
Method Article
Nuclei Isolation from Adult Mouse Kidney for Single-Nucleus RNA-Sequencing
* These authors contributed equally
In This Article
Summary
Here, we present a protocol to isolate high-quality nuclei from frozen mouse kidneys that improve the representation of medullary kidney cell types and avoids the gene expression artifacts from enzymatic tissue dissociation.
Abstract
The kidneys regulate diverse biological processes such as water, electrolyte, and acid-base homeostasis. Physiological functions of the kidney are executed by multiple cell types arranged in a complex architecture across the corticomedullary axis of the organ. Recent advances in single-cell transcriptomics have accelerated the understanding of cell type-specific gene expression in renal physiology and disease. However, enzyme-based tissue dissociation protocols, which are frequently utilized for single-cell RNA-sequencing (scRNA-seq), require mostly fresh (non-archived) tissue, introduce transcriptional stress responses, and favor the selection of abundant cell types of the kidney cortex resulting in an underrepresentation of cells of the medulla.
Here, we present a protocol that avoids these problems. The protocol is based on nuclei isolation at 4 °C from frozen kidney tissue. Nuclei are isolated from a central piece of the mouse kidney comprised of the cortex, outer medulla, and inner medulla. This reduces the overrepresentation of cortical cells typical for whole-kidney samples for the benefit of medullary cells such that data will represent the entire corticomedullary axis at sufficient abundance. The protocol is simple, rapid, and adaptable and provides a step towards the standardization of single-nuclei transcriptomics in kidney research.
Introduction
Kidneys display a highly complex tissue architecture. They consist of functionally and anatomically distinct segments along a corticomedullary axis and mediate biological functions, such as regulation of extracellular fluid volume, electrolyte balance, or acid-base homeostasis1.
Advances in single-cell transcriptomics have enabled the in-depth characterization of complex tissues and accelerated the understanding of segment and cell type-specific gene expression in renal physiology, development, and disease2,3,4.
However, the enzyme-based dissociation protocols that are frequently utilized for scRNA-seq display several drawbacks and constraints. Depending on the protocol, they generate transcriptional stress responses and tissue dissociation bias towards easier-to-dissociate cortical cell types5,6. Although protocols using cold-active proteases for embryonic kidneys are able to mitigate stress-related transcriptional alterations, they fail to overcome the dissociation bias towards cortical cells and might not be readily adaptable to different kinds of diseased kidney tissues7. In addition, single-cell approaches are not easily compatible with frozen tissue samples, limiting their application mostly to non-archived, fresh tissue, thus making the tissue collection a restricting factor6.
Single-nuclei RNA sequencing (snRNA-seq) can circumvent these limitations8,9. Here, we present a protocol for nuclei isolation from a central slice of frozen adult mouse kidney tissue (Figure 1)10. Our protocol is simple and provides a standardized approach to obtain RNA sequencing libraries with a balanced representation of diverse kidney cell types for experimental models that do not involve strong regional tissue changes. In the latter case, our protocol can also be performed with whole kidneys.
Protocol
All animal experiments were conducted in accordance with the Animal Welfare Act (TierSchG) and the Animal Welfare Experimental Animal Regulation (TierSchVersV) and were authorized by local authorities and the Animal Welfare Officers at our institution (MDC).
1. Tissue preparation
- Prepare a 6-well plate containing 2 mL of 1x phosphate-buffered saline (PBS) per well for each kidney that will be obtained. Prepare a 6-well plate containing 2 mL of RNA stabilization solution per well and kidney. Pre-cool both plates on ice.
- Euthanize a 3- to 6-month-old male C57BL/6 mouse. Place the mouse on a dissecting tray, pin down the extremities and sterilize the abdomen with 70% ethanol.
- Open the abdomen up to the ribcage using forceps and scissors. Lift the intestine and other organs to the side and remove the kidneys by carefully cutting the ureter, renal artery, and vein with a scissor. Wash the kidney in the previously prepared, ice-cold 1x PBS and remove the renal fascia and any remaining fat from the kidney until all white tissue is removed (Figure 2A).
- Place the kidney on a cold dissecting plate and use a sharp scalpel or razor blade to obtain a middle slice of 1-2 mm. Make sure the tissue piece contains the entire corticomedullary axis (Figure 2B). Use microdissection scissors and forceps to carefully trim the cortex from the sides of the center piece. Within the dissected tissue piece, the three segments cortex, outer medulla, and inner medulla should be clearly visible (Figure 2C).
NOTE: The slice should not exceed a thickness of 2 mm or a weight of 20 mg to ensure sufficient buffer amounts for effective tissue lysis and in order to minimize ambient background RNA in the cDNA libraries. Ambient RNA wastes sequence capacity, as it is not associated with single nuclei. - Transfer the kidney piece to the previously prepared RNA stabilization solution and incubate for 24 h at 4 °C to avoid RNA degradation. After 24 h, remove the RNA stabilization solution and store the tissue at -80 °C until further use. Carefully remove the excess solution with tissue paper.
2. Nuclei isolation
- Cleaning and preparation steps
- Clean benchtops and pipettes with 70% ethanol and RNase decontamination solution.
- Clean a round-bottomed, 2 mL tissue grinder tube and matching pestle A and B with RNase decontamination solution, followed by 70% ethanol and RNase-free water (1 grinder tube and pestle set per sample). Let it dry completely.
- Pre-cool the centrifuge to 4 °C.
- Label and pre-cool three 15 mL collection tubes, a 1.5 mL collection tube, a 5 mL fluorescence-activated cell sorting (FACS) collection tube, and a dry grinder tube for each sample on ice.
- Buffer preparation
- Warm up the Ribonucleoside-vanadyl complex stock solution to 65 °C until reconstituted to a green-black clear solution according to manufacturer's instructions.11
- Prepare 1x PBS containing 4% bovine serum albumin (BSA) as described in Table 1A. Additionally, prepare 1x PBS with 0.04% BSA (Table 1B). Filter both solutions using a 0.2 µm surfactant-free cellulose acetate (SFCA) membrane syringe filter and keep on ice until further use.
- Prepare Nuclei Lysis Buffer 1 (NLB1, Table 1C). Add 4 mL of EZ lysis buffer for Nuclei Lysis Buffer 2 (NLB2, Table 1D) and 2 mL of 0.04 % BSA / PBS for the Nuclei Suspension Buffer (NSB, Table 1E) to 15 mL tubes. Add the RNase inhibitor solution to NLB2 and NSB directly before use as indicated below in the protocol. Keep on ice until further use.
- Prepare EZ lysis buffer with 10% sucrose (Sucrose Gradient Buffer, Table 1F). Mix well and filter the buffer into a fresh 15 mL tube using a 0.2 µm SFCA membrane syringe filter. Keep on ice until further use.
- Tissue homogenization and cell lysis
NOTE: In order to minimize RNA degradation, all steps are carried out on ice. The grinder tube, Petri dish, and all buffers need to be pre-cooled. All resuspension steps are done by carefully pipetting the nuclei suspension. Do not vortex the sample to avoid shearing forces and damage to nuclei.- Take the frozen kidney piece and transfer it to a 60 mm polystyrene Petri dish on ice containing 1 mL of NLB1.
- Mince the tissue thoroughly using a razor blade or scalpel (Figure 3A).
- Cut off the tip of a 1 mL pipette tip and transfer the minced tissue and buffer to the grinder tube. Make sure to transfer all tissue pieces. Wash the Petri dish 5-10 times with the buffer, if necessary.
- Homogenize the suspension on ice by slowly moving pestle A, 25x up and down in the grinder tube. Avoid air bubbles caused by rapid movement (Figure 3B).
- Pass the homogenate through a 100 µm strainer in a pre-cooled 15 mL collection tube and wash the filter with another 1 mL of NLB1.
- Wash the grinder tube with cold EZ nuclei lysis buffer and discard the buffer.
- Transfer the homogenate back into the grinder tube and homogenize the suspension on ice by slowly moving pestle B, 15x up and down in the grinder tube. Avoid air bubbles caused by rapid movement (Figure 3C).
- Transfer the homogenate to a pre-cooled 15 mL collection tube. Wash the grinder tube with another 2 mL of NLB1 and make sure to transfer all tissue fragments to the collection tube. Incubate the homogenate (total volume of 4 mL) for 5 min on ice to lyse the cells.
- Nuclei purification
- Pass the homogenate through a 40 µm strainer into a pre-cooled 15 mL collection tube. Spin the collection tube for 5 min at 500 x g at 4 °C in a centrifuge with a swinging-bucket rotor. In the meantime, add RNase inhibitor solution to NLB2 (Table 1D).
- Remove the supernatant without disturbing the pellet. Carefully resuspend the pellet in 4 mL of NLB2.
- Carefully underlay the suspension with a 1 mL cushion of Sucrose Gradient Buffer. Centrifuge at 500 x g for 5 min at 4 °C in a centrifuge with a swinging-bucket rotor. In the meantime, add RNase inhibitor solution to NSB (Table 1E).
- After centrifugation, gently remove the collection tube from the centrifuge and be careful not to disturb the two layers when handling the collection tube. Cell debris is visible between the two layers. Remove the supernatant carefully starting with the debris. Remove the remaining supernatant without disturbing the nuclei pellet and carefully resuspend the pellet in 1 mL of NSB.
NOTE: The resuspension volume depends on the amount of tissue used for the isolation and pellet size gained after the last centrifugation step. The volume might need to be adapted to the expected number of nuclei. - Pass the homogenate through a 20 µm strainer into the pre-cooled 5 mL FACS collection tube.
3. Nuclei sorting
- Add 20 µL of 4′,6-diamidino-2-phenylindole (DAPI) per mL of NSB to a final concentration of 2 µM to the homogenate in the FACS collection tube and mix carefully. Incubate for 5 min on ice.
- Prepare the pre-cooled 1.5 mL collection tube with 20 µL of 4% BSA /1x PBS and add 0.5 µL RNase inhibitor solution to a final concentration of 1 U/µL. Immediately proceed to sorting.
- Sort the nuclei using a cell sorter.
- Mix the nuclei suspension briefly before inserting the FACS collection tube into the sorter.
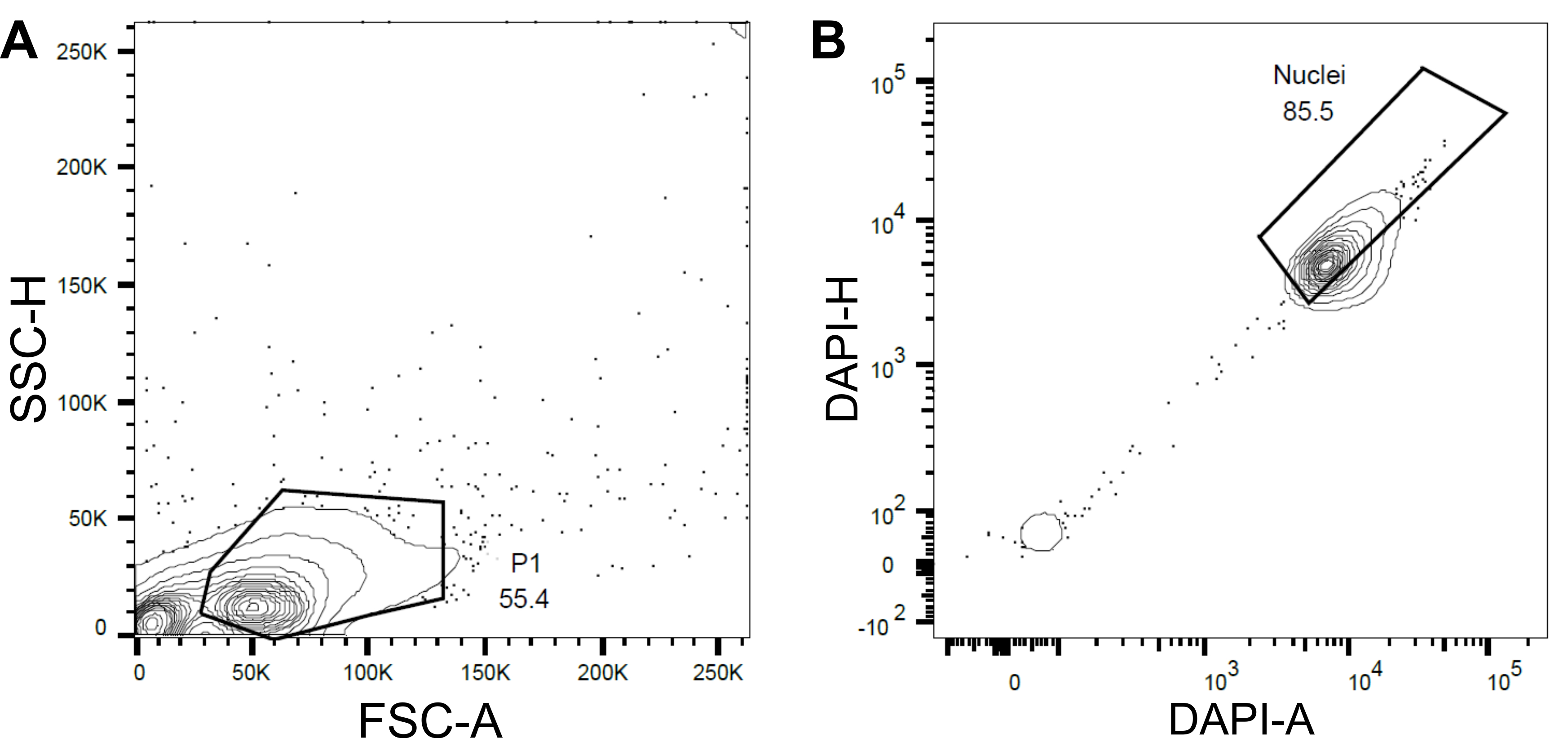
- Set a first gate P1 based on the forward scatter (FSC) and side scatter (SSC) to exclude debris and aggregates (Figure 4A).
- To exclude empty or damaged nuclei and multiplets, set a subsequent gate based on the DAPI-Area versus DAPI-Height (DAPI-A vs DAPI-H) (Figure 4B).
- Sort single nuclei into the 1.5 mL collection tube containing 4% BSA /1x PBS with 1 U/µL RNase inhibitor solution prepared in 3.2.
4. Quality control
- Measure the final nuclei concentration under a fluorescence microscope or in an automated counting chamber in at least two independent counts and assess the suspension quality (Figure 5).
NOTE: Optimal concentrations are between 700 - 1,200 nuclei/µL. Lower cell concentrations such as 700 nuclei/µL may be preferable as resulting cDNA libraries contained less ambient background RNA (transcripts not associated with individual nuclei). - Calculate the required volume of nuclei suspension for the desired recovery of sequenced single nuclei. In order to avoid nuclei aggregation and RNA degradation, proceed immediately to library preparation.
Results
To determine the performance of our protocol, we used the 10x Genomics Chromium Single Cell 3' Gene Expression Kit v3.1 for library preparation and analyzed the snRNA-seq data with the Seurat package12,13.
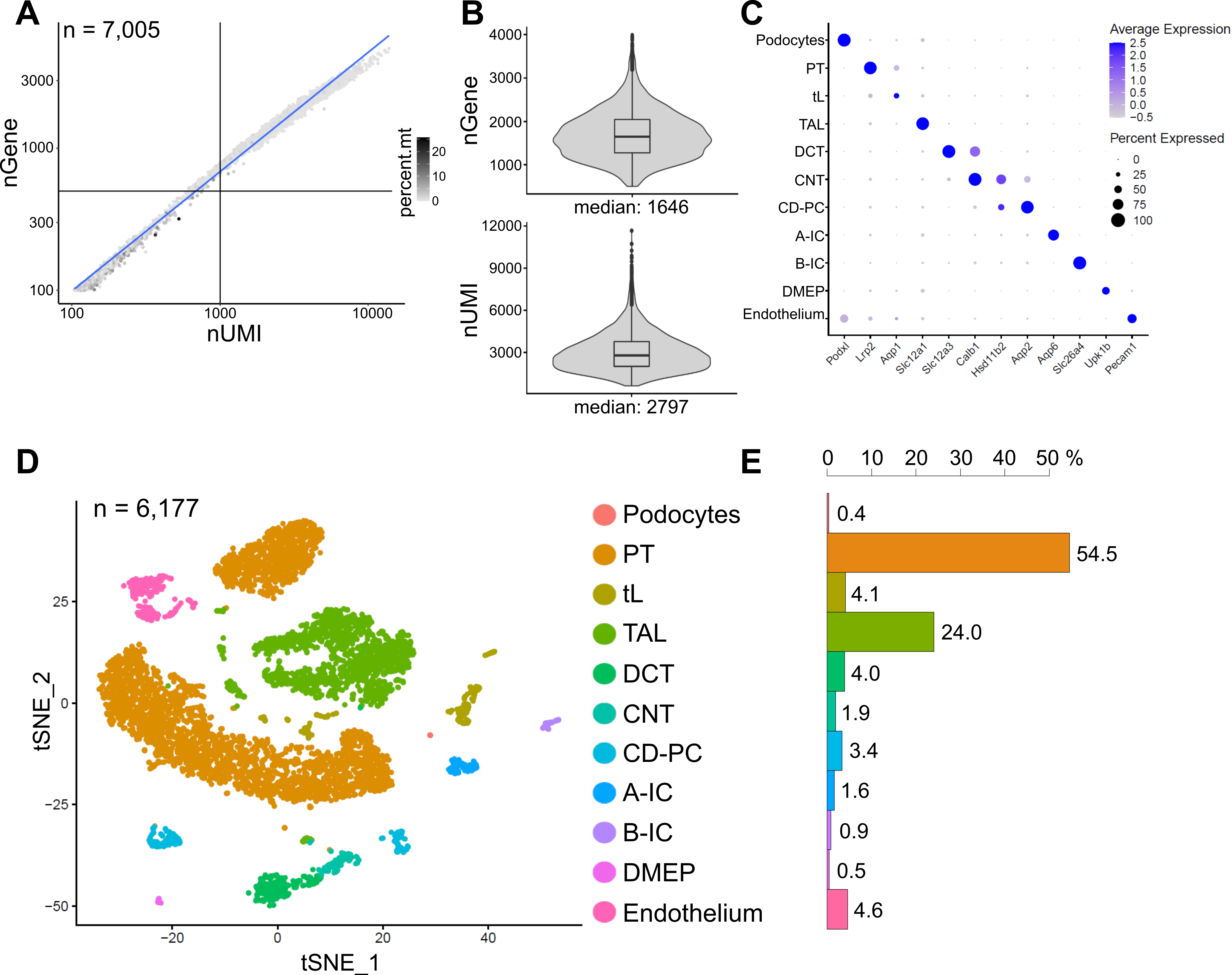
Figure 6 shows results from a representative snRNA-seq library. To assess the quality of our nuclei, we plotted the number of genes against the number of transcripts (defined by unique molecular identifiers (UMIs)) colored by the fraction of mitochondrial reads (Figure 6A). Nuclei of good quality generally show higher numbers of reads, correlating UMI and gene numbers, and low mitochondrial read fractions.
For the subsequent analysis, nuclei with less than 500 or more than 4000 counted genes, or more than 5% of mitochondrial RNA were excluded (n = 828). Only genes expressed in a minimum of three nuclei were included. We detected about 20,000 genes in total in the remaining 6,000 nuclei with 1,600 median genes and 2,800 median UMIs per nucleus (Figure 6B).
Clustering was based on highly variable genes. We identified a total of 18 clusters. Cell identities were annotated based on known marker genes (not shown). Subclusters of one cell type were summarized to one cluster resulting in a total of 11 distinct cell types: podocytes, proximal tubule (PT), thin limb (tL), thick ascending limb (TAL), distal convoluted tubule (DCT), connecting tubule (CNT), collecting duct principal and intercalated cells (CD-PC, A-IC, B-IC), deep medullary epithelium of the pelvis (DMEP) and endothelium. Gene expression patterns of cluster-enriched markers were visualized in a dot plot (Figure 6C) and cell type clusters in a t-distributed stochastic neighbor embedding (t-SNE) plot (Figure 6D).
To assess cell type distributions in our sample, the percentage of each cell type was calculated (Figure 6E) and used to determine the ratio of PT to TAL. The PT is mainly located in the kidney cortex and frequently overrepresented in kidney single-cell datasets as cells of the PT are easy to dissociate and highly abundant in whole kidney samples. The TAL on the other hand stretches across the whole outer medulla14. Thus, the ratio of PT and TAL fractions represents a good measure for the enrichment of medullary cell types in a kidney single-cell dataset. In general, the PT/TAL ratio in single-cell whole kidney datasets ranged from 8 (unpublished data from cold-protease-treated whole kidney tissue) to 45 for enzymatically dissociated tissue10,14,15. In the snRNA-seq dataset presented here we were able to reach a PT/TAL ratio of 2. This result illustrates that removal of excess cortex during tissue dissection combined with snRNA-seq results in a strikingly improved kidney cell type representation.

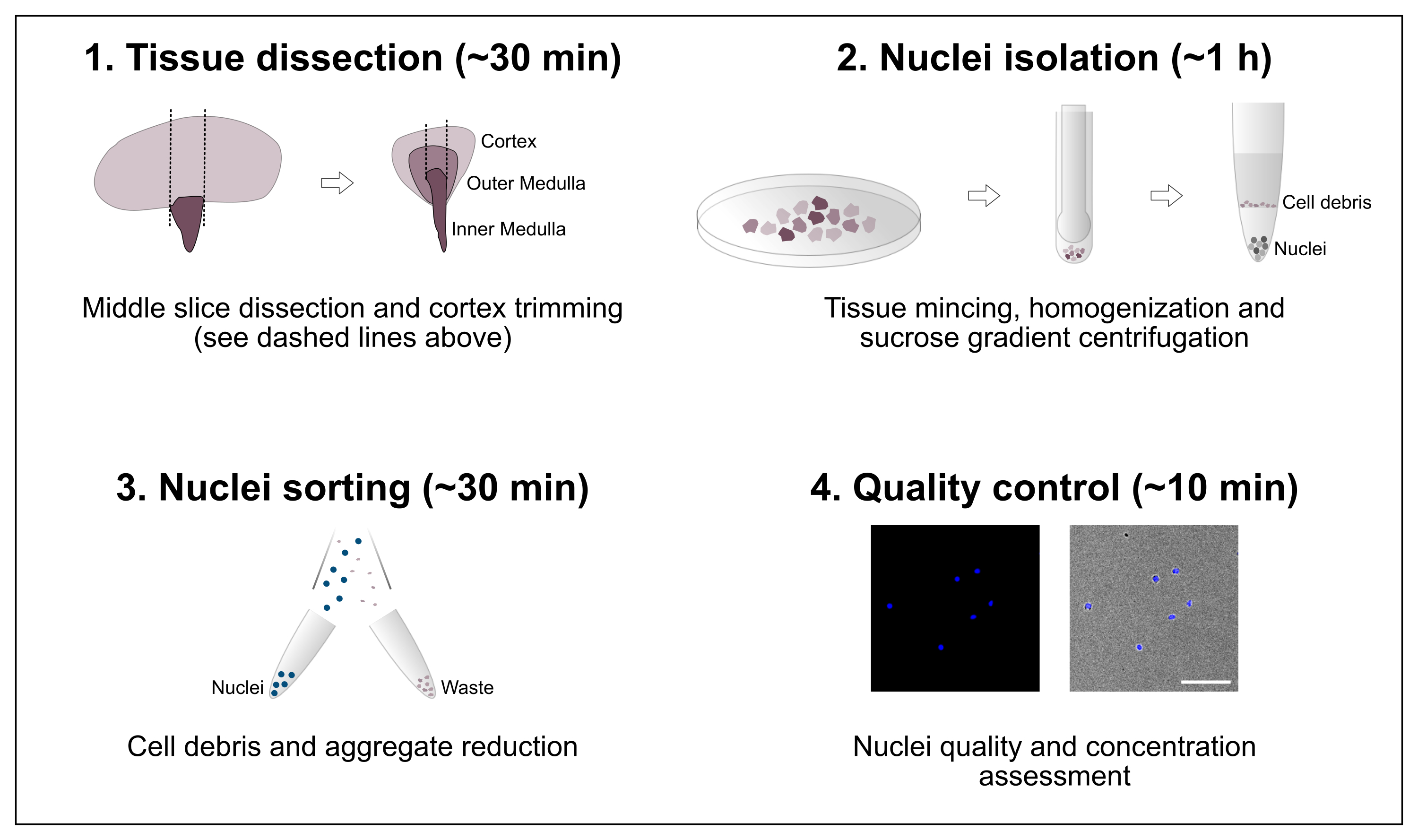
Figure 1: Schematic overview of the workflow. The protocol consists of four major steps that include tissue dissection followed by nuclei isolation, nuclei sorting and a final purity and concentration assessment. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

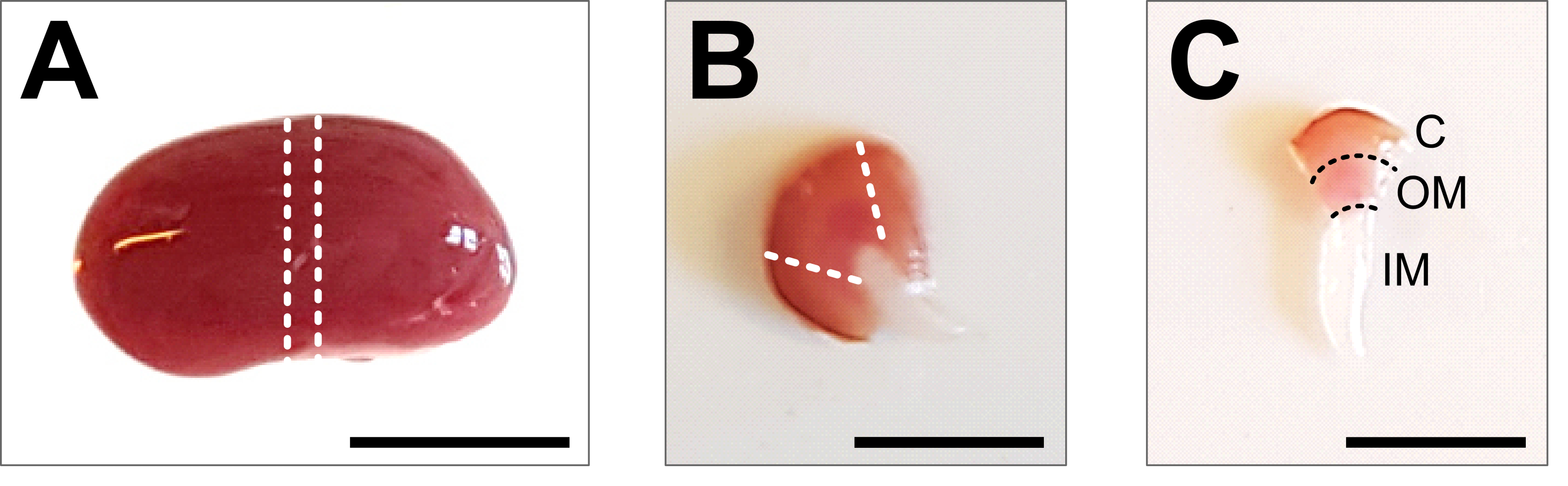
Figure 2: Kidney dissection and tissue preparation. (A) Representative image of dissected whole kidney. The dotted lines indicate the cuts required to obtain a middle slice of 1-2 mm with a representation of all renal cell types. (B) Representative image of obtained middle slice. The dotted lines indicate the cuts for cortex trimming from the side. (C) Representative image of central kidney piece with trimmed cortex. Cortex (C), outer medulla (OM) and inner medulla (IM) are clearly visible. Scale bar = 500 µm. Please click here to view a larger version of this figure.
{kind=link}

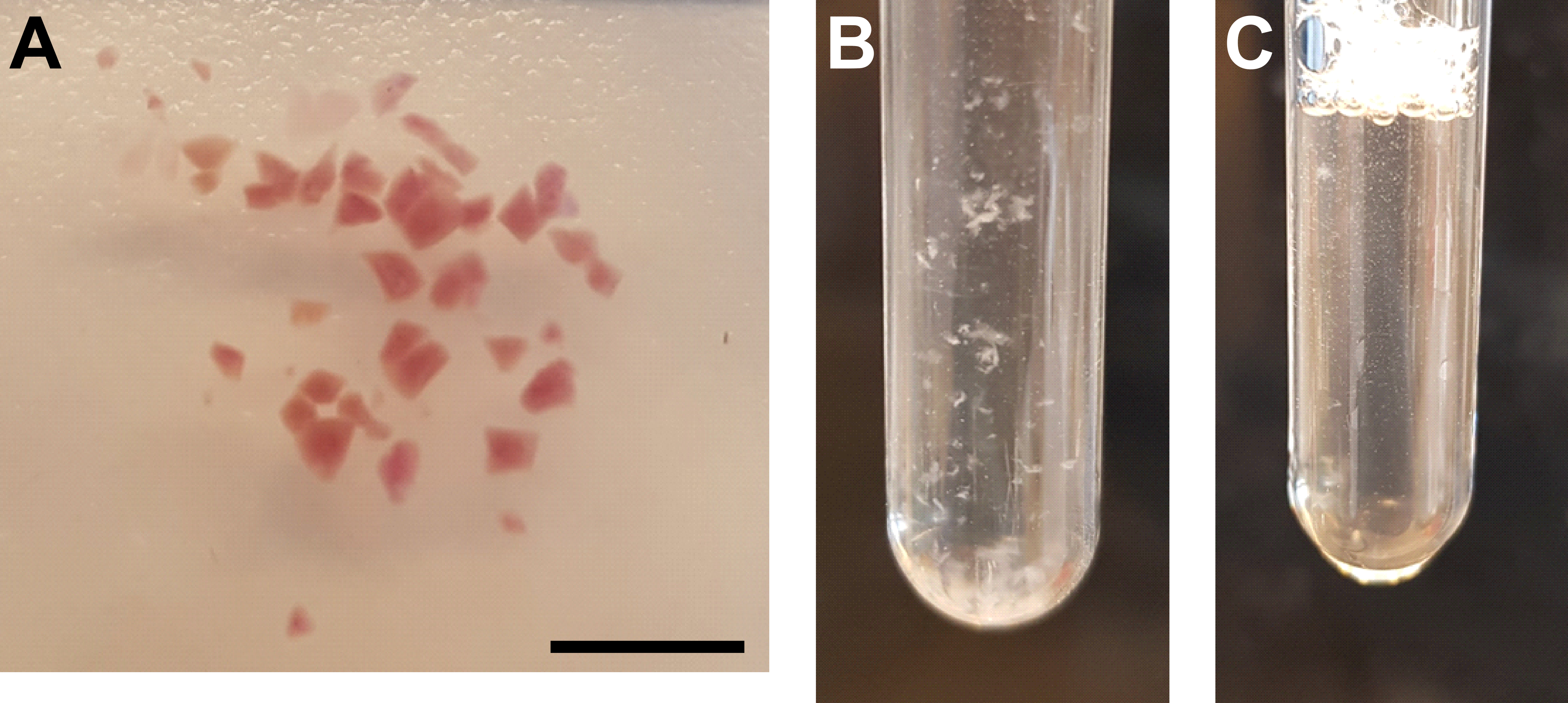
Figure 3: Tissue homogenization and nuclei purification. (A) Representative image showing sufficiently minced kidney tissue. Scale bar = 500 µm. (B) Homogenate after first homogenization step (25 strokes with pestle A, 2 mL grinder tube). (C) Homogenate after second homogenization step (15 strokes with pestle B, 2 mL grinder tube). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Gating strategy for nuclei sorting. (A) A first gate P1 was set based on forward scatter (FSC) vs side scatter (SSC) to exclude debris and aggregates. (B) A subsequent gate based on DAPI-Area (DAPI-A) vs DAPI-Height (DAPI-H) excluded empty or damaged nuclei and multiplets. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Nuclei suspension before and after nuclei sorting. (A, C) DAPI-stained nuclei (blue). (B, D) Overlay of DAPI and brightfield (BF) channel. Before sorting (upper panel) the nuclei suspension contains cell debris and aggregates (labeled with white arrowheads). After sorting (lower panel) the nuclei suspension appears much cleaner. Examples of DAPI-stained nuclei are labeled with black arrowheads. Good quality nuclei appear round and smooth with an intact membrane and are well-separated, whereas nuclei of poor quality appear wrinkled and show loss of the nuclear membrane. Scale bar = 250 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Quality control and analysis of a representative snRNA-seq dataset. (A) Number of genes (nGene) plotted against the number of unique molecular identifiers (nUMI) colored by the fraction of mitochondrial reads (percent.mt). Low quality nuclei correspond to the bottom left quadrant of the plot (n = 828) and were excluded from subsequent analysis. (B) Distribution and median of nGene and nUMI detected per nucleus in the snRNA-seq dataset, representing 6,177 nuclei (> 500 genes). Libraries were sequenced to a median depth of ~ 8,200 mapped reads per nucleus. (C) Dot plot showing gene expression patterns of cluster-enriched markers (x-axis) for individual cell types (y-axis). The size of the dot corresponds to the proportion of cells expressing the indicated gene. The color corresponds to the average expression. (D) T-distributed stochastic neighbor embedding (t-SNE) plot of identified cell types. (E) Cell type distribution in snRNA-seq dataset. PT, proximal tubule; tL, thin limb; TAL, thick ascending limb, DCT, distal convoluted tubule; CNT, connecting tubule; CD-PC, collecting duct principal cells; A-IC, type A intercalated cells; B-IC, type B intercalated cells; DMEP, deep medullary epithelium of pelvis. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Final concentration | Volume (mL) | ||
| (A) 4 % BSA / PBS | ||||
| Phosphate-Buffered Saline (PBS) with 10% Bovine Albumin | 4% | 2 | ||
| PBS (Phosphate-Buffered Saline) 1X without calcium or magnesium | - | 3 | ||
| (B) 0.04 % BSA / PBS | ||||
| 4 % BSA / PBS | 0.04 % | 0.5 | ||
| PBS (Phosphate-Buffered Saline) 1X without calcium or magnesium | - | 49.5 | ||
| (C) Nuclei Lysis Buffer 1 (NLB1) | ||||
| Nuclear EZ Lysis Buffer | - | 4 | ||
| RiboLock RNase Inhibitor (40 U/µL) | 1 U/µL | 0.1 | ||
| Ribonucleoside-vanadyl complex (200 mM) | 10 mM | 0.2 | ||
| (D) Nuclei Lysis Buffer 2 (NLB2) | ||||
| Nuclear EZ Lysis Buffer | - | 4 | ||
| RiboLock RNase Inhibitor (40 U/µL) | 1 U/µL | 0.1 | ||
| (E) Nuclei Suspension Buffer (NSB) | ||||
| 0.04 % BSA / PBS | - | 2 | ||
| RiboLock RNase Inhibitor (40 U/µL) | 1 U/µL | 0.05 | ||
| (F) Sucrose Gradient Buffer (10 % Sucrose) | ||||
| Weight 1 g of Sucrose | ||||
| Dissolve in 6 ml of Nuclear EZ Lysis Buffer | ||||
| Fill up to 10 mL with Nuclear EZ Lysis Buffer | ||||
| Filter through a 0.2 µm syringe filter into a fresh tube | ||||
Table 1: Solution recipes: (A) Preparation of 4% BSA/1x PBS. Filter using a 0.2 µm SFCA membrane syringe filter and keep on ice until use. (B) Preparation of 0.04% BSA/1 x PBS. Filter using a 0.2 µm SFCA membrane syringe filter and keep on ice until use. (C) Preparation of Nuclei Lysis Buffer 1 (NLB1). Indicated volumes are provided per sample. Keep on ice until use. (D) Preparation of Nuclei Lysis Buffer 2 (NLB2). Indicated volumes are provided per sample. Add RiboLock RNase Inhibitor to NLB2 directly before use as noted in the protocol. Keep on ice until use. (E) Preparation of Nuclei Suspension Buffer (NSB). Indicated volumes are provided per sample. Add RiboLock RNase Inhibitor to NSB directly before use as noted in the protocol. Keep on ice until use. (F) Preparation of Sucrose Gradient Buffer. Filter using a 0.2 µm SFCA membrane syringe filter and keep on ice until use.
Discussion
Single-cell transcriptomics advance the understanding of cell type-specific gene expression in renal physiology and disease. Here, we provided a simple and reproducible method to isolate high-quality single nuclei from frozen mouse kidney tissue for snRNA-seq in a standardized way.
For snRNA-seq, it is critical to use high-quality nuclei as input for library generation and to avoid RNA degradation during tissue processing. Therefore, the incubation of tissue pieces in RNA stabilization solution immediately after dissection is essential to protect and stabilize cellular RNA and allows to store samples at - 80 °C indefinitely. When applying this protocol to frozen tissue without RNA stabilization solution treatment, such as archival material, a trial run is required, and the RNA quality needs to be assessed as we observed a significant loss of RNA integrity in snap-frozen tissue without prior incubation in RNA stabilization solution.
In general, appropriate sample handling is crucial to maximizing the recovery of intact, single nuclei. All resuspension steps should be carried out by pipetting carefully to avoid shear stress and physical damage. Buffers for the final nuclei resuspension and nuclei sorting should contain BSA to avoid nuclei loss and aggregation.
Buffer volumes in this protocol are optimized for very small tissue samples (~15 mg). It is critical to ensure complete cell lysis and sufficient washing to generate high-quality suspensions. Larger tissue blocks or whole kidney samples will result in excessive nuclei concentrations that lead to clumping and aggregation, high abundance of ambient RNA, and overall poor suspension quality. If larger samples or other tissues are processed, it is highly recommended to perform trial runs to determine optimal buffer volumes for minimal ambient RNA levels. Nuclei and RNA quality and concentrations need to be examined carefully as overloading results in overall poor performance.
In addition, large amounts of cell debris, causing high levels of ambient RNA not associated with single nuclei influence the sequencing results negatively. Clarification of the nuclei suspension by centrifugation through a sucrose cushion mitigates this problem to some extent, but it can also lead to bias in cell type representation by counter selecting against dense, small nuclei present, for instance, in immune cells16. If this is of concern, the sucrose gradient should be omitted. By contrast, we found that flow cytometry based on DAPI staining was critical to reduce the amount of cell debris in order to produce a high-quality single nuclei suspension.
The isolation of single nuclei has considerable advantages when compared to single-cell approaches8. It is compatible with properly frozen tissue, making the tissue collection more flexible, and circumvents the need of enzyme-based tissue dissociation, which can introduce transcriptional stress responses6,17. Furthermore, it overcomes the dissociation bias that favors the selection of easily dissociable cell types of the renal cortex, which may lead to an underrepresentation of medullary cell types in some enzyme-based approaches5,6,10.
Using a central kidney piece instead of whole kidney tissue further saves resources and corrects for the overrepresentation of abundant cell types as described earlier10. However, depending on the mouse model or phenotype investigated, it may be beneficial to use whole kidney samples instead of a single middle slice. Whole kidney samples may be more representative of true cell proportions, or changes occurring in the whole kidney, whereas a trimmed middle slice proved advantageous for medullary phenotypes or when sample material was limited. This decision, therefore, is highly user-specific and should be considered carefully.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank the Scientific Genomics Platform at the Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin for technical support.
JL and KMSO were supported by the German Research Foundation (DFG) Research Training Group GRK 2318 and by Research Unit FOR 2841. KMSO was supported by Collaborative Research Grant 1365. AB was supported by funding from the Gottfried Wilhelm Leibniz Prize of the DFG awarded to NR.
Materials
| Name | Company | Catalog Number | Comments |
| Cell sorter | - | - | For fluorescence-activated cell sorting (FACS); e.g. BD FACSAria Cell Sorter. |
| Centrifuge 5810 R | Eppendorf | 5811000015 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL Automated Cell Counter | Invitrogen | AMQAF1000 | Needs to contain DAPI light cube to count nuclei. Alternatively, nuclei can be counted manually under fluorescence microscope. |
| 4′,6-Diamidino-2-phenyl-indol-dihydrochlorid (DAPI) | Biotrend | 40043/b | Stock solution prepared with a concentration of 100 µM. Used for nuclei staining in a final concentration of 2 µM. |
| D(+)-Sucrose ≥99.9%, ultrapure DNAse-, RNAse-free | VWR | 0335-500G | |
| DNA LoBind Microcentrifuge Tubes (1.5 mL) | Eppendorf | 22431021 | |
| Ethanol, 70 % | - | - | |
| FACS tubes | pluriSelect | 43-10100-46 | |
| KIMBLE Dounce tissue grinder set 2 mL complete | Sigma-Aldrich | D8938-1SET | |
| Minisart Syringe Filters 0.2 µm | Sartorius | 16534-GUK | |
| Nuclease-free Water | Invitrogen | AM9937 | |
| Nuclei EZ Prep Nuclei Isolation Kit | Sigma-Aldrich | NUC-101 | Nuclei EZ Lysis Buffer (Product No. N3408) needed for buffer preparation. |
| PBS (Phosphate-Buffered Saline) 1X without calcium or magnesium | Corning | 21-040-CV | |
| Petri dishes, polystyrene 60 mm | Sigma-Aldrich | P5481 | |
| Phosphate-Buffered Saline (PBS) with 10% Bovine Albumin | Sigma-Aldrich | SRE0036 | |
| pluriStrainer Mini 100 µm | pluriSelect | 43-10100-46 | |
| pluriStrainer Mini 20 µm | pluriSelect | 43-10020-40 | |
| pluriStrainer Mini 40 µm | pluriSelect | 43-10040-40 | |
| Polystyrene Centrifuge Tube (15 mL) | Falcon | 352099 | |
| Razor blades | - | - | |
| RiboLock RNase Inhibitor (40 U/µL) | Thermo Fisher | EO0384 | |
| Ribonucleoside-vanadyl complex | New England Biolabs | S1402S | Follow manufacturer's instructions (https://international.neb.com/products/s1402-ribonucleoside-vanadyl-complex#Product%20Information). Upon use the 200 mM stock solution is reconstituted to a green-black clear solution by incubating at 65 °C. |
| RNAlater Stabilization Solution | Invitrogen | AM7020 | |
| RNase AWAY | Fisher Scientific | 11952385 |
References
- Thomas, R. S. Kidney modeling and systems physiology. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 1 (2), 172-190 (2009).
- Potter, S. S. Single-cell RNA sequencing for the study of development, physiology and disease. Nature Reviews Nephrology. 14 (8), 479-492 (2018).
- Park, J., Liu, C. L., Kim, J., Susztak, K. Understanding the kidney one cell at a time. Kidney International. 96 (4), 862-870 (2019).
- Clark, A. R., Greka, A. The power of one: advances in single-cell genomics in the kidney. Nature Reviews Nephrology. 16 (2), 73-74 (2020).
- Lake, B. B., et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nature Communications. 10 (1), 2832(2019).
- Wu, H., Kirita, Y., Donnelly, E. L., Humphreys, B. D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. Journal of the American Society of Nephrology. 30 (1), 23-32 (2019).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144 (19), 3625-3632 (2017).
- Habib, N., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 14 (10), 955-958 (2017).
- Muto, Y., et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nature Communications. 12 (1), 2190(2021).
- Hinze, C., et al. Kidney single-cell transcriptomes predict spatial corticomedullary gene expression and tissue osmolality gradients. Journal of the American Society of Nephrology. 32 (2), 291(2021).
- Berger, S. L. Isolation of cytoplasmic RNA: ribonucleoside-vanadyl complexes. Methods in Enzymology. 152, 227-234 (1987).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 36 (5), 411-420 (2018).
- Stuart, T., et al. Comprehensive Integration of single-cell data. Cell. 177 (7), 1888-1902 (2019).
- Park, J., et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 360 (6390), 758-763 (2018).
- Kirita, Y., Wu, H., Uchimura, K., Wilson, P. C., Humphreys, B. D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proceedings of the National Academy of Sciences of the United States of America. 117 (27), 15874-15883 (2020).
- Schneeberger, S., et al. The neuroinflammatory interleukin-12 signaling pathway drives Alzheimer's disease-like pathology by perturbing oligodendrocyte survival and neuronal homeostasis. bioRxiv. , 441313(2021).
- Nguyen, Q. H., Pervolarakis, N., Nee, K., Kessenbrock, K. experimental considerations for single-cell RNA sequencing approaches. Frontiers in Cell and Developmental Biology. 6, 108(2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved