, use uma sonda contra o ITR descrito em outro lugar25. Certifique-se de que o DNA plasmídeo selecionado como padrão contém sequência de poliA SV40.
- Preparação padrão de estoque
- Diluir o padrão de DNA plasmídeo de estoque (pTransgene plasmid contendo sequência de sv40 polyA) para uma concentração final de 10 μg/μL e armazenar a -20 °C em 6 alíquotas μL.
- Determine o número de cópia presente no padrão de DNA plasmídeo usando a seguinte calculadora on-line26.
NOTA: Use um DNA plasmídeo usado para o padrão produzido por um fornecedor comercial para garantir a qualidade e a correta concentração. Prepare uma grande quantidade de padrão (por exemplo, 10 mL) para realizar estudos de ponte na transição para um padrão recém-preparado.
- Prepare a seguinte mistura de reagente mencionada na Tabela 4 para ambas as amostras e o padrão em um tubo centrífuga de 1,5 mL.
NOTA: Prepare o excesso suficiente de mistura mestra. Consulte tabela 5 para obter sequências de primer/sonda.
| Componente | Volume |
| Mix mestre qPCR universal (2X) | 10 μL |
| Água de grau molecular | 4,5 μL |
| 40x Primer/sonda polyA SV40 | 0,5 μL |
| Final Volume | 15 μL |
Tabela 4: mix mestre qPCR para titulação AAV. Componentes e volumes recomendados necessários para qPCR de DNA extraído de vetores virais AAV.
| Componente | Seqüenciar |
| Primer para a frente | 5'-AGCAATAGCATCAAATTTCACAA-3' |
| Primer reverso | 5'-CCAGACATGATAAGATACATTGATGAGTT-3' |
| Sondar | /56-FAM/AGCATTTTT/Zen/TTCACTGCATTCTAGTTGTGGTTTGTC/3IABkFQ |
Tabela 5: Sequências de primer contra a sequência de DNA SV40 polyA. Sequências dos primers e sondas usadas para titulação qPCR, que se ligam a áreas específicas de vetores virais AAV que contêm a sequência de poliA SV40.
- Pipeta o mestre misturar para cima e para baixo para misturar.
- Configure a placa de diluição.
- Use uma placa clara de 96 poços para preparar diluições padrão e amostra, adicione 45 μL de água de grau molecular a cada poço em cada outra coluna começando com a coluna 1 (colunas 1, 3, 5, 7, 9 e 11).
- Adicione 5 μL do padrão ao bem A1 e pipeta para misturar.
- Use uma nova ponta de pipeta filtrada para criar uma diluição de 1/10 do poço A1 para B1.
- Continue uma série de diluições de 10 vezes na coluna até chegar ao G1.
- Não adicione ao H1, pois isso funcionará como um controle negativo.
- Aplique a primeira amostra (S1) adicionando-a ao bem A3, formando uma diluição de 1/10. Pipeta esta mistura e transfira 5 μLpara bem B3. Descarte a ponta da pipeta após esta transferência.
- Com uma nova ponta de pipeta, misture a solução em bem B3 e forme uma diluição de 1/100. Transfira 5 μL dessa mistura para bem C3 e descarte a ponta após a transferência.
- Com uma nova ponta de pipeta, pipeta para cima e para baixo a solução em bem C3 para fazer uma diluição de 1/1000. Descarte a ponta.
- Continue diluindo amostras sem adicionar amostras às colunas 2, 4, 6, 8, 10 ou 12.
- Uma vez que todas as amostras sejam diluídas, misture o conteúdo em poços da coluna 1 e, em seguida, transfira 20 μL para a coluna 2.
- Repita isso para as colunas 3, 5, 7, 9 e 11 para criar réplicas de cada padrão e diluição amostral. Consulte a Figura 3 para o layout da placa.
NOTA: Ao seguir a configuração da placa na Figura 3,as amostras diluídas nas linhas G e H terão apenas 1/10 e 1/100 diluições.

Figura 3: Layout da placa para titulação aav qPCR. Azul indica a colocação da diluição seriada do padrão; verde indica a colocação do controle negativo; roxo indica a colocação da diluição das amostras. Cada padrão, negativo ou amostra é adicionado em reprodução. Um exemplo para a concentração da norma foi adicionado para mostrar a série de diluição do padrão, e a colocação de diluições amostrais foram adicionadas aos seus respectivos poços. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Titulação por QPCR baseado em detecção de poliA SV40
- Adicione 15 μL de mix mestre qPCR a cada poço de uma placa de 96 qPCR de 96 poços.
- Transfira 5 μL de cada amostra da placa clara de 96 poços para a placa de 96 bem-contornada de 96 bem qPCR.
- Use uma pipeta multicanal para garantir a mistura adequada da mistura e amostra mestre qPCR.
- Sele a placa com uma máquina de vedação e centrifugar a placa qPCR a 1500 x g por 30 s.
- Execute a reação qPCR no instrumento de amplificação e detecção de PCR em tempo real baseado em placa, usando as condições sugeridas na Tabela 6.
| Secção | Ciclos | Hora | Temperatura | Descrição |
| Pré-incubação | 1x | 5 min. | 95 °C | Desnaturação de DNA. |
| Amplificação | 38x | 15 s | 95 °C | Amplificação do DNA. As configurações podem ser modificadas se usar primers alternativos com diferentes temperaturas de ressarção. |
| 60 s | 60 °C | |||
| Arrefecimento | 1x | 60 s | 40 °C | Resfriamento da placa. Fim da corrida. |
Tabela 6: Protocolo termocicr para titulação qPCR baseada em sonda de hidrólise. Protocolo termocicro recomendado para o uso de titulação qPCR baseada em sonda de vetores AAV extraídos de DNA.
NOTA: Para a planilha de titulação qPCR AAV consulte Tabela 7.
- Análise de dados para determinar números de cópia do genoma AAV.
- Preencha as células de dados da planilha(Tabela 7A)com os valores de concentração obtidos a partir do qPCR executados tanto para diluições padrão quanto amostral.
- Use valores de concentração da Tabela 7A para produzir uma curva padrão(Tabela 7B).
NOTA: A curva padrão será mostrada como um logaritmo natural (y = a ln(x) + b) juntamente com a eficiência R2. Uma curva padrão deve ter uma eficiência próxima de 100 % e R2 perto de 1,0 (≥0,99). - Preencha a eficiência da inclinação preenchendo esta calculadora online27.
NOTA: Uma eficiência entre 90%-110% é aceitável. Se a eficiência do qPCR estiver fora desse intervalo, re-execute o qPCR. - Use valores de concentração da Tabela 7A para mediar as diluições de cada amostra e determinar o desvio padrão de cada amostra(Tabela 7C).
NOTA: Exclua diluições de amostras que estejam a mais de um desvio padrão da média das diluições amostrais. - Usando a concentração média de cada diluição, multiplique pelo fator de diluição e, em seguida, divida por cinco para obter os genomas vetoriais (vg)/μL de cada amostra(Tabela 7C).
- Calcule o vg/mL de cada amostra multiplicando a média das concentrações de cada amostra por 80.000(Tabela 7C).
- Média do vg/mL de cada diluição para produzir o vg/mL final de cada amostra(Tabela 7C).
NOTA: O usuário deve dividir a concentração média de cada diluição por um fator cinco para explicar os 5 μL carregados em cada poço para que o qPCR corra para produzir a concentração em vg/μL. O fator de 80.000 contabiliza a transição do valor médio de concentração de cada amostra para vg/mL. Em primeiro lugar, a média do valor de concentração de cada amostra deve ser multiplicada por 2 para dar conta dos genomas de uma única-fixação, uma vez que o conjunto de primer-probe apenas quantifica o DNA de sentido positivo, de uma única cadeia ( ssDNA), e o genoma AAV existe em uma proporção aproximada de 1:1 entre o SSDNA positivo e negativo25,28. A média do valor de concentração de cada amostra deve ser multiplicada x40 para contabilizar a diluição amostral de 5 μL de vetor purificado (seção 4.1) a 200 μL de DNA extraído (seção 4.6.12). Por fim, a média do valor de concentração de cada amostra deve ser multiplicada x1000 para converter de vg/μL para vg/mL.
6 Avaliação da qualidade do vetor e pureza
- Controle de Qualidade - Mancha Ocidental
- Prepare um gel de 12% de PAGE SDS.
- Realize eletroforese de gel de poliacrilamida.
NOTA: Carga 6 x 1010 vg de amostras por poço. - Transfira as proteínas para a membrana de difluoreto de polivinida (PVDF).
- Bloqueando a membrana PVDF
- Remova a membrana do aparelho de transferência e enxágue em 0,1% de PBST para remover acrilamida solta.
- Coloque a membrana na solução de bloqueio por pelo menos 1 h em temperatura ambiente ou durante a noite a 4 °C.
NOTA: O tampão de bloqueio pode ser complementado com 2% de soro de cabra.
- Incubação com anticorpo primário
- Decante o tampão de bloqueio e adicione o anticorpo primário, um anticorpo monoclonal do rato anti-AAV a uma diluição de 1:200.
- Incubar durante a noite a 4 °C.
- Decante o anticorpo primário e lave cinco vezes com 0,1% de PBST para 5 min em temperatura ambiente com agitação.
- Incubação com anticorpo secundário
- Decante a solução de lavagem e adicione o anticorpo secundário conjugado HRP, diluído a uma 1:7500 no buffer de bloqueio, e incubar por 1h à temperatura ambiente com agitação.
- Decante o anticorpo secundário e lave cinco vezes com 0,1% de PBST para 5 min em temperatura ambiente com agitação.
- Realize uma lavagem final com PBS em temperatura ambiente com agitação.
- Detecte as proteínas usando substrato de chemiluminescente (ECL) aprimorado.
- Imagem do gel para visualizar as proteínas virais (subunidades VP1, VP2 e VP3)(Figura 4).

Figura 4: Mancha ocidental mostrando proteínas capsidas AAV. Pista A; Escada MW, Pista B; AAV6.2FF-hIgG01, Pista C; AAV6.2FF-hIgG02, Lane D; AAV6.2FF-hIgG03 e Lane E; AAV6.2FF-hIgG04. 6 x 1010 vg de cada AAV6.2FF-hIgG foi carregado em suas respectivas pistas. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Controle de Pureza - SDS PAGE e Mancha de Coomassie
- Prepare gel E amostras SDS-PAGE conforme descrito da etapa 6.1.1 e 6.1.2.
- Fixar o gel na solução de fixação por 1h ou durante a noite com agitação suave. Altere a solução de fixação uma vez durante a primeira hora.
- Manche o gel na solução de coloração por 2-4 h com agitação suave.
- Destain o gel com uma solução desemining. Reabastecer a solução de desestabilização várias vezes até que o fundo do gel esteja totalmente detido (4-24 h).
- Armazene o gel detido em uma solução de armazenamento.
- Imagem do gel para visualizar todas as proteínas manchadas pela solução de coloração Coomassie.

Figura 5: Gel manchado de coomassie. Pista A; Escada MW, Pista B; AAV6.2FF-hIgG01, Pista C; AAV6.2FF-hIgG02, Lane D; AAV6.2FF-hIgG03, Lane E; AAV6.2FF-hIgG04, Pista F; AAV6.2FF-hIgG05 e Lane G; AAV6.2FF-hIgG06. 6 x 1010 vg de cada AAV6.2FF-hIgG foi carregado em suas respectivas pistas. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Ensaio alternativo de controle de pureza - HEK293 detecção de proteínas celulares hospedeiras ELISA
- Realize a detecção de proteína de célula hospedeira HEK293 via ELISA de acordo com as instruções do fabricante.
NOTA: Use diluições de 5 x 10-2 e 1 x 10-3 para amostras purificadas de rAAV. Uma vez que o TMB seja adicionado ao poço, incubar longe da luz. A regressão linear não pode ser usada para analisar os resultados. - Realize uma análise ponto a ponto, spline cúbico ou método de ajuste logístico de quatro parâmetros para interpolar concentrações de desconhecidos e multiplicar pelo fator de diluição para determinar a concentração amostral original.
- Realize a detecção de proteína de célula hospedeira HEK293 via ELISA de acordo com as instruções do fabricante.
Resultados
A tradução de pequenos modelos de roedores para modelos animais maiores e eventual aplicação clínica apresenta um desafio significativo devido à grande quantidade de AAV necessária para transduzir animais maiores e alcançar efeitos terapêuticos. Para comparar a eficiência de transdução do capsídeo AAV6.2FF projetado racionalmente, anteriormente demonstrou um aumento de 101 vezes na eficiência de transdução em células musculares murinas em comparação com AAV63,ratos, hamsters e c...
Discussão
A produção de vetores AAV (rAAV) recombinantes descritos neste artigo utiliza materiais, reagentes e equipamentos comuns encontrados na maioria dos laboratórios e instalações de pesquisa de biologia molecular. Este artigo permite que o rAAV in vitro e in vivo de alta qualidade seja produzido pelo leitor. Acima de tudo, este protocolo para produção de rAAV, comparado a protocolos mais tediosos envolvendo purificação de cloreto de césio, é eficiente e evita o uso de ultracentrifugação. Uma ve...
Divulgações
Sarah K. Wootton é uma inventora de uma patente americana US10806802B2 para o capsid AAV6.2FF.
Agradecimentos
Amira D. Rghei, Brenna A. Y. Stevens, Sylvia P. Thomas e Jacob G. E. Yates foram beneficiários do Ontário Veterinary College Student Stipends, bem como de Bolsas de Pós-Graduação em Ontário. Amira D. Rghei foi a beneficiária de um Mitacs Accelerate Studentship. Este trabalho foi financiado pelo Canadian Institutes for Health Research (CIHR) Project Grant (#66009) e uma bolsa colaborativa de Projetos de Pesquisa em Saúde (NSERC) (#433339) para a SKW.
Materiais
| Name | Company | Catalog Number | Comments |
| 0.22 μm filter | Millipore Sigma | S2GPU05RE | |
| 0.25% Trypsin | Fisher Scientific | SM2001C | |
| 1-Butanol | Thermo Fisher Scientific | A399-4 | CAUTION. Use under a laminar flow hood. Wear gloves |
| 10 chamber cellstack | Corning | 3271 | |
| 1L PETG bottle | Thermo Fisher Scientific | 2019-1000 | |
| 30% Acrylamide/Bis Solution | Bio-Rad | 1610158 | |
| 96-well skirted plate | FroggaBio | FS-96 | |
| Adhesive plate seals | Thermo Fisher Scientific | 08-408-240 | |
| Ammonium persulfate (APS) | Bio-Rad | 161-0700 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Blood and Tissue Clean up Kit | Qiagen | 69506 | Use for DNA clean up in section 4.6 of protocol |
| Bromophenol blue | Fisher Scientific | B392-5 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Cell Culture Dishes | Greiner bio-one | 7000232 | 15 cm plates |
| Culture Conical Tube | Thermo Fisher Scientific | 339650 | 15 mL conical tube |
| Culture Conical Tube | Fisher Scientific | 14955240 | 50 mL conical tube |
| Dulbecco's Modified Eagle Medium (DMEM) with 1000 mg/L D-glucose, L-glutamine | Cytiva Life Sciences | SH30022.01 | |
| ECL Western Blotting Substrate | Thermo Fisher Scientific | 32209 | |
| Ethanol | Greenfield | P016EA95 | Dilute ethyl alcohol(95% vol) to 20% for section 3.7.4 and 70% for section 6.1.1.1 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | SH30396.03 | |
| Glacial acetic acid | Fisher Scientific | A38-500 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | Fisher Scientific | BP381-500 | |
| HBSS with Mg2+ and Ca2+ | Thermo Fisher Scientific | SH302268.02 | |
| HBSS without Mg2+ and Ca2+ | Thermo Fisher Scientific | SH30588.02 | |
| HEK293 cells | American Tissue Culture Collection | CRL-1573 | Upon receipt, thaw the cells and culture as described in manufacturer’s protocol. Once cells have been minimally passaged and are growing well, freeze a subfraction for future in aliquots and store in liquid nitrogen. Always use cells below passage number 30. Once cultured cells have been passaged more than 30 times, it is recommended to restart a culture from the stored aliquots |
| HEK293 host cell protein ELISA kit | Cygnus Technologies | F650S | Follow manufacturer’s instructions |
| Heparin sulfate column | Cytiva Life Sciences | 17040703 | |
| Kimwipe | Thermo Fisher Scientific | KC34120 | |
| L-glutamine (200 mM) | Thermo Fisher Scientific | SH30034.02 | |
| Large Volume Centrifuge Tube Support Cushion | Corning | CLS431124 | Support cushion must be used with large volume centrifuge tubes uless the centrifuge rotor has the approriate V-bottom cushions |
| Large Volume Centrifuge Tubes | Corning | CLS431123-6EA | 500 mL centrifuge tubes |
| MgCl2 | Thermo Fisher Scientific | 7791-18-6 | |
| Microcentrifuge tube | Fisher Scientific | 05-408-129 | 1.5 mL microcentrifuge tube, sterilize prior to use |
| Molecular Grade Water | Cytiva Life Sciences | SH30538.03 | |
| N-Lauroylsarcosine sodium salt | Sigma Aldrich | L5125 | CAUTION. Wear gloves |
| NaCl | Thermo Fisher Scientific | BP35810 | |
| Optimem, reduced serum medium | Thermo Fisher Scientific | 31985070 | |
| Pasteur pipets | Fisher Scientific | 13-678-20D | Sterilize prior to use |
| PBS (10x) | Thermo Fisher Scientific | 70011044 | Dilute to 1x for use on cells |
| Penicillin-Streptomycin Solution | Cytiva Life Sciences | SV30010 | |
| pHelper plasmid | De novo design or obtained from plasmid repository | NA | |
| Pipet basin | Thermo Fisher Scientific | 13-681-502 | Purchase sterile pipet basins |
| Polyethylene glycol tert-octylphenyl ether (Triton X-100) | Thermo Fisher Scientific | 9002-93-1 | CAUTION. Wear gloves |
| Polyethylenimine (PEI) | Polyscience | 24765-1 | Follow manufacturer’s instructions to produce a 1L solution. 0.22μm filter and store at 4°C |
| Polypropylene semi-skirted PCR Plate | FroggaBio | WS-96 | |
| Polysorbate 20 (Tween 20) | Thermo Fisher Scientific | BP337-100 | CAUTION. Wear gloves |
| polyvinylidene difluoride (PVDF) membrane | Cytiva Life Sciences | 10600023 | Use forceps to manipulate. Wear gloves. |
| Primary antibody | Progen | 65158 | |
| Protein Ladder | FroggaBio | PM008-0500 | |
| Proteinase K | Thermo Fisher Scientific | AM2546 | |
| pTrangene plasmid | De novo design or obtained from plasmid repository | NA | Must contain SV40 polyA in genome to be compatible with AAV titration in section 5.0 |
| Pump tubing | Cole-Parmer | RK-96440-14 | Optimize length of tubing and containment of virus in fractions E1-E5 |
| RQ1 Dnase 10 Reaction Buffer | Promega | M6101 | Use at 10x concentration in protocol from section 4.0 |
| RQ1 Rnase-free Dnase | Promega | M6101 | |
| Sample dilutent | Cygnus Technologies | I700 | Must be purchased separately for use with HEK293 host cell protein ELISA kit |
| Secondary antibody, HRP | Thermo Fisher Scientific | G-21040 | |
| Skim milk powder | Oxoid | LP0033B | |
| Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 28312 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Sodium hydroxide (NaOH) | Thermo Fisher Scientific | SS266-4 | |
| SV40 polyA primer probe | IDT | Use sequence in Table X for quote from IDT for synthesis | |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | 15524010 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Tris Base | Fisher Scientific | BP152-5 | |
| Trypan blue | Bio-Rad | 1450021 | |
| Ultra-Filter | Millipore Sigma | UFC810024 | Ultra-4 Centrifugal 10K device must be used, as it has a 10000 molecular weight cutoff |
| Universal Nuclease for cell lysis | Thermo Fisher Scientific | 88702 | |
| Universal qPCR master mix | NEB | M3003L | |
| Whatman Paper | Millipore Sigma | WHA1001325 | |
| β-mercaptoethanol | Fisher Scientific | 21985023 | CAUTION. Use under a laminar flow hood. Wear gloves |
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. |
Referências
- Hastie, E., Samulski, R. J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success-a personal perspective. Human Gene Therapy. 26 (5), 257-265 (2015).
- Wang, D., Tai, P. W. L., Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. 18 (5), 358-378 (2019).
- Nathwani, A. C., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England Journal of Medicine. 371 (21), 1994-2004 (2014).
- Kuzmin, D. A., et al. The clinical landscape for AAV gene therapies. Nature Reviews Drug Discovery. 20 (3), 173-174 (2021).
- Ylä-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (10), 1831-1832 (2012).
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020)
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (2020)
- Statement from FDA Commissioner Scott Gottlieb, MD and Peter Marks, MD Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. FDA Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (2020)
- Asokan, A., Schaffer, D. V., Samulski, R. J. The AAV vector toolkit: poised at the clinical crossroads. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (4), 699-708 (2012).
- Rose, J. A., Hoggan, M. D., Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proceedings of the National Academy of Sciences of the United States of America. 56 (1), 86-92 (1966).
- Lusby, E., Fife, K. H., Berns, K. I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. Journal of Virology. 34 (2), 402-409 (1980).
- Masat, E., Pavani, G., Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discovery Medicine. 15 (85), 379-389 (2013).
- Ling, C. Enhanced Transgene Expression from Recombinant Single-Stranded D-Sequence-Substituted Adeno-Associated Virus Vectors in Human Cell Lines In Vitro and in Murine Hepatocytes In Vivo. Journal of Virology. 89 (2), 952-961 (2014).
- Cathomen, T., Stracker, T. H., Gilbert, L. B., Weitzman, M. D. A genetic screen identifies a cellular regulator of adeno-associated virus. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 14991-14996 (2001).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Aschauer, D. F., Kreuz, S., Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLOS One. 8 (9), 76310 (2013).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Pillay, S., et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. Journal of Virology. 91 (18), (2017).
- Merkel, S. F. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. Journal of Neurochemistry. 140 (2), 216-230 (2017).
- van Lieshout, L. P., et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Molecular Therapy. Methods & Clinical Development. 9, 323-329 (2018).
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M., Samulski, R. J. single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. Journal of Virology. 80 (22), 11393-11397 (2006).
- Liu, J., Moon, Y. -. A. Simple purification of adeno-associated virus-DJ for liver-specific gene expression. Yonsei Medical Journal. 57 (3), 790-794 (2016).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adeno-associated virus vectors. Human Gene Therapy. 9 (18), 2745-2760 (1998).
- Kimura, T., et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Scientific Reports. 9 (1), 13601 (2019).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2012).
- . Paramyxoviridae: The viruses and their replication. Fields Virology Available from: https://www.scholars.northwestern.edu/en/publications/paramyxoviridae-the-viruses-and-their-replication (1996)
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Kaludov, N., Brown, K. E., Walters, R. W., Zabner, J., Chiorini, J. A. Adeno-associated virus serotype 4 (AAV4) and AAV5 Both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of Virology. 75 (15), 6884-6893 (2001).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Human Gene Therapy Methods. 26 (6), 228-242 (2015).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Backovic, A., et al. Capsid protein expression and adeno-associated virus like particles assembly in Saccharomyces cerevisiae. Microbial Cell Factories. 11, 124 (2012).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados