
Method Article
Zellkernisolierung aus der Niere adulter Mäuse für die Einzelkern-RNA-Sequenzierung
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Hier stellen wir ein Protokoll zur Isolierung hochwertiger Kerne aus gefrorenen Mausnieren vor, das die Repräsentation von medullären Nierenzelltypen verbessert und die Genexpressionsartefakte aus der enzymatischen Gewebedissoziation vermeidet.
Zusammenfassung
Die Nieren regulieren verschiedene biologische Prozesse wie Wasser, Elektrolyt und Säure-Basen-Homöostase. Physiologische Funktionen der Niere werden von mehreren Zelltypen ausgeführt, die in einer komplexen Architektur über die kortikomedulläre Achse des Organs angeordnet sind. Jüngste Fortschritte in der Einzelzell-Transkriptomik haben das Verständnis der zelltypspezifischen Genexpression in der Nierenphysiologie und -erkrankung beschleunigt. Enzymbasierte Gewebedissoziationsprotokolle, die häufig für die Einzelzell-RNA-Sequenzierung (scRNA-seq) verwendet werden, erfordern jedoch meist frisches (nicht archiviertes) Gewebe, führen zu transkriptionellen Stressreaktionen und begünstigen die Auswahl reichlich vorhandener Zelltypen der Nierenrinde, was zu einer Unterrepräsentation der Zellen der Medulla führt.
Hier stellen wir ein Protokoll vor, das diese Probleme vermeidet. Das Protokoll basiert auf der Isolierung von Zellkernen bei 4 °C aus gefrorenem Nierengewebe. Kerne werden aus einem zentralen Stück der Mausniere isoliert, das aus dem Kortex, der äußeren Medulla und der inneren Medulla besteht. Dies reduziert die für Ganznierenproben typische Überrepräsentation kortikaler Zellen zugunsten der Markzellen, so dass die Daten die gesamte kortikomedulläre Achse in ausreichender Menge repräsentieren. Das Protokoll ist einfach, schnell und anpassungsfähig und stellt einen Schritt in Richtung Standardisierung der Einzelkern-Transkriptomik in der Nierenforschung dar.
Einleitung
Nieren weisen eine hochkomplexe Gewebearchitektur auf. Sie bestehen aus funktionell und anatomisch unterschiedlichen Segmenten entlang einer kortikomedullären Achse und vermitteln biologische Funktionen, wie z.B. die Regulation des extrazellulären Flüssigkeitsvolumens, des Elektrolythaushalts oder der Säure-Basen-Homöostase1.
Fortschritte in der Einzelzell-Transkriptomik haben die eingehende Charakterisierung komplexer Gewebe ermöglicht und das Verständnis der segment- und zelltypspezifischen Genexpression in der Nierenphysiologie, -entwicklung und -erkrankung beschleunigt 2,3,4.
Die enzymbasierten Dissoziationsprotokolle, die häufig für scRNA-seq verwendet werden, weisen jedoch mehrere Nachteile und Einschränkungen auf. Je nach Protokoll erzeugen sie transkriptionelle Stressreaktionen und Gewebedissoziationsverzerrungen hin zu leichter dissoziierbaren kortikalen Zelltypen 5,6. Obwohl Protokolle, die kälteaktive Proteasen für embryonale Nieren verwenden, in der Lage sind, stressbedingte Transkriptionsveränderungen zu mildern, können sie die Dissoziationsverzerrung gegenüber kortikalen Zellen nicht überwinden und sind möglicherweise nicht ohne weiteres an verschiedene Arten von erkranktem Nierengewebe anpassbar7. Darüber hinaus sind Einzelzellansätze nicht ohne weiteres mit gefrorenen Gewebeproben kompatibel, so dass ihre Anwendung meist auf nicht archiviertes, frisches Gewebe beschränkt ist, was die Gewebeentnahme zu einem einschränkenden Faktor6 macht.
Die Einzelkern-RNA-Sequenzierung (snRNA-seq) kann diese Einschränkungen umgehen 8,9. Hier stellen wir ein Protokoll für die Isolierung von Zellkernen aus einer zentralen Scheibe gefrorenen Nierengewebes adulter Mäuse vor (Abbildung 1)10. Unser Protokoll ist einfach und bietet einen standardisierten Ansatz, um RNA-Sequenzierungsbibliotheken mit einer ausgewogenen Darstellung verschiedener Nierenzelltypen für experimentelle Modelle zu erhalten, die keine starken regionalen Gewebeveränderungen beinhalten. Im letzteren Fall kann unser Protokoll auch mit ganzen Nieren durchgeführt werden.
Protokoll
Alle Tierversuche wurden nach dem Tierschutzgesetz (TierSchG) und der Tierschutzversuchstierverordnung (TierSchVersV) durchgeführt und von den örtlichen Behörden und den Tierschutzbeauftragten unserer Einrichtung (MDC) genehmigt.
1. Gewebevorbereitung
- Bereiten Sie eine 6-Well-Platte vor, die 2 ml 1x phosphatgepufferte Kochsalzlösung (PBS) pro Vertiefung für jede erhaltene Niere enthält. Bereiten Sie eine 6-Well-Platte vor, die 2 ml RNA-Stabilisierungslösung pro Well und Niere enthält. Beide Platten auf Eis vorkühlen.
- Euthanasieren Sie eine 3 bis 6 Monate alte männliche C57BL/6-Maus. Legen Sie die Maus auf ein Präpariertablett, stecken Sie die Extremitäten fest und sterilisieren Sie den Bauch mit 70% Ethanol.
- Öffnen Sie den Bauch mit einer Pinzette und einer Schere bis zum Brustkorb. Heben Sie den Darm und andere Organe zur Seite und entfernen Sie die Nieren, indem Sie den Harnleiter, die Nierenarterie und die Vene vorsichtig mit einer Schere durchschneiden. Waschen Sie die Niere in der zuvor zubereiteten, eiskalten 1x PBS und entfernen Sie die Nierenfaszie und das restliche Fett aus der Niere, bis das gesamte weiße Gewebe entfernt ist (Abbildung 2A).
- Legen Sie die Niere auf eine Kaltpräparierplatte und verwenden Sie ein scharfes Skalpell oder eine Rasierklinge, um eine mittlere Scheibe von 1-2 mm zu erhalten. Stellen Sie sicher, dass das Gewebestück die gesamte kortikomedulläre Achse enthält (Abbildung 2B). Verwenden Sie eine Mikrodissektionsschere und eine Pinzette, um den Kortex vorsichtig von den Seiten des Mittelstücks abzuschneiden. Innerhalb des präparierten Gewebestücks sollten die drei Segmente Kortex, äußere Medulla und innere Medulla deutlich sichtbar sein (Abbildung 2C).
HINWEIS: Die Scheibe sollte eine Dicke von 2 mm oder ein Gewicht von 20 mg nicht überschreiten, um ausreichende Puffermengen für eine effektive Gewebelyse zu gewährleisten und die Hintergrund-RNA in den cDNA-Bibliotheken zu minimieren. Umgebungs-RNA verschwendet Sequenzkapazität, da sie nicht mit einzelnen Kernen assoziiert ist. - Das Nierenstück wird in die zuvor hergestellte RNA-Stabilisierungslösung überführt und 24 h bei 4 °C inkubiert, um einen RNA-Abbau zu vermeiden. Entfernen Sie nach 24 h die RNA-Stabilisierungslösung und lagern Sie das Gewebe bis zur weiteren Verwendung bei -80 °C. Entfernen Sie die überschüssige Lösung vorsichtig mit Seidenpapier.
2. Isolierung von Kernen
- Reinigungs- und Vorbereitungsschritte
- Reinigen Sie Tischplatten und Pipetten mit 70% Ethanol und RNase-Dekontaminationslösung.
- Reinigen Sie ein 2-ml-Gewebemahlröhrchen mit rundem Boden und den passenden Stößel A und B mit RNase-Dekontaminationslösung, gefolgt von 70 % Ethanol und RNase-freiem Wasser (1 Mahlröhrchen und Stößelset pro Probe). Lassen Sie es vollständig trocknen.
- Die Zentrifuge wird auf 4 °C vorgekühlt.
- Beschriften und kühlen Sie drei 15-ml-Sammelröhrchen, ein 1,5-ml-Sammelröhrchen, ein 5-ml-FICS-Sammelröhrchen (Fluoreszenz-aktivierte Zellsortierung) und ein Trockenmahlröhrchen für jede Probe auf Eis vor.
- Vorbereitung des Puffers
- Die Ribonukleosid-Vanadyl-Komplex-Stammlösung wird auf 65 °C erwärmt, bis sie gemäß den Anweisungen des Herstellers zu einer grün-schwarzen klaren Lösung rekonstituiert ist. 11 Preise
- Bereiten Sie 1x PBS mit 4% Rinderserumalbumin (BSA) vor, wie in Tabelle 1A beschrieben. Bereiten Sie zusätzlich 1x PBS mit 0,04% BSA vor (Tabelle 1B). Filtern Sie beide Lösungen mit einem 0,2 μm tensidfreien Celluloseacetat (SFCA)-Membranspritzenfilter und halten Sie sie bis zur weiteren Verwendung auf Eis.
- Bereiten Sie den Kernlysepuffer 1 vor (NLB1, Tabelle 1C). 4 ml EZ-Lysepuffer für den Nuklei-Lysepuffer 2 (NLB2, Tabelle 1D) und 2 ml 0,04 % BSA/PBS für den Nuklei-Suspensionspuffer (NSB, Tabelle 1E) werden in 15-ml-Röhrchen gegeben. Fügen Sie die RNase-Inhibitorlösung direkt vor der Verwendung zu NLB2 und NSB hinzu, wie unten im Protokoll angegeben. Bis zur weiteren Verwendung auf Eis aufbewahren.
- EZ-Lysepuffer mit 10% Saccharose (Saccharosegradientenpuffer, Tabelle 1F) herstellen. Gut mischen und den Puffer mit einem 0,2 μm SFCA-Membranspritzenfilter in ein frisches 15-ml-Röhrchen filtrieren. Bis zur weiteren Verwendung auf Eis aufbewahren.
- Gewebehomogenisierung und Zelllyse
HINWEIS: Um den RNA-Abbau zu minimieren, werden alle Schritte auf Eis durchgeführt. Das Mahlrohr, die Petrischale und alle Puffer müssen vorgekühlt werden. Alle Resuspensionsschritte werden durch vorsichtiges Pipettieren der Kernsuspension durchgeführt. Verwirbeln Sie die Probe nicht, um Scherkräfte und Schäden an den Kernen zu vermeiden.- Nehmen Sie das gefrorene Nierenstück und geben Sie es in eine 60-mm-Styropor-Petrischale auf Eis, die 1 ml NLB1 enthält.
- Zerkleinern Sie das Gewebe gründlich mit einer Rasierklinge oder einem Skalpell (Abbildung 3A).
- Schneiden Sie die Spitze einer 1-ml-Pipettenspitze ab und geben Sie das Hackfleisch und den Puffer in das Mühlenröhrchen. Stellen Sie sicher, dass Sie alle Gewebestücke übertragen. Waschen Sie die Petrischale bei Bedarf 5-10 Mal mit dem Puffer.
- Homogenisieren Sie die Suspension auf Eis, indem Sie den Stößel A langsam 25x im Mahlrohr auf und ab bewegen. Vermeiden Sie Luftblasen, die durch schnelle Bewegungen verursacht werden (Abbildung 3B).
- Das Homogenisat wird in einem vorgekühlten 15-ml-Auffangröhrchen durch ein 100-μm-Sieb geleitet und der Filter mit weiteren 1 ml NLB1 gewaschen.
- Waschen Sie das Mahlröhrchen mit kaltem EZ-Kernlysepuffer und entsorgen Sie den Puffer.
- Übertragen Sie das Homogenisat zurück in das Mahlrohr und homogenisieren Sie die Suspension auf Eis, indem Sie den Stößel B langsam 15x im Mahlrohr auf und ab bewegen. Vermeiden Sie Luftblasen, die durch schnelle Bewegungen verursacht werden (Abbildung 3C).
- Das Homogenisat wird in ein vorgekühltes 15-ml-Sammelröhrchen überführt. Waschen Sie das Mühlenröhrchen mit weiteren 2 ml NLB1 und stellen Sie sicher, dass alle Gewebefragmente in das Sammelröhrchen übertragen werden. Inkubieren Sie das Homogenat (Gesamtvolumen von 4 ml) 5 Minuten lang auf Eis, um die Zellen zu lysieren.
- Reinigung von Kernen
- Das Homogenisat wird durch ein 40-μm-Sieb in ein vorgekühltes 15-ml-Auffangröhrchen geleitet. Das Auffangröhrchen wird 5 min lang bei 500 x g bei 4 °C in einer Zentrifuge mit Schaufelrotor geschleudert. In der Zwischenzeit fügen Sie NLB2 eine RNase-Inhibitorlösung hinzu (Tabelle 1D).
- Entfernen Sie den Überstand, ohne das Pellet zu stören. Resuspendieren Sie das Pellet vorsichtig in 4 ml NLB2.
- Legen Sie die Aufhängung vorsichtig mit einem 1-ml-Kissen aus Saccharose-Gradientenpuffer unter. Zentrifugieren bei 500 x g für 5 min bei 4 °C in einer Zentrifuge mit Schaufelrotor. In der Zwischenzeit fügen Sie RNase-Inhibitorlösung zu NSB hinzu (Tabelle 1E).
- Entfernen Sie nach der Zentrifugation vorsichtig das Auffangröhrchen aus der Zentrifuge und achten Sie darauf, die beiden Schichten beim Umgang mit dem Auffangröhrchen nicht zu stören. Zwischen den beiden Schichten sind Zelltrümmer sichtbar. Entfernen Sie den Überstand vorsichtig, beginnend mit den Trümmern. Entfernen Sie den verbleibenden Überstand, ohne das Kernpellet zu stören, und resuspendieren Sie das Pellet vorsichtig in 1 ml NSB.
HINWEIS: Das Resuspensionsvolumen hängt von der Menge des Gewebes ab, das für die Isolierung verwendet wird, und von der Pelletgröße, die nach dem letzten Zentrifugationsschritt gewonnen wird. Möglicherweise muss das Volumen an die erwartete Anzahl von Kernen angepasst werden. - Das Homogenisat wird durch ein 20-μm-Sieb in das vorgekühlte 5-ml-FACS-Sammelröhrchen geleitet.
3. Sortierung der Kerne
- 20 μl 4′,6-Diamidino-2-phenylindol (DAPI) pro ml NSB bis zu einer Endkonzentration von 2 μM in das Homogenat im FACS-Sammelröhrchen geben und vorsichtig mischen. 5 Minuten auf Eis inkubieren.
- Bereiten Sie das vorgekühlte 1,5-ml-Sammelröhrchen mit 20 μl 4% BSA /1x PBS vor und fügen Sie 0,5 μl RNase-Inhibitorlösung zu einer Endkonzentration von 1 U/μl hinzu.
- Sortieren Sie die Kerne mit einem Zellsortierer.
- Mischen Sie die Kernsuspension kurz, bevor Sie das FACS-Sammelröhrchen in den Sortierer einsetzen.
- Legen Sie ein erstes Gate P1 basierend auf der Vorwärtsstreuung (FSC) und der Seitenstreuung (SSC) fest, um Trümmer und Aggregate auszuschließen (Abbildung 4A).
- Um leere oder beschädigte Kerne und Multiplets auszuschließen, legen Sie ein nachfolgendes Gatter basierend auf der DAPI-Fläche im Vergleich zur DAPI-Höhe (DAPI-A vs. DAPI-H) fest (Abbildung 4B).
- Sortieren Sie einzelne Kerne in das 1,5-ml-Sammelröhrchen mit 4% BSA /1x PBS mit 1 U/μl RNase-Inhibitorlösung, die in 3.2 hergestellt wurde.
4. Qualitätskontrolle
- Messen Sie die endgültige Kernkonzentration unter einem Fluoreszenzmikroskop oder in einer automatischen Zählkammer in mindestens zwei unabhängigen Zählungen und beurteilen Sie die Suspensionsqualität (Abbildung 5).
HINWEIS: Optimale Konzentrationen liegen zwischen 700 und 1.200 Kernen/μl. Niedrigere Zellkonzentrationen wie 700 Kerne/μl können vorzuziehen sein, da die resultierenden cDNA-Bibliotheken weniger Hintergrund-RNA aus der Umgebung enthielten (Transkripte, die nicht mit einzelnen Kernen assoziiert sind). - Berechnen Sie das erforderliche Volumen der Kernsuspension für die gewünschte Gewinnung sequenzierter Einzelkerne. Um die Aggregation von Kernen und den RNA-Abbau zu vermeiden, fahren Sie sofort mit der Vorbereitung der Bibliothek fort.
Ergebnisse
Um die Leistung unseres Protokolls zu bestimmen, verwendeten wir das 10x Genomics Chromium Single Cell 3' Gene Expression Kit v3.1 für die Bibliotheksvorbereitung und analysierten die snRNA-seq-Daten mit dem Seurat-Paket12,13.
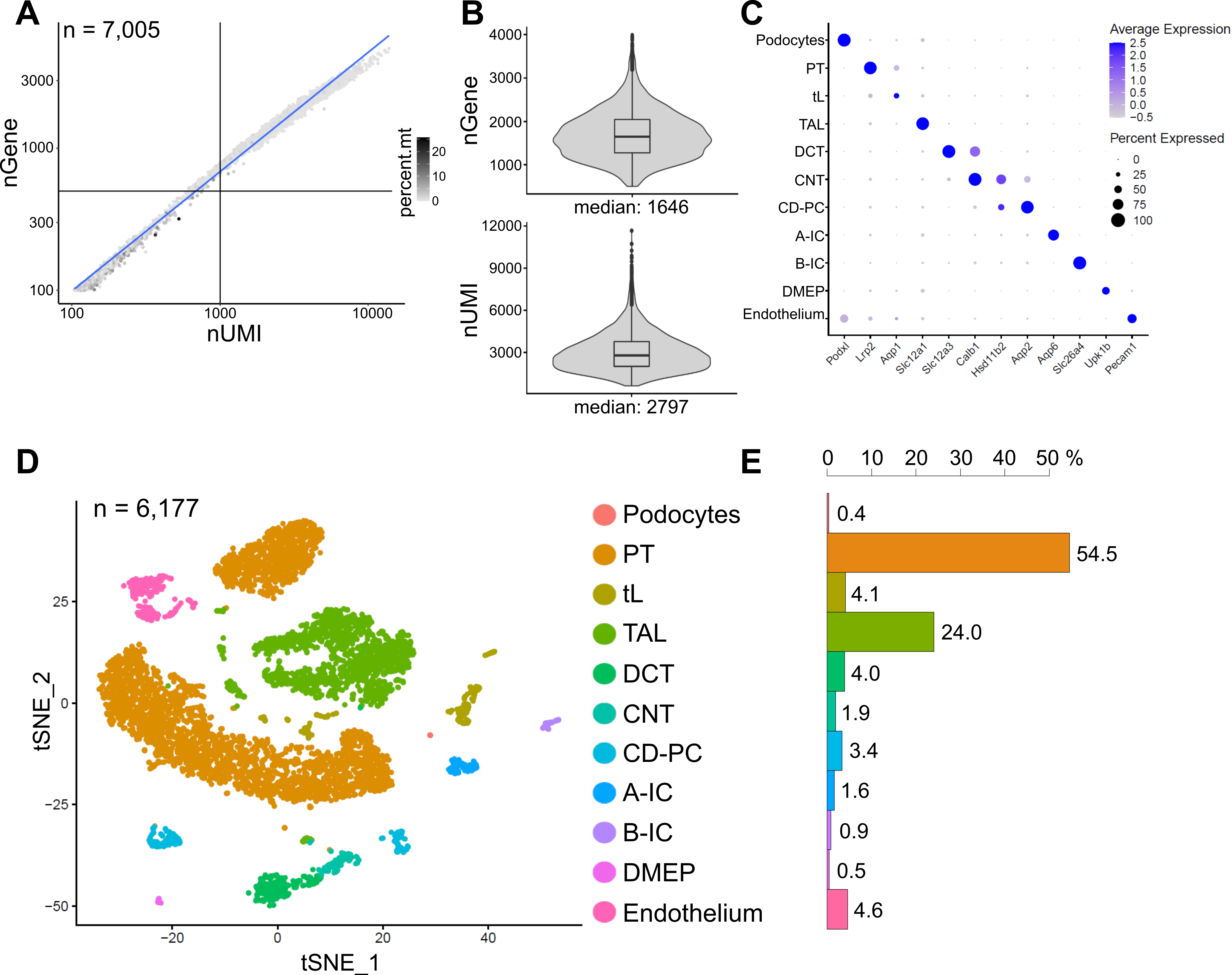
Abbildung 6 zeigt die Ergebnisse einer repräsentativen snRNA-seq-Bibliothek. Um die Qualität unserer Kerne zu beurteilen, haben wir die Anzahl der Gene gegen die Anzahl der Transkripte (definiert durch eindeutige molekulare Identifikatoren (UMIs)) aufgetragen, die durch den Anteil der mitochondrialen Reads gefärbt sind (Abbildung 6A). Kerne von guter Qualität zeigen im Allgemeinen eine höhere Anzahl von Lesevorgängen, die mit UMI- und Genzahlen korrelieren, und niedrige mitochondriale Lesefraktionen.
Für die anschließende Analyse wurden Kerne mit weniger als 500 oder mehr als 4000 gezählten Genen oder mehr als 5% mitochondrialer RNA ausgeschlossen (n = 828). Es wurden nur Gene eingeschlossen, die in mindestens drei Kernen exprimiert wurden. Wir haben insgesamt etwa 20.000 Gene in den verbleibenden 6.000 Kernen mit 1.600 medianen Genen und 2.800 medianen UMIs pro Zellkern nachgewiesen (Abbildung 6B).
Das Clustering basierte auf sehr variablen Genen. Wir identifizierten insgesamt 18 Cluster. Die Zellidentitäten wurden auf der Grundlage bekannter Markergene annotiert (nicht gezeigt). Subcluster eines Zelltyps wurden zu einem Cluster zusammengefasst, was zu insgesamt 11 verschiedenen Zelltypen führte: Podozyten, proximaler Tubulus (PT), dünne Extremität (tL), dicke aufsteigende Extremität (TAL), distale gewundene Tubuli (DCT), Verbindungstubuli (CNT), Sammelkanalhaupt- und interkalierte Zellen (CD-PC, A-IC, B-IC), tiefes Markepithel des Beckens (DMEP) und Endothel. Genexpressionsmuster von Cluster-angereicherten Markern wurden in einem Punktdiagramm (Abbildung 6C) und Zelltyp-Cluster in einem t-verteilten stochastischen Nachbareinbettungsdiagramm (t-SNE) visualisiert (Abbildung 6D).
Um die Zelltypverteilungen in unserer Stichprobe zu bestimmen, wurde der Prozentsatz jedes Zelltyps berechnet (Abbildung 6E) und zur Bestimmung des Verhältnisses von PT zu TAL verwendet. Der PT befindet sich hauptsächlich in der Nierenrinde und ist in den Einzelzell-Datensätzen der Nieren häufig überrepräsentiert, da Zellen des PT leicht zu dissoziieren sind und in ganzen Nierenproben sehr häufig vorkommen. Das TAL hingegen erstreckt sich über die gesamte äußere Medulla14. Somit stellt das Verhältnis von PT- und TAL-Fraktionen ein gutes Maß für die Anreicherung von Markzelltypen in einem Nieren-Einzelzell-Datensatz dar. Im Allgemeinen reichte das PT/TAL-Verhältnis in einzelligen Ganznierendatensätzen von 8 (unveröffentlichte Daten aus mit kalter Protease behandeltem ganzem Nierengewebe) bis 45 für enzymatisch dissoziiertes Gewebe10,14,15. In dem hier vorgestellten snRNA-seq-Datensatz konnten wir ein PT/TAL-Verhältnis von 2 erreichen. Dieses Ergebnis zeigt, dass die Entfernung von überschüssigem Kortex während der Gewebedissektion in Kombination mit snRNA-seq zu einer auffallend verbesserten Darstellung des Nierenzelltyps führt.

Abbildung 1: Schematische Übersicht über den Workflow. Das Protokoll besteht aus vier Hauptschritten, darunter die Gewebedissektion, gefolgt von der Isolierung der Kerne, der Sortierung der Kerne und einer abschließenden Bewertung der Reinheit und Konzentration. Maßstabsbalken = 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
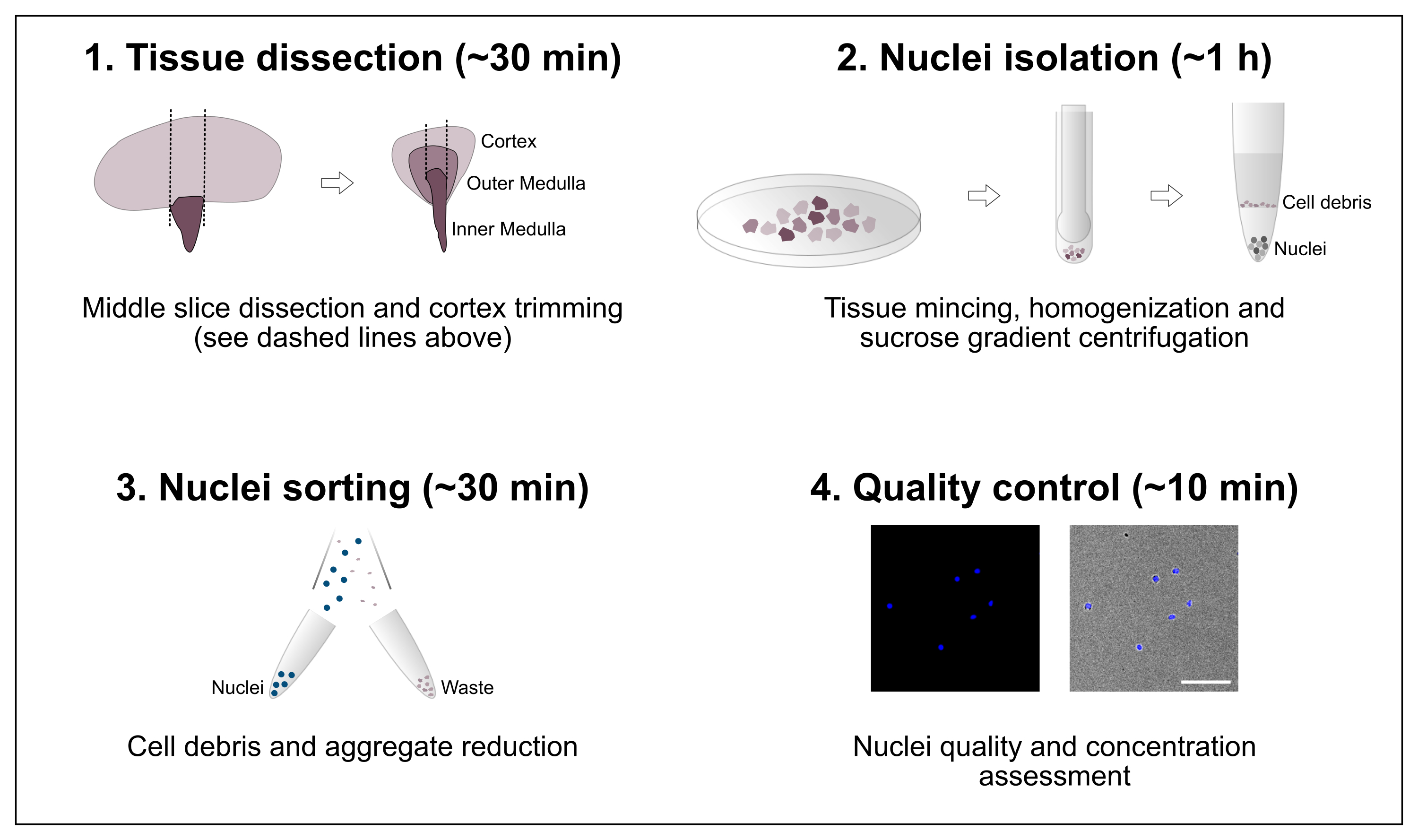{kind=link}

Abbildung 2: Nierendissektion und Gewebepräparation . (A) Repräsentatives Bild der präparierten gesamten Niere. Die gestrichelten Linien zeigen die Schnitte an, die erforderlich sind, um eine mittlere Scheibe von 1-2 mm mit einer Darstellung aller Nierenzelltypen zu erhalten. (B) Repräsentatives Bild der erhaltenen mittleren Scheibe. Die gestrichelten Linien zeigen die Schnitte für das Kortex-Trimmen von der Seite an. (C) Repräsentatives Bild des zentralen Nierenstücks mit beschnittenem Kortex. Kortex (C), äußere Medulla (OM) und innere Medulla (IM) sind deutlich sichtbar. Maßstabsbalken = 500 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
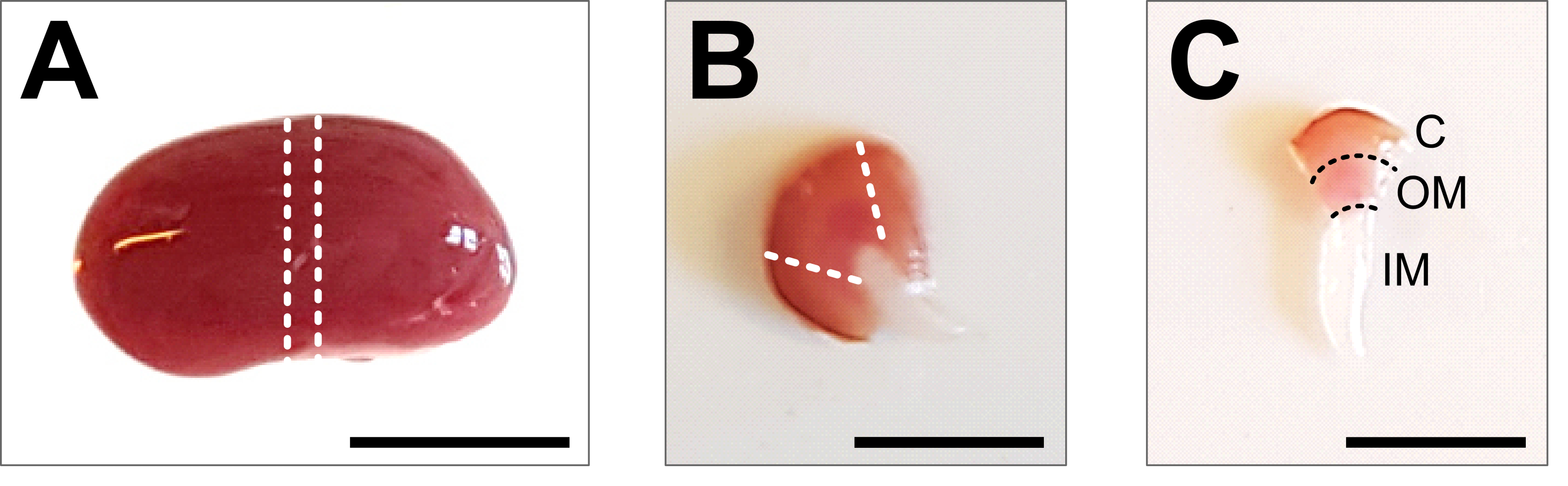{kind=link}

Abbildung 3: Gewebehomogenisierung und Zellkernreinigung . (A) Repräsentatives Bild, das ausreichend zerkleinertes Nierengewebe zeigt. Maßstabsbalken = 500 μm. (B) Homogenisieren nach dem ersten Homogenisierungsschritt (25 Hübe mit Stößel A, 2 mL Mahlrohr). (C) Homogenisieren nach dem zweiten Homogenisierungsschritt (15 Hübe mit Stößel B, 2 ml Mahlrohr). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
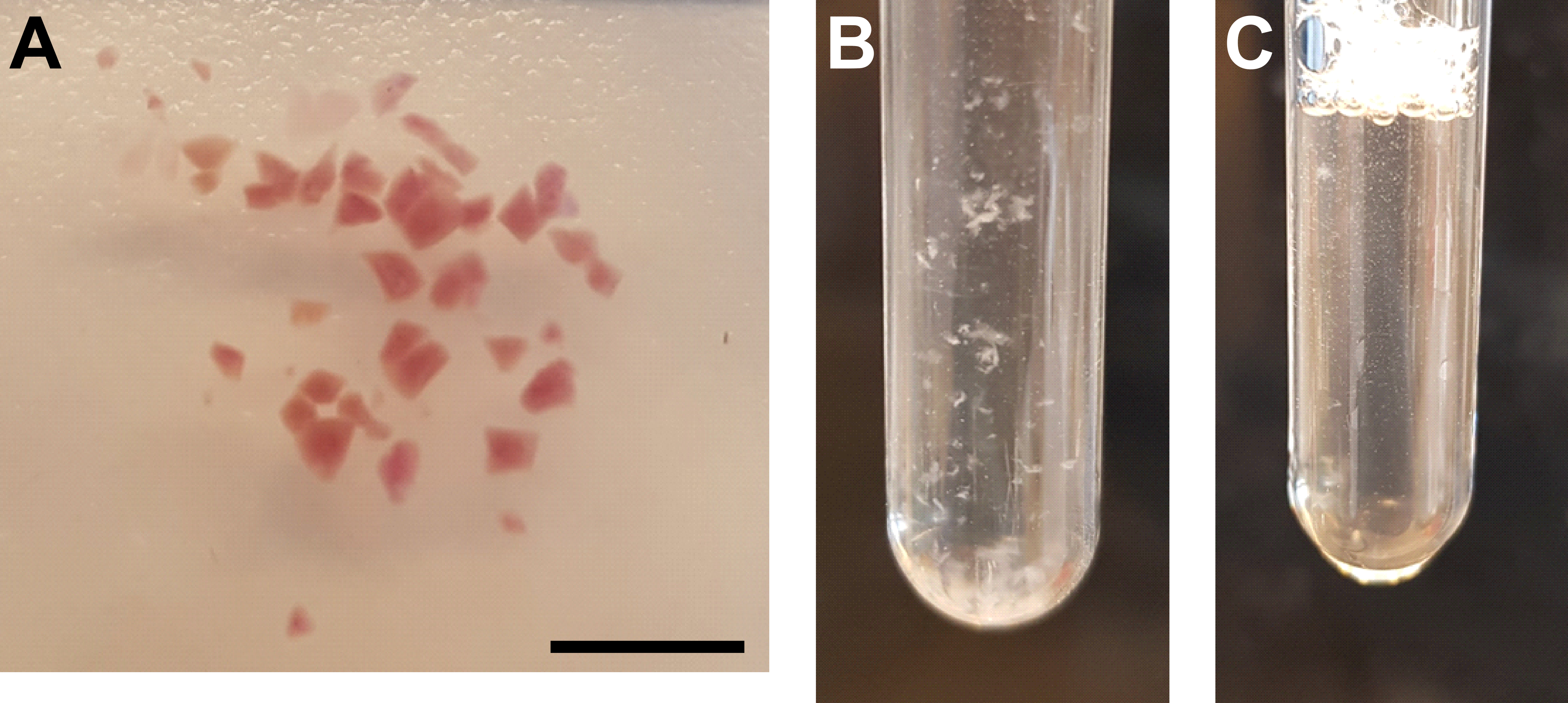{kind=link}

Abbildung 4: Gating-Strategie für die Sortierung von Kernen. (A ) Ein erstes Gate P1 wurde basierend auf Vorwärtsstreuung (FSC) vs. Seitenstreuung (SSC) gesetzt, um Trümmer und Aggregate auszuschließen. (B ) Ein nachfolgendes Gatter basierend auf DAPI-Area (DAPI-A) vs. DAPI-Height (DAPI-H) schloss leere oder beschädigte Kerne und Multiplets aus. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
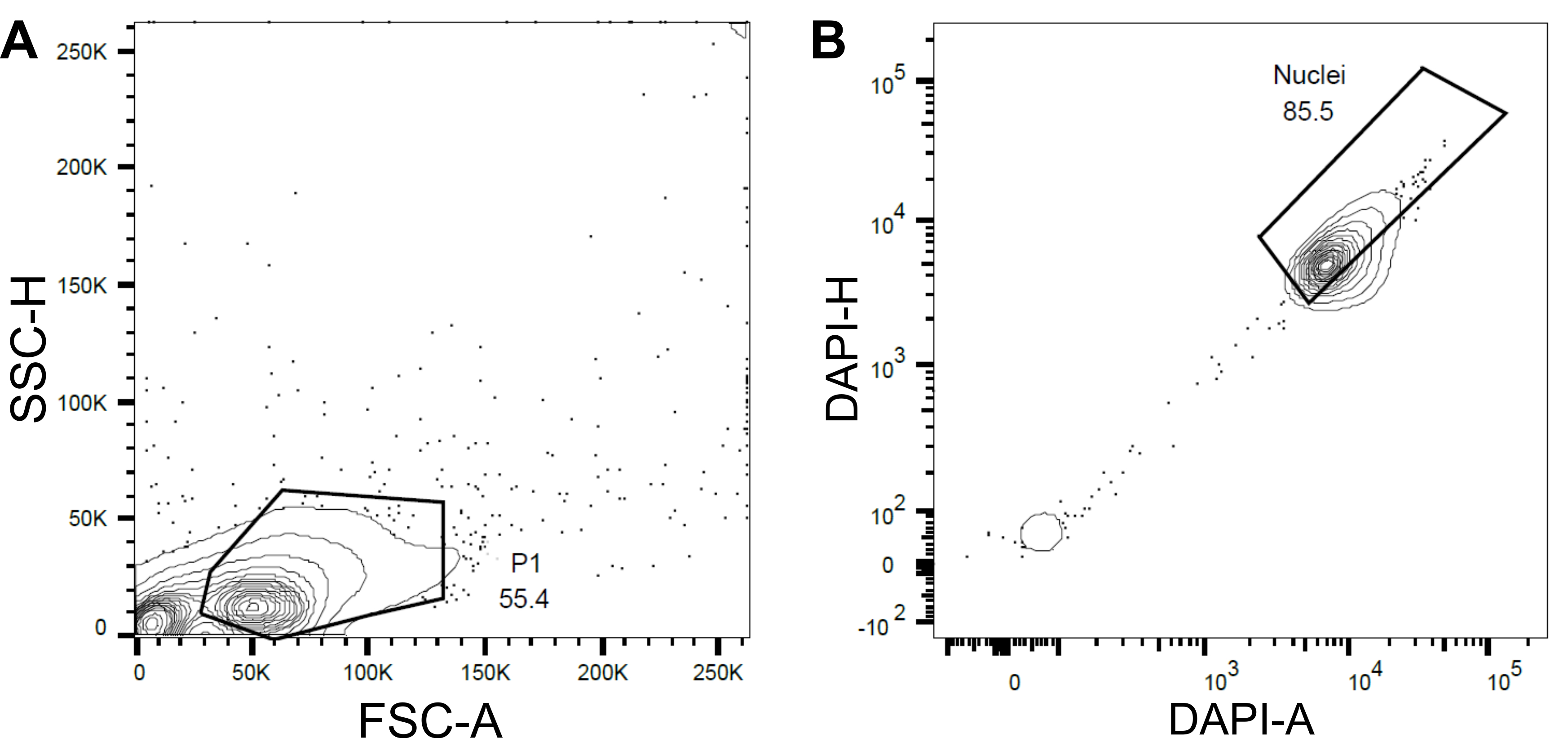{kind=link}

Abbildung 5: Kernsuspension vor und nach der Zellkernsortierung. (A, C) DAPI-gefärbte Kerne (blau). (B, D) Überlagerung von DAPI und Hellfeldkanal (BF). Vor dem Sortieren (oberes Bild) enthält die Kernsuspension Zelltrümmer und Aggregate (markiert mit weißen Pfeilspitzen). Nach dem Sortieren (unteres Bild) erscheint die Kernsuspension viel sauberer. Beispiele für DAPI-gefärbte Kerne sind mit schwarzen Pfeilspitzen markiert. Kerne von guter Qualität erscheinen rund und glatt mit einer intakten Membran und sind gut getrennt, während Kerne von schlechter Qualität faltig erscheinen und einen Verlust der Kernmembran zeigen. Maßstabsbalken = 250 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 6: Qualitätskontrolle und Analyse eines repräsentativen snRNA-seq-Datensatzes. (A) Anzahl der Gene (nGen) aufgetragen gegen die Anzahl der eindeutigen molekularen Identifikatoren (nUMI), die durch den Anteil der mitochondrialen Reads (percent.mt) gefärbt sind. Kerne niedriger Qualität entsprechen dem unteren linken Quadranten des Diagramms (n = 828) und wurden von der nachfolgenden Analyse ausgeschlossen. (B) Verteilung und Median von nGen und nUMI pro Zellkern im snRNA-seq-Datensatz, der 6.177 Kerne (> 500 Gene) repräsentiert. Die Bibliotheken wurden bis zu einer mittleren Tiefe von ~ 8.200 kartierten Lesevorgängen pro Kern sequenziert. (C) Punktdiagramm, das Genexpressionsmuster von clusterangereicherten Markern (x-Achse) für einzelne Zelltypen (y-Achse) zeigt. Die Größe des Punktes entspricht dem Anteil der Zellen, die das angegebene Gen exprimieren. Die Farbe entspricht dem durchschnittlichen Ausdruck. (D) T-verteiltes stochastisches Neighbor Embedding (t-SNE) Diagramm der identifizierten Zelltypen. (E) Zelltypverteilung im snRNA-seq-Datensatz. PT, proximaler Tubulus; tL, dünnes Glied; TAL, dickes aufsteigendes Glied, DCT, distaler gewundener Tubulus; CNT, Verbindungstubuli; CD-PC, Sammelkanal-Hauptzellen; A-IC, interkalierte Zellen vom Typ A; B-IC, Typ B interkalierte Zellen; DMEP, tiefes Markepithel des Beckens. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Reagenz | Endkonzentration | Volumen (ml) | ||
| (A) 4 % BSA / PBS | ||||
| Phosphatgepufferte Kochsalzlösung (PBS) mit 10% Rinderalbumin | 4% | 2 | ||
| PBS (phosphatgepufferte Kochsalzlösung) 1X ohne Calcium oder Magnesium | - | 3 | ||
| (B) 0,04 % BSA / PBS | ||||
| 4 % BSA / PBS | 0.04 % | 0.5 | ||
| PBS (phosphatgepufferte Kochsalzlösung) 1X ohne Calcium oder Magnesium | - | 49.5 | ||
| (C) Kern-Lysepuffer 1 (NLB1) | ||||
| Nuklearer EZ-Lysepuffer | - | 4 | ||
| RiboLock RNase-Inhibitor (40 U/μl) | 1 U/μL | 0.1 | ||
| Ribonukleosid-Vanadyl-Komplex (200 mM) | 10 mM | 0.2 | ||
| (D) Kern-Lysepuffer 2 (NLB2) | ||||
| Nuklearer EZ-Lysepuffer | - | 4 | ||
| RiboLock RNase-Inhibitor (40 U/μl) | 1 U/μL | 0.1 | ||
| e) Kern-Suspensionspuffer (NSB) | ||||
| 0,04 % BSA / PBS | - | 2 | ||
| RiboLock RNase-Inhibitor (40 U/μl) | 1 U/μL | 0.05 | ||
| f) Saccharose-Gradientenpuffer (10 % Saccharose) | ||||
| Gewicht 1 g Saccharose | ||||
| In 6 ml Nuclear EZ Lysepuffer auflösen | ||||
| Füllen Sie bis zu 10 ml mit Nuclear EZ Lysepuffer | ||||
| Filtrieren Sie durch einen 0,2 μm Spritzenfilter in ein frisches Röhrchen | ||||
Tabelle 1: Lösungsrezepte: (A) Herstellung von 4% BSA/1x PBS. Filtern Sie mit einem 0,2 μm SFCA-Membranspritzenfilter und halten Sie ihn bis zur Verwendung auf Eis. (B) Herstellung von 0,04% BSA/1 x PBS. Filtern Sie mit einem 0,2 μm SFCA-Membranspritzenfilter und halten Sie ihn bis zur Verwendung auf Eis. (c) Herstellung des Kernlysepuffers 1 (NLB1). Die angegebenen Volumina werden pro Probe angegeben. Bis zum Gebrauch auf Eis aufbewahren. (D) Herstellung von Kernlysepuffer 2 (NLB2). Die angegebenen Volumina werden pro Probe angegeben. Fügen Sie den RiboLock RNase-Inhibitor direkt vor der Anwendung zu NLB2 hinzu, wie im Protokoll angegeben. Bis zum Gebrauch auf Eis aufbewahren. e) Herstellung von Kernsuspensionspuffer (NSB). Die angegebenen Volumina werden pro Probe angegeben. Fügen Sie den RiboLock RNase-Inhibitor direkt vor der Anwendung zu NSB hinzu, wie im Protokoll angegeben. Bis zum Gebrauch auf Eis aufbewahren. (f) Herstellung von Saccharosegradientenpuffer. Filtern Sie mit einem 0,2 μm SFCA-Membranspritzenfilter und halten Sie ihn bis zur Verwendung auf Eis.
Diskussion
Die Einzelzell-Transkriptomik fördert das Verständnis der zelltypspezifischen Genexpression in der Nierenphysiologie und -erkrankung. Hier haben wir eine einfache und reproduzierbare Methode zur Verfügung gestellt, um hochwertige Einzelkerne aus gefrorenem Mausnierengewebe auf standardisierte Weise für snRNA-seq zu isolieren.
Für snRNA-seq ist es entscheidend, qualitativ hochwertige Kerne als Input für die Bibliotheksgenerierung zu verwenden und den RNA-Abbau während der Gewebeverarbeitung zu vermeiden. Daher ist die Inkubation von Gewebestücken in RNA-Stabilisierungslösung unmittelbar nach der Dissektion unerlässlich, um die zelluläre RNA zu schützen und zu stabilisieren, und ermöglicht es, Proben unbegrenzt bei - 80 °C zu lagern. Bei der Anwendung dieses Protokolls auf gefrorenes Gewebe ohne Behandlung mit RNA-Stabilisierungslösungen, wie z. B. Archivmaterial, ist ein Probelauf erforderlich, und die RNA-Qualität muss bewertet werden, da wir einen signifikanten Verlust der RNA-Integrität in schockgefrorenem Gewebe ohne vorherige Inkubation in RNA-Stabilisierungslösung beobachtet haben.
Im Allgemeinen ist eine geeignete Probenhandhabung entscheidend für die Maximierung der Gewinnung intakter Einzelkerne. Alle Resuspensionsschritte sollten durch sorgfältiges Pipettieren durchgeführt werden, um Scherbeanspruchungen und physische Schäden zu vermeiden. Puffer für die endgültige Kernresuspension und die Kernsortierung sollten BSA enthalten, um den Verlust und die Aggregation von Kernen zu vermeiden.
Die Puffervolumina in diesem Protokoll sind für sehr kleine Gewebeproben (~15 mg) optimiert. Es ist wichtig, eine vollständige Zelllyse und eine ausreichende Wäsche sicherzustellen, um qualitativ hochwertige Suspensionen zu erzeugen. Größere Gewebeblöcke oder ganze Nierenproben führen zu übermäßigen Kernkonzentrationen, die zu Verklumpung und Aggregation, einer hohen Häufigkeit von Umgebungs-RNA und einer insgesamt schlechten Suspensionsqualität führen. Wenn größere Proben oder andere Gewebe verarbeitet werden, wird dringend empfohlen, Testläufe durchzuführen, um optimale Puffervolumina für minimale RNA-Konzentrationen in der Umgebung zu bestimmen. Die Qualität und Konzentration von Kernen und RNA muss sorgfältig untersucht werden, da eine Überlastung zu einer insgesamt schlechten Leistung führt.
Darüber hinaus beeinflussen große Mengen an Zelltrümmern, die zu hohen Mengen an Umgebungs-RNA führen, die nicht mit einzelnen Kernen assoziiert sind, die Sequenzierungsergebnisse negativ. Die Klärung der Kernsuspension durch Zentrifugation durch ein Saccharosekissen mildert dieses Problem bis zu einem gewissen Grad, kann aber auch zu Verzerrungen in der Zelltypdarstellung führen, indem sie gegen dichte, kleine Kerne selektiert, die beispielsweise in Immunzellen vorhanden sind16. Wenn dies von Belang ist, sollte der Saccharosegradient weggelassen werden. Im Gegensatz dazu stellten wir fest, dass die Durchflusszytometrie auf der Grundlage der DAPI-Färbung entscheidend ist, um die Menge an Zelltrümmern zu reduzieren und eine qualitativ hochwertige Einzelkernsuspension herzustellen.
Die Isolierung einzelner Kerne hat erhebliche Vorteile im Vergleich zu Einzelzellansätzen8. Es ist mit richtig gefrorenem Gewebe kompatibel, wodurch die Gewebeentnahme flexibler wird und die Notwendigkeit einer enzymbasierten Gewebedissoziation umgangen wird, die zu transkriptionellen Stressreaktionen führen kann 6,17. Darüber hinaus überwindet es die Dissoziationsverzerrung, die die Selektion leicht dissoziierbarer Zelltypen der Nierenrinde begünstigt, was zu einer Unterrepräsentation von Markzelltypen in einigen enzymbasierten Ansätzen führen kann 5,6,10.
Die Verwendung eines zentralen Nierenstücks anstelle des gesamten Nierengewebes spart weitere Ressourcen und korrigiert die Überrepräsentation von reichlich vorhandenen Zelltypen, wie zuvor beschrieben10. Je nach untersuchtem Mausmodell oder Phänotyp kann es jedoch vorteilhaft sein, ganze Nierenproben anstelle einer einzelnen mittleren Scheibe zu verwenden. Ganze Nierenproben können repräsentativer für die tatsächlichen Zellproportionen oder Veränderungen in der gesamten Niere sein, während sich eine getrimmte mittlere Scheibe für medulläre Phänotypen oder wenn das Probenmaterial begrenzt war, als vorteilhaft erwies. Diese Entscheidung ist daher sehr benutzerspezifisch und sollte sorgfältig abgewogen werden.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Wir danken der Scientific Genomics Platform am Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft, Berlin für die technische Unterstützung.
JL und KMSO wurden gefördert durch das Graduiertenkolleg GRK 2318 der Deutschen Forschungsgemeinschaft (DFG) und die Forschergruppe FOR 2841. KMSO wurde durch den Collaborative Research Grant 1365 unterstützt. AB wurde gefördert durch den Gottfried Wilhelm Leibniz-Preis der DFG, der an NR.
Materialien
| Name | Company | Catalog Number | Comments |
| Cell sorter | - | - | For fluorescence-activated cell sorting (FACS); e.g. BD FACSAria Cell Sorter. |
| Centrifuge 5810 R | Eppendorf | 5811000015 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL Automated Cell Counter | Invitrogen | AMQAF1000 | Needs to contain DAPI light cube to count nuclei. Alternatively, nuclei can be counted manually under fluorescence microscope. |
| 4′,6-Diamidino-2-phenyl-indol-dihydrochlorid (DAPI) | Biotrend | 40043/b | Stock solution prepared with a concentration of 100 µM. Used for nuclei staining in a final concentration of 2 µM. |
| D(+)-Sucrose ≥99.9%, ultrapure DNAse-, RNAse-free | VWR | 0335-500G | |
| DNA LoBind Microcentrifuge Tubes (1.5 mL) | Eppendorf | 22431021 | |
| Ethanol, 70 % | - | - | |
| FACS tubes | pluriSelect | 43-10100-46 | |
| KIMBLE Dounce tissue grinder set 2 mL complete | Sigma-Aldrich | D8938-1SET | |
| Minisart Syringe Filters 0.2 µm | Sartorius | 16534-GUK | |
| Nuclease-free Water | Invitrogen | AM9937 | |
| Nuclei EZ Prep Nuclei Isolation Kit | Sigma-Aldrich | NUC-101 | Nuclei EZ Lysis Buffer (Product No. N3408) needed for buffer preparation. |
| PBS (Phosphate-Buffered Saline) 1X without calcium or magnesium | Corning | 21-040-CV | |
| Petri dishes, polystyrene 60 mm | Sigma-Aldrich | P5481 | |
| Phosphate-Buffered Saline (PBS) with 10% Bovine Albumin | Sigma-Aldrich | SRE0036 | |
| pluriStrainer Mini 100 µm | pluriSelect | 43-10100-46 | |
| pluriStrainer Mini 20 µm | pluriSelect | 43-10020-40 | |
| pluriStrainer Mini 40 µm | pluriSelect | 43-10040-40 | |
| Polystyrene Centrifuge Tube (15 mL) | Falcon | 352099 | |
| Razor blades | - | - | |
| RiboLock RNase Inhibitor (40 U/µL) | Thermo Fisher | EO0384 | |
| Ribonucleoside-vanadyl complex | New England Biolabs | S1402S | Follow manufacturer's instructions (https://international.neb.com/products/s1402-ribonucleoside-vanadyl-complex#Product%20Information). Upon use the 200 mM stock solution is reconstituted to a green-black clear solution by incubating at 65 °C. |
| RNAlater Stabilization Solution | Invitrogen | AM7020 | |
| RNase AWAY | Fisher Scientific | 11952385 |
Referenzen
- Thomas, R. S. Kidney modeling and systems physiology. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 1 (2), 172-190 (2009).
- Potter, S. S. Single-cell RNA sequencing for the study of development, physiology and disease. Nature Reviews Nephrology. 14 (8), 479-492 (2018).
- Park, J., Liu, C. L., Kim, J., Susztak, K. Understanding the kidney one cell at a time. Kidney International. 96 (4), 862-870 (2019).
- Clark, A. R., Greka, A. The power of one: advances in single-cell genomics in the kidney. Nature Reviews Nephrology. 16 (2), 73-74 (2020).
- Lake, B. B., et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nature Communications. 10 (1), 2832(2019).
- Wu, H., Kirita, Y., Donnelly, E. L., Humphreys, B. D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. Journal of the American Society of Nephrology. 30 (1), 23-32 (2019).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144 (19), 3625-3632 (2017).
- Habib, N., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 14 (10), 955-958 (2017).
- Muto, Y., et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nature Communications. 12 (1), 2190(2021).
- Hinze, C., et al. Kidney single-cell transcriptomes predict spatial corticomedullary gene expression and tissue osmolality gradients. Journal of the American Society of Nephrology. 32 (2), 291(2021).
- Berger, S. L. Isolation of cytoplasmic RNA: ribonucleoside-vanadyl complexes. Methods in Enzymology. 152, 227-234 (1987).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 36 (5), 411-420 (2018).
- Stuart, T., et al. Comprehensive Integration of single-cell data. Cell. 177 (7), 1888-1902 (2019).
- Park, J., et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 360 (6390), 758-763 (2018).
- Kirita, Y., Wu, H., Uchimura, K., Wilson, P. C., Humphreys, B. D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proceedings of the National Academy of Sciences of the United States of America. 117 (27), 15874-15883 (2020).
- Schneeberger, S., et al. The neuroinflammatory interleukin-12 signaling pathway drives Alzheimer's disease-like pathology by perturbing oligodendrocyte survival and neuronal homeostasis. bioRxiv. , 441313(2021).
- Nguyen, Q. H., Pervolarakis, N., Nee, K., Kessenbrock, K. experimental considerations for single-cell RNA sequencing approaches. Frontiers in Cell and Developmental Biology. 6, 108(2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten