
Method Article
In situ Visualisierung des Axonwachstums und der Wachstumskegeldynamik in akuten ex vivo embryonalen Hirnschnittkulturen
In diesem Artikel
Zusammenfassung
Dieses Protokoll demonstriert eine einfache und robuste Methode zur Untersuchung des In-situ-Axonwachstums und der Wachstumskegeldynamik. Es beschreibt, wie ex vivo physiologisch relevante akute Hirnschnitte hergestellt werden können und stellt eine benutzerfreundliche Analysepipeline zur Verfügung.
Zusammenfassung
Während der neuronalen Entwicklung navigieren Axone durch die kortikale Umgebung, um ihre endgültigen Ziele zu erreichen und synaptische Verbindungen herzustellen. Wachstumskegel - die sensorischen Strukturen, die sich an den distalen Spitzen der sich entwickelnden Axone befinden - führen diesen Prozess aus. Die Untersuchung der Struktur und Dynamik des Wachstumskegels ist entscheidend für das Verständnis der axonalen Entwicklung und der Wechselwirkungen mit dem umgebenden Zentralnervensystem (ZNS), die es ihm ermöglichen, neuronale Schaltkreise zu bilden. Dies ist unerlässlich bei der Entwicklung von Methoden zur Reintegration von Axonen in neuronale Schaltkreise nach Verletzungen in der Grundlagenforschung und in präklinischen Kontexten. Bisher basiert das allgemeine Verständnis der Wachstumskegeldynamik in erster Linie auf Untersuchungen von Neuronen, die in zwei Dimensionen kultiviert sind (2D). Obwohl zweifellos grundlegend für das aktuelle Wissen über die strukturelle Dynamik von Wachstumskegeln und die Reaktion auf Reize, stellen 2D-Studien die physiologische dreidimensionale (3D) Umgebung, auf die neuronale Wachstumskegel in intaktem ZNS-Gewebe stoßen, falsch dar. In jüngerer Zeit wurden Kollagengele eingesetzt, um einige dieser Einschränkungen zu überwinden und die Untersuchung der neuronalen Entwicklung in 3D zu ermöglichen. Sowohl synthetischen 2D- als auch 3D-Umgebungen fehlen jedoch Signalsignale innerhalb des ZNS-Gewebes, die die Erweiterung und Wegfindung von sich entwickelnden Axonen steuern. Dieses Protokoll bietet eine Methode zur Untersuchung von Axonen und Wachstumskegeln unter Verwendung organotypischer Gehirnschnitte, bei denen sich entwickelnde Axone auf physiologisch relevante physikalische und chemische Hinweise treffen. Durch die Kombination von Feinabstimmung in utero und ex utero Elektroporation zur spärlichen Lieferung von Fluoreszenzreportern zusammen mit hochauflösender Mikroskopie stellt dieses Protokoll eine methodische Pipeline für die Visualisierung der Axon- und Wachstumskegeldynamik in situ dar. Darüber hinaus ist eine detaillierte Toolkit-Beschreibung der Analyse von Langzeit- und Lebendzellbilddaten enthalten.
Einleitung
Neuronen sind stark polarisierte Zellen, die die grundlegende Recheneinheit im Nervensystem darstellen. Sie empfangen und emittieren Informationen, die auf der Kompartimentierung von Input- und Output-Standorten beruhen: Dendriten bzw.Axone 1. Während der Entwicklung dehnen sich Axone aus, während sie durch eine unglaublich komplexe Umgebung navigieren, um ihr Ziel zu erreichen. Die Axonnavigation wird vom Wachstumskegel geleitet, einer sensorischen Struktur, die sich an der Spitze des sich entwickelnden Axons befindet. Der Wachstumskegel ist dafür verantwortlich, Umwelthinweise zu erkennen und in die dynamische räumliche Reorganisation seines Zytoskeletts zu übersetzen 2,3. Die daraus resultierenden morphomechanischen Reaktionen weisen den Wachstumskegel an, sich vom auslösenden Hinweis auszudehnen oder zurückzuziehen, was zu spezifischen Axonmanövern führt.
Das aktuelle Verständnis der Axonausdehnung und Wachstumskegeldynamik stammt aus Studien, die das Axonwachstum überzweidimensionale (2D) Substrate 2,4,5,6,7 untersuchen. Diese bahnbrechenden Studien identifizierten ein ausgeklügeltes Zusammenspiel zwischen Wachstumskegeln und Wachstumssubstraten und zeigten auffällige Unterschiede, die von Substrateigenschaften wie Haft- und Steifigkeit abhängig sind 8,9. Geleitet von diesen Erkenntnissen wurde angenommen, dass extrazelluläre Umwelthinweise das Axonwachstum diktieren sollten, wobei das Wachstumskegel-Zytoskelett dieses Wachstumausführte 2,10,11,12. Insbesondere können Neuronen Axone in nicht adhäsiven Substraten (z. B. Polylysin, Polyornithin) erweitern13. Darüber hinaus kann die Substratsteifigkeit die Axonwachstumsrate unabhängig von Zelladhäsionskomplexen beeinflussen8. Daher kann die Untersuchung der Wachstumskegeldynamik in 2D-Substraten allein das Kräftegleichgewicht, das sich aus der Wechselwirkung von axonalen Wachstumskegeln mit physiologisch relevanten dreidimensionalen (3D) Umgebungen, wie sie in vivo gefunden werden, ergeben, nicht genau modellieren.
Um die Einschränkungen der 2D-Assays zu überwinden, wurden Axonwachstum und Wachstumskegeldynamik in 3D-Matrizen 8,9 untersucht. Diese Matrizen stellen einen physiologischeren Kontext dar, ermöglichen jedoch die Untersuchung zellintrinsischer Mechanismen des Axonwachstums. Es ermöglicht die Einzelzelluntersuchung von Wachstumszapfen bei einer Vielzahl von Erkrankungen und pharmakologischen Behandlungen9. In solchen 3D-Umgebungen zeigten Axone eine ausgeprägte zytoskelettale Dynamik und wuchsen schneller als diejenigen, die in 2D-kultivierten Neuronen beobachtet wurden9. Diese eleganten Studien zeigten den Einfluss einer zusätzlichen Dimension auf die Reorganisation des Wachstumskegel-Zytoskeletts und damit auf sein Verhalten.
Trotz der offensichtlichen Vorteile, die 3D-Matrizen gegenüber 2D-Oberflächen bei der Unterstützung der nativen neuronalen Entwicklung und des Axonwachstums bieten, bleiben sie ein vereinfachtes synthetisches Gerüst, das die Komplexität der Dynamik, die im Gewebe des Zentralnervensystems (ZNS) beobachtet wird, nicht widerspiegeln kann. Hier wurde die Abgabe von Reporterplasmiden durch ex utero und in utero Elektroporation mit organotypischer Scheibenkultur des Gehirns und in situ hochauflösender Live-Bildgebung kombiniert, um die Wachstumskegeldynamik in einem physiologischen Kontext zu analysieren. Diese Methodik ermöglicht die Visualisierung der sich entwickelnden Axone bei gleichzeitiger Erfahrung der 3-dimensionalen In-vivo-Umgebungen und der Komplexität ihrer physikalisch-chemischen Zusammensetzung. Schließlich werden benutzerfreundliche Verfahren zur Messung des Axonwachstums und der Wachstumskegeldynamik mit allgemein lizenzierter und öffentlich verfügbarer Software beschrieben.
Protokoll
Tierversuche müssen den einschlägigen institutionellen und bundesstaatlichen Vorschriften entsprechen. In diesem Protokoll wurden am Embryonaltag 15,5 und 12,5 (E15,5 und E12,5) schwangere weibliche C57BL/6JRj-Mäuse verwendet. Die Versuche wurden nach dem Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV) durchgeführt.
1. Herstellung von Plasmiden zur Injektion
- Isolieren Sie die DNA mit dem endotoxinfreien Maxiprep-Kit gemäß dem Protokoll des Herstellers (siehe Materialtabelle).
- Mischen Sie ausgewählte DNA in der gewünschten Konzentration (Tabelle 1) und 10% Fast Green-Lösung (siehe Materialtabelle), um die Abgabe von DNA-Mischung in die Hirnventrikel zu visualisieren.
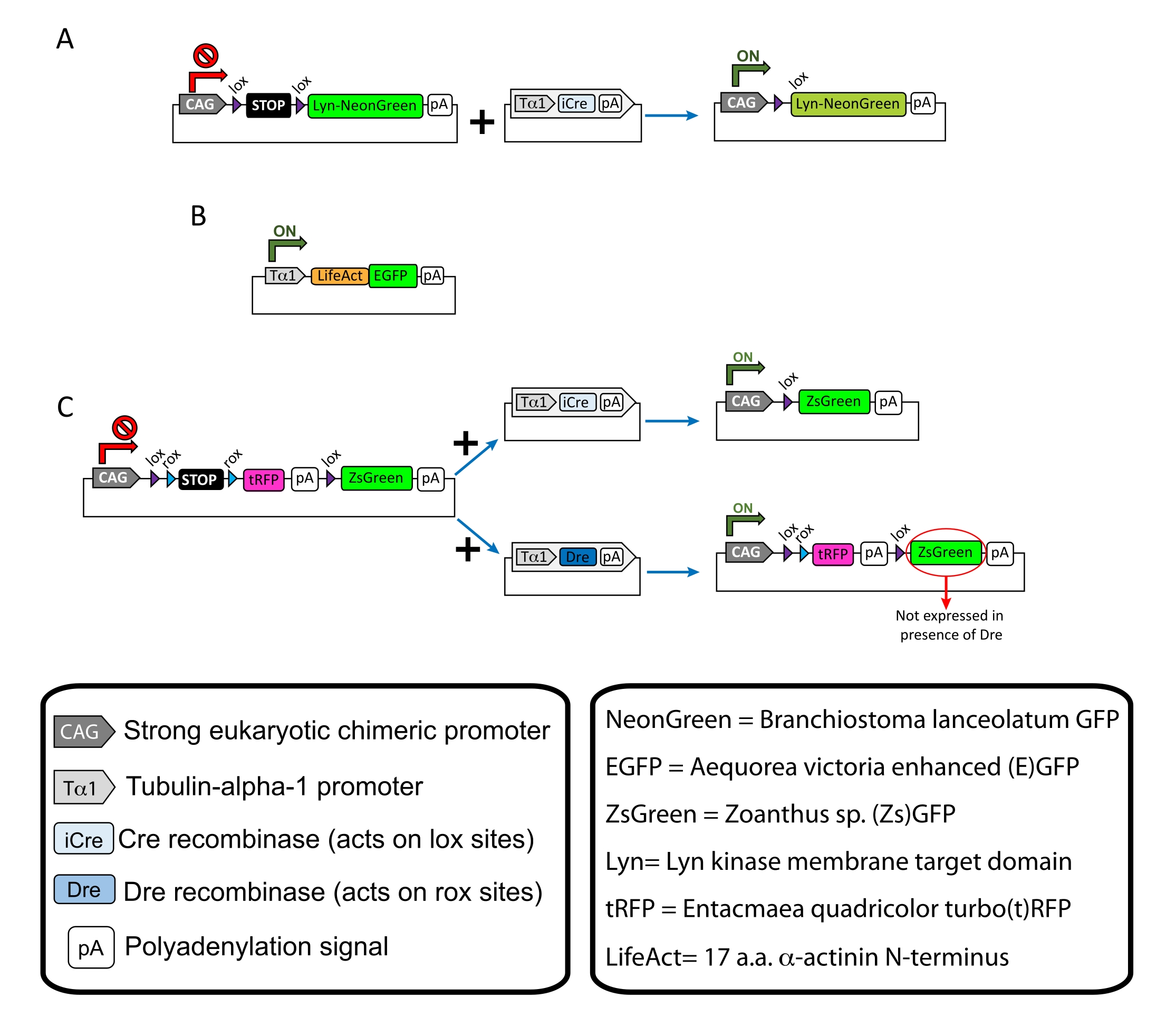
HINWEIS: Spezifische Plasmide wurden für die spärliche Markierung der kortikalen Neuronen (Abbildung 1A), filamentöse Aktinstrukturen (F-Aktin) im Wachstumskegel (Abbildung 1B) und die Doppelmarkierung der Wachstumskegel innerhalb desselben Kortex verwendet (Abbildung 1C). Alle in diesem Protokoll verwendeten Plasmide (Tabelle 1) wurden in Addgene abgeschieden (siehe Materialtabelle). - Bereiten Sie Glaskapillaren mit einem Kapillarelektrodenzieher-Set gemäß dem folgenden Programm vor: Druck: 500, Wärme: 800, Zug: 30 und Geschwindigkeit: 40.
- Laden Sie 15 μL DNA / Fast Green-Mischung mit Mikrolader-Pipettenspitzen in jede Glaskapillare.
HINWEIS: Stellen Sie sicher, dass sich keine Blasen bilden. - Bewahren Sie DNA-gefüllte Kapillaren in einer 10 cm großen Schale mit einem Stück Modelliermasse über den Durchmesser der Schale auf. Kapillaren können am Tag vor dem Experiment bei 4 °C geladen und gelagert werden. Versiegeln Sie das hintere Ende der Kapillare mit flexibler Folie, um ein Austrocknen zu verhindern.
2. Vorbereitung von Lösungen
- Bereiten Sie Hank's Buffered Salt Solution vor, die mit Glukose (HBSS-G) ergänzt ist.
- Fügen Sie 0,5% von 20% Glukosevorrat zu einer Flasche 1x HBSS hinzu. Gut mischen und bei 4 °C bis zu 2 Wochen lagern. Für die Embryoextraktion HBSS-G-Lösung mit Carbogen (95%O2 und 5% CO2) unter Verwendung von sprudelndem Stein kurz vor der Embryonenentnahme aufblasen.
- Slice-Media-Lösung
- Bereiten Sie frische Scheibenmedien mit Neurobasal 1x, 5% Pferdeserum, 5% fötalem Kalbsserum, B27-Ergänzung 1:50, L-Glutamin-Ergänzung 1:400, Penicillin-Streptomycin 1:200 und Neuropan-2-Ergänzung 1:100 (bei pH = 7,3) unter sterilen Bedingungen vor (siehe Materialtabelle).
- Bereiten Sie 3 cm große Gerichte mit je 1 ml Scheibenmedien zu. Vor dem Experiment mindestens 1 h bei 35 °C mit 5%CO2 in den Inkubator geben, um den pH-Wert des Mediums durch Gasaustausch auszugleichen.
HINWEIS: Das pH-Gleichgewicht der Medien wird durch die Ansäuerung der Medien durch das CO2 aus dem Inkubator verursacht. Schnittmedien können bis zu 1 Woche bei 4 °C gelagert werden.
- Agaroselösung mit niedrigem Schmelzpunkt (3%)
- Wiegen Sie die gewünschte Menge an niedrigschmelzpunktigem Agarosepulver und lösen Sie es in einem geeigneten Volumen von 1x HBSS-G in einer Glasflasche auf. Ungefähr 7 ml Agaroselösung pro Gehirn werden benötigt.
- Legen Sie die Flasche für 2-3 min in eine Mikrowelle, wobei die Kappe locker platziert ist, und schütteln Sie sie alle 10-20 s.
- Sobald sich das Pulver vollständig aufgelöst hat, stellen Sie die Flasche mindestens 1 h vor dem Experiment in ein Wasserbad oder ein auf 37 ° C eingestelltes Perlenbad, damit Agarose abkühlen kann.
HINWEIS: Es wird empfohlen, die Agarose zweimal über 15 Minuten zu erhitzen, um sicherzustellen, dass das Agarosepulver gelöst wird. Dies ist entscheidend für die richtige Adhäsion von Agarose im Hirngewebe. Ein Thermometer sollte verwendet werden, um die Temperatur der Agaroselösung während der Einbettung von Gehirnen zu messen und sicherzustellen, dass sie zwischen 37-40 ° C liegt. Gehirne von unterschiedlich alten Tieren haben unterschiedliche Steifheit. Es wird empfohlen, eine Reihe von Agarosekonzentrationen zu testen, um eine Homogenität zwischen Gewebe und Agarose zu finden. - Herstellung von phosphatgepufferter Kochsalzlösung mit 0,3% Triton X-100 (PBS-T).
- Zubereitung von phosphatgepufferter Kochsalzlösung mit 0,2% Natriumazid (PBS-NaN 3).
HINWEIS: Die in den Schritten 2.4-2.5 beschriebenen Lösungen sind für den späteren immunhistochemischen Schritt vorgesehen.
3. Vorbereitung der Operationsstation
- Reinigen Sie die Operationsstation mit 70% -96% Ethanol und legen Sie die Operationsunterlage auf die Stationsoberfläche.
- Sterilisieren Sie die chirurgischen Instrumente durch Spülen mit 70% -96% Ethanol, gefolgt von einer Trockensterilisation in einem heißen Perlensterilisator.
- Reinigen Sie Platinpinzettenelektroden (siehe Materialtabelle) mit 70% -96% Ethanol, bevor Sie sie an den Impulsgeber anschließen.
- Führen Sie eine DNA/Fast Green-gefüllte Glaskapillare in den Kapillarhalter ein. Unmittelbar vor dem Gebrauch die Kapillarspitze vorsichtig mit einer feinen Schere abbrechen und die Testlösung fließt in einem 1,5 ml Mikrozentrifugenröhrchen, das mit vorgewärmter Kochsalzlösung oder Wasser gefüllt ist.
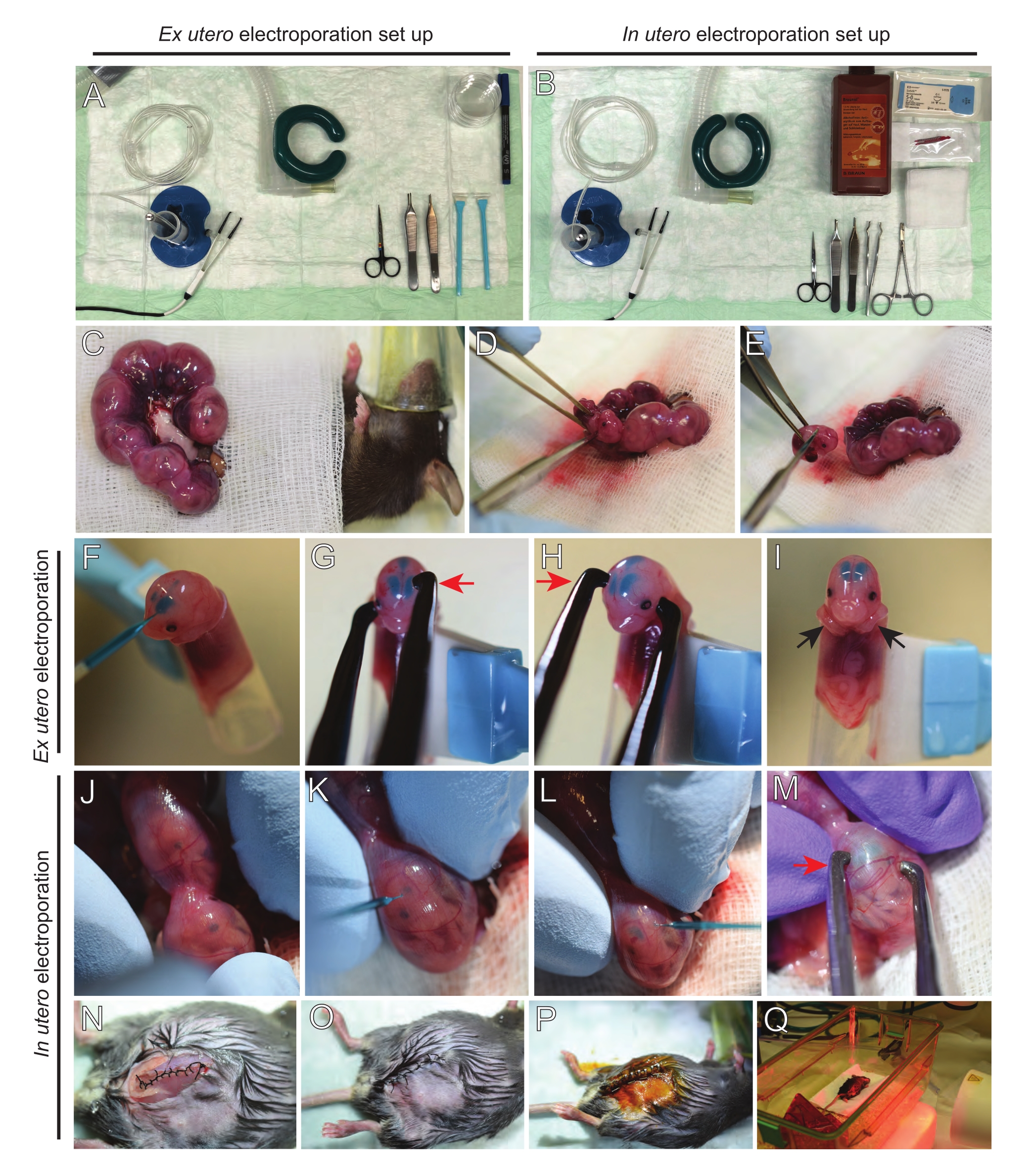
HINWEIS: Abbildung 2A, B zeigt den Aufbau einer Operationsstation und Werkzeuge, die für die Ex-utero-Elektroporation (EUE) und die utero-Elektroporation (IUE) verwendet werden. - Für IUE Kochsalzlösung auf 37 °C in einem Wasserbad aufwärmen.
4. Embryonengewinnung
- Legen Sie die schwangere Maus in die Anästhesieinduktorkammer mit 5% Isofluran, bis die Maus tief betäubt ist. Bestätigen Sie die Anästhesie durch das Fehlen eines Pedalentzugsreflexes.
- Übertragen Sie die Maus auf eine Operationsunterlage und halten Sie Isofluran bei 1,5% -2% durch einen Nasenkegel.
- Tragen Sie Salbe auf beide Augen auf, um das Austrocknen der Hornhaut zu verhindern.
- Rasieren Sie den Bauch der Maus und verwenden Sie dann 70% -96% ethanolgetränkte Gaze, um rasierte Haare zu entfernen. Reinigen Sie den Bereich mit Betadin.
- Machen Sie mit einer sterilen kleinen chirurgischen Schere einen 2 cm langen Hautschnitt entlang der Bauchmittellinie, gefolgt von einem 1,5 cm Muskelschnitt.
HINWEIS: Die Schnittgröße hängt von der Embryonengröße ab. In der Tat benötigen größere Embryonen einen größeren Einschnitt, um ihre Extraktion unterzubringen. - Schneiden Sie ein Loch in der Mitte der Gaze so breit, dass es zum Hautschnitt passt (~ 2 cm Durchmesser), tränken Sie es mit warmer Kochsalzlösung und legen Sie es um die Bauchöffnung.
- Ziehen Sie beide Uterushörner mit einer Wattestäbchen heraus, die in warmer Kochsalzlösung getränkt ist, oder mit einer Pinzette und greifen Sie vorsichtig die Zwischenräume zwischen den Embryonen an, um sie herauszuziehen. Setzen Sie Embryonen auf feuchte Gaze (Abbildung 2C).
HINWEIS: Selbst eine kleine Schädigung der Blutgefäße und Kapillaren um die Gebärmutterhörner führt wahrscheinlich zu starken Blutungen. Vermeiden Sie daher jederzeit den direkten Umgang mit diesen vaskularisierten Bereichen. - Schneiden Sie den Gebärmuttersack auf und entfernen Sie jeden Embryo (Abbildung 2D).
- Euthanasieren Sie jeden Embryo unmittelbar nach der Extraktion über einen absteigenden diagonalen Schnitt, um eine vollständige Rückenmarkstransektion zu gewährleisten (Abbildung 2E).
- Legen Sie die Embryonen in eine 10 cm große Schale, die HBSS-G enthält, auf Eis.
HINWEIS: Die Enthauptung des Embryos wird vermieden, um das Austreten von DNA / Fast Green-Gemisch aus dem Gehirn zu verhindern und eine einfache Positionierung des Embryos im Halter zu erleichtern (siehe ex utero-Elektroporation , Schritt 5). - Opfern Sie die Mutter unmittelbar nach der Entnahme von Embryonen, indem Sie eine Zervixdislokation durchführen.
HINWEIS: Hier wird die Mutter unter Narkose eingeschläfert, um ihr weitere Schmerzen oder Leiden nach dem Eingriff gemäß dem vom Tierschutzgesetz des Landesumweltamtes Nordrhein-Westfalen (LANUV) genehmigten Protokoll zu ersparen.
5. Ex-utero-Elektroporation (EUE)
- Nehmen Sie einen Embryo auf und legen Sie ihn in den Halter.
HINWEIS: Eine geschnittene 1 ml Pipettenspitze, die am Ende eines Wolkenkratzers befestigt ist, wird als Embryohalter verwendet. Es ist wichtig, die Arme der Embryonen während des Eingriffs außerhalb der Spitze zu halten, um zu verhindern, dass sie in die Spitze rutschen (Abbildung 2F-I). Der Durchmesser der Pipettenspitze ist leicht einstellbar, um Embryonen unterschiedlicher Größe aufzunehmen. Schneiden Sie eine zweite Spitze lang ab, wo der Durchmesser der Spitze der Embryogröße entspricht, und verwenden Sie sie als Adaptereinsatz für den oben genannten Halter. - Führen Sie vorsichtig eine mit DNA/Fast Green gefüllte Glaskapillare durch den Schädel des Embryos in den lateralen Ventrikel ein und injizieren Sie 2-3 μL DNA-Plasmidmischung (Abbildung 1A,B; Tabelle 1) in jeden Ventrikel (Abbildung 2F).
HINWEIS: Verwenden Sie die lambdoidalen und sagittalen Nähte als Leitfaden für den Ort der DNA-Injektion. Die lambdoidalen und sagittalen Nähte sind faserige Gelenke, die die Knochenplatte des Schädels verbinden. Ersteres verbindet den Parietalknochen mit dem Hinterhauptbein, und letzteres verbindet die beiden Parietalknochen. - Halten Sie den Kopf des Embryos zwischen Platinpinzettenelektroden im entsprechenden Winkel, um den gewünschten Hirnbereich (in diesem Fall 60 ° -Winkel) anzuvisieren, wobei die Kathode dem Bereich zugewandt ist, in dem der DNA-Transfer beabsichtigt ist (Abbildung 2G-H).
- Wenden Sie fünf Impulse bei 30 mV mit einem Intervall von 1 s und einer Dauer von 50 ms mit einem Rechteckwellenimpulsgenerator an.
HINWEIS: Denken Sie daran, dass Gehirne in EUE ein effektiveres elektrisches Feld erfahren als die in IUE. Daher führt EUE bei einer gegebenen DNA-Konzentration zu einer höheren Effizienz des DNA-Transfers als IUE, und die DNA-Konzentrationen müssen entsprechend angepasst werden. - Wenn eine bilaterale Elektroporation gewünscht ist, wiederholen Sie die Schritte 5.3-5.4, wobei die Kathode und die Anode die vorherige Position widerspiegeln, um den kontralateralen Kortex anzusprechen.
HINWEIS: Da beiden Ventrikeln DNA injiziert wurde, wurden Kortexe beider Hemisphären ins Visier genommen. - Geben Sie den elektroporigen Embryo in eine 6 cm große Schale, die eiskaltes HBSS-G enthält. Wiederholen Sie die Schritte 5.1-5.6 für alle erforderlichen Embryonen.
6. In-utero-Elektroporation (IUE)
- Injizieren Sie schwangere Maus mit Analgetikum; 50 μL Buprenorphin (0,1 mg/kg) (siehe Materialtabelle) subkutan, 20 Minuten vor dem Eingriff.
- Führen Sie die Schritte 4.1-4.8 aus dem Abschnitt Embryoextraktion aus.
HINWEIS: Vermeiden Sie es, Embryonen unnötig freizulegen, indem Sie sie mit steriler Gaze bedecken, die in warmer Kochsalzlösung getränkt ist. - Drehen Sie den Embryo mit den Fingerspitzen vorsichtig in der Gebärmutter, bis sich lambdoidale und sagittale Nähte befinden (Abbildung 2J). Führen Sie vorsichtig DNA/Fast Green-Glaskapillare durch die Gebärmutterwand und den Schädel des Embryos in den lateralen Ventrikel ein und injizieren Sie 2-3 μL DNA-Plasmidmischung (Abbildung 1A, C) nach Belieben in einen oder beide Ventrikel (Abbildung 2K-L).
HINWEIS: Übermäßiger Fingerdruck auf Gebärmutterhörner könnte zum Kollaps des Fruchtsacks führen. - Halten Sie den Kopf des Embryos zwischen Platinpinzettenelektroden im entsprechenden Winkel, um den gewünschten Hirnbereich (in diesem Fall 60 ° -Winkel) anzuvisieren, wobei die Kathode dem Bereich zugewandt ist, in dem der DNA-Transfer beabsichtigt ist. Vermeiden Sie es, die Gebärmutter zu quetschen, da dies zum Kollaps des Fruchtsacks führen kann (Abbildung 2M).
- Wenden Sie fünf Impulse bei 35 mV mit einem Intervall von 600 ms und einer Dauer von 50 ms mit einem Rechteckwellenimpulsgenerator an.
- Wenn beide lateralen Ventrikel injiziert wurden, wiederholen Sie die Schritte 6.5-6.6, wobei die Kathode und die Anode die vorherige Position widerspiegeln, um den kontralateralen Kortex anzuvisieren.
- Wiederholen Sie die Schritte 6.3-6.6 für alle erforderlichen Embryonen.
- Sobald alle erforderlichen Embryonen elektroporiert wurden, verwenden Sie eine salzhaltige Wattestäbchen, um die Uterushörner vorsichtig wieder in die Bauchhöhle zu legen.
HINWEIS: Die Zugabe von Kochsalzlösung in die Peritonealhöhle hilft den Uterushörnern, wieder in Position zu rutschen. - Nähen Sie Muskel- und Hautschnitte mit 5-0 Nahtmaterial. Verwenden Sie Nahtclips, um die Wunde zu sichern und die Nahtwunde zu desinfizieren, indem Sie sie mit Betadin besprühen (Abbildung 2N-P).
- Injizieren Sie der Maus subkutan 200 μL 5% Glukose.
- Injizieren Sie der Maus ein Antibiotikum; 50 μL Enrofloxacin (5 mg/kg) subkutan (siehe Materialtabelle).
- Legen Sie die Maus zurück in den Aufwachkäfig und halten Sie die Wärme mit einem Ferninfrarot-Wärmelicht oder einem Heizkissen für mindestens 20 Minuten nach dem Eingriff aufrecht (Abbildung 2Q).
- Überwachen Sie die Maus täglich und injizieren Sie Meloxicam nach dem Eingriff zur Schmerzlinderung gemäß institutionellen und bundesstaatlichen Richtlinien.
- Embryonen werden 2 Tage nach dem Eingriff (d. h. E17.5) nach Schritt 4 extrahiert.
7. Gehirnextraktion und Einbettung in Agarose
HINWEIS: Es wird empfohlen, die folgenden Schritte unter einem Dissektionsmikroskop durchzuführen, um eine bessere Präzision zu erzielen. Die Vermeidung von Hirnschäden ist entscheidend für den Erfolg des Eingriffs.
- Richten Sie Extraktionswerkzeuge in einem sterilen Arbeitsbereich unter einer Dissektionshaube ein (Abbildung 3A).
- Trennen Sie den Kopf eines Embryos mit einer Schere vom Rest des Körpers.
- Fixieren Sie den Kopf, wie in Abbildung 3C dargestellt, und entfernen Sie dann die Haut und den Schädel, indem Sie entlang der Mittellinie schneiden, beginnend von der Basis des Kopfes in Richtung Nase (Abbildung 3D).
- Schälen Sie die Haut und den Schädel seitlich und machen Sie eine ausreichend große Lücke (~ 1 cm), damit das Gehirn herausgeschnitten werden kann.
- Um das Gehirn zu entfernen, setzen Sie die geschlossene Spitze der sterilen Dissektionsschere ein, beginnend unter dem Riechkolben, der sich in Richtung Hirnstamm bewegt (Abbildung 3E).
- Schneiden Sie den Hirnstamm ab und schneiden Sie alle losen Hirnhäute um das Gehirn herum ab (Abbildung 3F).
HINWEIS: Lose Hirnhäute führen oft dazu, dass Scheiben nach dem Schneiden am Agaroseblock haften, was zur Ablösung von Gewebe von der Agarose während der Scheibenentnahme führt. - Wiederholen Sie die Schritte 7.1-7.6 für alle Embryonen und halten Sie das Gehirn bis zum Einbettungsschritt auf Eis (idealerweise nicht länger als 30 min).
HINWEIS: Die folgenden Schritte 7.7.1-7.7.4 beziehen sich auf die Gehirnextraktion von E12.5-Gehirnen.- Isolieren Sie die Oberseite des Kopfes direkt unter dem Auge, wie in Abbildung 3G dargestellt.
- Schneiden Sie die Haut und den Schädel oben auf dem Hirnstamm nach der gestrichelten Linie wie in Abbildung 3H gezeigt, ohne den Hirnstamm zu entfernen.
- Machen Sie einen 2 mm langen Haut-Schädel-Schnitt am Hinterkopf, wie in Abbildung 3I gezeigt (zur Verdeutlichung siehe Zeichnungen).
HINWEIS: Dieser Schnitt bietet erste Greifpunkte, um die Haut- und Schädelschichten abzuziehen. Typischerweise wirken sie sich als eine Schicht ab. Die typische Schnittgröße beträgt 2 mm, entsprechend der Länge der Schneidkante der verwendeten Mikrofederschere. - Beginnen Sie mit dem Abziehen von Hautschädelschichten, indem Sie eine Seite des Einschnitts befestigen und die andere vorsichtig ziehen. Schließen Sie mit der gleichen Sorgfalt ab, indem Sie die Basis des Kopfes abziehen, bis das Gehirn befreit ist (Abbildung 3J).
HINWEIS: Dies muss mit großer Sorgfalt erfolgen, wobei zu beachten ist, dass das Gehirn nicht entlang der Gewebeschichten gezogen wird. Wechseln Sie die Seiten, um das Gewebe zu entfernen, das das Gehirn bedeckt.
- Gießen Sie warme Agarose (bei 37-40 °C) in eine 3 cm große Schale.
- Nehmen Sie das Gehirn mit einem perforierten Löffel auf und entfernen Sie überschüssige Flüssigkeit, indem Sie den Boden des Löffels gegen trockenes Seidenpapier tupfen. Legen Sie das Gehirn in eine Agaroseschale.
HINWEIS: Es ist wichtig, so viel Flüssigkeit wie möglich aus dem Gehirn zu entfernen, um eine bessere Haftung von Agarose im Gewebe zu ermöglichen. - Das Gericht mit flüssiger Agarose auf Eis legen. Mit einem kleineren Löffel Agarose für 10 s mischen, um sie gleichmäßig abzukühlen. Manövrieren Sie das Gehirn in die Mitte der Schale. Platzieren Sie das Gehirn horizontal in der Schale mit der dorsalen Seite nach oben und stellen Sie sicher, dass es vollständig mit Agarose aus allen Richtungen bedeckt ist (Abbildung 3K).
HINWEIS: Gehirne sinken oft auf den Boden des Gerichts, sobald es in Agarose gegeben wird; Heben Sie das Gehirn mit einem kleinen Löffel an, bis eine Lücke von 1-2 mm unter dem Gehirn entsteht. - Wiederholen Sie die Schritte 7.8-7.10 für alle Gehirne.
- Sobald Agarose polymerisiert ist, fügen Sie 500 μL HBSS-G auf den Agaroseblock hinzu, um ein Austrocknen zu verhindern. Dann bedecken Sie die Schüssel mit Eis.
HINWEIS: Bewahren Sie die Probe vor dem Schneiden 5 Minuten lang auf Eis auf, damit die Gehirntemperatur 4 ° C erreichen kann.
8. Organotypische Schnittkultur
HINWEIS: Reinigen Sie Vibratom und umgebende Oberflächen mit 70% -96% Ethanol, um eine Kontamination der Scheiben zu vermeiden. Der Aufbau der Vibratom-Workstation (siehe Materialtabelle) ist in Abbildung 3B dargestellt.
- Füllen Sie die Vibratom-Pufferschale mit kaltem HBSS-G und die äußere Schale mit Eis, um das HBSS-G während des gesamten Verfahrens kalt zu halten.
- Kontinuierliche Versorgung von HBSS-G in der Pufferschale mit Carbogen unter Verwendung eines sprudelnden Steins.
- Machen Sie mit einer frischen Klinge einen großen Schnitt (~ 2 x 2 cm) um das Gehirn und entfernen Sie einen Agaroseblock, der das Gehirn enthält, mit genügend umgebender Agarose, um die Agarose in einen kleinen Rechteckblock zu schneiden.
HINWEIS: Dieser Schritt ermöglicht es, den Winkel des Blocks so einzustellen, dass die Sagittalachse des Gehirns senkrecht zur Vibratomplatte steht und die koronale Achse parallel zur Klinge ausgerichtet ist. Lassen Sie etwa 5 mm Agarose an der dorsalen Seite des Gehirns für eine einfache Handhabung der Scheiben. - Legen Sie einen kleinen Tropfen schnell klebenden lösungsmittelfreien Sekundenklebers in die Mitte des Probenhalters und verteilen Sie ihn auf einen Bereich, der den Boden des Agaroseblocks bedeckt.
- Nehmen Sie vorsichtig den Agaroseblock auf und trocknen Sie den Boden ab, indem Sie gegen Seidenpapier tupfen. Legen Sie den Block auf den geklebten Bereich des Probenhalters, wobei die rostrale Seite des Gehirns nach oben zeigt. Den Probenhalter auf Eis legen und den Kleber 1 min trocknen lassen.
- Sobald der Kleber getrocknet ist, legen Sie den Probenhalter in die Pufferschale.
- Schneiden Sie das Gehirn in koronale Scheiben in einem Winkel von 15°.
HINWEIS: Die Dicke der Scheiben kann je nach Anwendung variieren. Hier wurden Gehirne mit einer Dicke von 150 μm geschnitten. Stellen Sie die Vibratomgeschwindigkeit auf 1,0-1,5 mm / s ein, um überschüssige Agarose oben zu trimmen und Riechkolben zu trimmen. Reduzieren Sie die Schnittgeschwindigkeit auf 0,5 mm/s für das Sammeln von kortikalen Scheiben für die Analyse. Die meisten Vibratome können pausiert werden, um jede Scheibe zu sammeln. Wenn eine verminderte Qualität der Scheiben oder die Ablösung von Gewebe von Agarose auftreten, kann es hilfreich sein, die Schnittgeschwindigkeit zu reduzieren oder die Vibratomklinge zu ersetzen. - Sammeln Sie mit sauberen Spateln Gehirnscheiben und legen Sie sie auf die Polytetrafluorethylen (PTFE) -Membran, die in einer 35 mm großen Glasbodenschale mit Paraffin (bis zu fünf Gehirnscheiben / Membran) immobilisiert ist (Abbildung 3L-M).
HINWEIS: Befestigen Sie die PTFE-Membran in einer 35 mm Glasbodenschale mit Wachs. Dadurch stabilisiert sich die Membran bei Zugabe der Schnittkulturmedien und auch während der Bildgebung. - Entfernen Sie mit einer 200-μL-Pipette überschüssiges HBSS-G aus der Umgebung der Scheiben auf der PTFE-Membran und lassen Sie die Scheiben halbtrocken.
- Geben Sie 500 μL Schichtmedien (vorgewärmt auf 35 °C) direkt in den Raum unter der PTFE-Membran.
HINWEIS: Unter der Membran dürfen sich beim Hinzufügen des Mediums keine Blasen bilden. Dadurch bleiben ganze oder teilweise Slices ohne Medienaustausch. Ersetzen Sie 200 μL Medien alle 2 Tage in Kultur oder nach jeder Bildgebungssitzung. - Die Scheiben bei 35 °C mit 5% CO2 inkubieren.
9. Immunhistochemie
- Fixieren Sie die Scheiben mit 1 ml 4% Paraformaldehyd (PFA) - ergänzt mit 4% Saccharose pro Gericht. 30 min bei RT inkubieren.
ACHTUNG: Tragen Sie beim Umgang mit PFA einen Laborkittel und Handschuhe. Führen Sie Fixierungsschritte unter einer chemischen Haube durch und entsorgen Sie PFA-Abfälle entsprechend. - Waschen Sie die Scheiben zweimal mit 300 μL PBS für 5 min. Die Scheiben auf eine 24-Well-Platte geben.
HINWEIS: Das Experiment kann in diesem Stadium pausiert werden. PBS-NaN 3 zu den Scheiben geben und bei 4 °C lagern. NaN3 ist eine toxische Verbindung; Tragen Sie bei der Handhabung von Lösungen damit einen Laborkittel und Handschuhe. Die Schritte 9.3-9.10 sind in einem Orbitalschüttler durchzuführen. - Die Scheiben mit 300 μL 0,1 M Glycin bei 4 °C über Nacht abschrecken.
- Glycin mit PBS bei RT 3x für 10 min auswaschen.
- Permeabilisieren Sie die Scheiben mit 300 μL PBS-T bei RT für 2 h.
- Block mit 10% Ziegenserum in PBS-T bei RT für 2 h.
- 300 μL primärer Antikörper (Anti-Vimentin-Antikörper bei einer Verdünnung von 1:200; siehe Materialverzeichnis), verdünnt in 10%igem Ziegenserum in PBS-T-Lösung bei 4 °C über Nacht.
HINWEIS: In den Schritten 9.8-9.12 wurden die Scheiben lichtgeschützt, um einen Verlust der Fluoreszenz zu verhindern. - Primärer Antikörper mit PBS bei RT 3x 20 min waschen.
HINWEIS: PBS wurde anstelle von PBS-T verwendet, um den Triton X-100 auszuwaschen. - Fügen Sie 300 μL sekundären Antikörper (entweder Alexa Fluor 488 oder 647 bei einer Verdünnung von 1:400; siehe Tabelle der Materialien) in PBS bei RT für 2 h hinzu.
HINWEIS: DAPI wird unmittelbar nach dem Entfernen des sekundären Antikörpers bei einer Verdünnung von 1:10.000 für 5 min zugegeben. - Waschen Sie den sekundären Antikörper mit PBS bei RT 3x für 20 min. Mit destilliertem Wasser 2x 1 min waschen.
- Die Scheiben mit einer feinen Bürste auf einen Glasschieber geben und dann 20 min bei 30 °C trocknen.
- Montieren Sie die Scheiben mit einem wässrigen Montagemedium. Halten Sie die Folien über Nacht bei RT, damit die Montagemedien kuratiert werden können.
10. Bilderfassung
HINWEIS: Unabhängig vom DNA-Delivery-Ansatz (IUE oder EUE) wurden die Schnitte im gleichen Entwicklungsalter (E17.5-E18.5) analysiert. IUE ermöglicht es neuronalen Vorläufern, sich für zwei weitere Tage in vivo zu teilen und zu entwickeln. EUE hingegen ermöglicht die Verfolgung früher Entwicklungsereignisse.
- Schalten Sie den Kammerinkubator ein und stellen Sie ihn auf 35 °C mit 5% CO2 - idealerweise 4 h vor der Bildgebung - ein, damit sich die Mikroskopkomponenten bei 35 °C ausgleichen können.
- Verwenden Sie für die Tiefenbildgebung von Scheiben Wasser-Tauchobjektive, um die Diskrepanz des Brechungsindex zwischen Gewebe und Objektiv zu reduzieren.
HINWEIS: Hier wurde der hochauflösende Bildgebungsmodus verwendet. Die Bildgebung durch die PTFE-Membran erfordert ein Objektiv mit einem langen Arbeitsabstand (~1 mm). Wenn ein Objektiv mit langem Arbeitsabstand nicht verfügbar ist, können die Scheiben in eine 8-Well-Glasbodenschüssel gegeben werden. Um Scheiben zu übertragen, fügen Sie 1 ml Schichtmedien an der Oberseite der Membran hinzu und verwenden Sie dann einen Spatel, um eine Scheibe anzuheben und in eine Vertiefung mit 200 μL Medien zu übertragen. Entfernen Sie überschüssige Medien mit einer 1 ml Pipettenspitze, so dass die Scheiben halbtrocken bleiben. - Für die Bildgebung des Axonwachstums lokalisieren Sie eine Region des Kortex mit niedriger bis mittlerer Zelldichte. Für die Bildgebung der Wachstumskegeldynamik lokalisieren Sie einen Wachstumskegel in der Zwischenzone oder subventrikulären Zone des Kortex.
- Definieren Sie eine Z-Stack-Größe in der Bildverarbeitungssoftware (siehe Materialverzeichnis). Für das Axonwachstum in einem großen Z-Stack legen Sie eine Schrittgröße von 2 μm fest. Für Wachstumskegel in einem kleineren Z-Stack stellen Sie eine Schrittgröße von 1 μm ein.
HINWEIS: Berücksichtigen Sie immer die potenzielle Bewegung des Wachstumskegels und des Axons durch die x-, y- und z-Ebenen. Axone wachsen in organotypischen Kulturen viel häufiger als in In-vitro-Kulturen . Hier reichte ein Z-Stack von rund 80 μm zur Abbildung des Axonwachstums aus. Für die Wachstumskegeldynamik war ein Z-Stack von ~6 μm ausreichend. - Für die Abbildung des Axonwachstums von Neuronen in einem größeren Bereich definieren Sie einen Kachelscan.
- Verwenden Sie die geringstmögliche Laserleistung, um die Wahrscheinlichkeit zu minimieren, dass Wachstumskegel während der Akquisition gebleicht werden.
- Für die Bildgebung des Axonwachstums erfassen Sie Zeitraffer für 2 h mit einem Intervall von 5 min. Für die Dynamik des Bildgebungswachstumskegels erfassen Sie Zeitraffer für 2-5 Minuten mit einem Intervall von 2,5-3 s.
11. Datenanalyse
- Messen Sie die Geschwindigkeit des Axonwachstums mit Kymographen
- Öffnen Sie die Bilddatei in Fidschi14 über Datei > Öffnen und wählen Sie das Bild aus.
- Erhalten Sie die maximale Intensität Projektion des Zeitraffers durch Image > Stacks > Z-Projektion > Maximum Intensity Projection.
- Gehen Sie durch den Zeitraffer und lokalisieren Sie ein wachsendes Axon.
- Zeichnen Sie nach dem Auffinden eine Linie durch das wachsende Axon. Beginnen Sie mit der Spitze des Axons im ersten Bild und folgen Sie dem Axon durch den gesamten Zeitraffer.
- Generieren Sie einen Kymographen mit dem Plugin KymoResliceWide.
- Legen Sie den Maßstab des Kymographen fest, indem Sie zu Image > Properties gehen. Stellen Sie den Abstand in μm in Pixelbreite und die Zeit in s oder min in Pixelhöhe ein.
- Wechseln Sie zu Analysieren > Maßnahme.
HINWEIS: Es wird ein Winkel relativ zur x-Achse angegeben. - Berechnen Sie die Geschwindigkeit des Axonwachstums, indem Sie den Winkel in der folgenden Gleichung ersetzen: SIN(BOGENMAẞ(θ) )/COS(BOGENMAẞ(θ)) in einer Tabelle.
- Messen Sie das Volumen des Wachstumskegels mit einer Bildanalysesoftware (siehe Materialtabelle).
- Öffnen Sie die Bilddatei in der Bildanalysesoftware über Datei > Öffnen und wählen Sie die gewünschte Datei aus.
- Wählen Sie den Assistenten Neue Flächen hinzufügen aus.
HINWEIS: Ein Abschnitt in der unteren linken Ecke wird mit sechs Schritten für die manuelle Bearbeitung angezeigt. - Wählen Sie in Schritt 1 unter Algorithmuseinstellungen die Option Nur eine Region von Interesse segmentieren. Schneiden Sie in Schritt 2 den Rahmen so zu, dass er in allen Rahmen den gesamten Wachstumskegel passt.
- Halten Sie den Schwellenwert in Schritt 3 auf die absolute Intensität und stellen Sie sicher, dass die gesamte Wachstumskegelregion in Schritt 4 gethresholdt wird.
- Wählen Sie in Schritt 5 unter Filtertyp die Option Anzahl der Voxel lmg = 1 aus.
HINWEIS: Im letzten Schritt können mehrere Messsätze erstellt werden. Hier wurde nur eine Messung für das Volumen erstellt. - Wählen Sie die Schaltfläche Ausführen, um alle Erstellungsschritte auszuführen und den Assistenten zum Hinzufügen neuer Flächen zu beenden.
- Wählen Sie auf der Registerkarte Statistik oben im Fenster des Assistenten auf der Registerkarte Detailliert die Option Bestimmte Werte und Volumen aus.
Ergebnisse
Es werden repräsentative Ergebnisse gezeigt, die mit dem beschriebenen Methoden-Workflow erzielt wurden. E15.5-Mäuse wurden in der vorliegenden Demonstration verwendet, obwohl dieses Protokoll leicht an praktisch alle embryonalen Altersgruppen von E11 bis Ende E17 angepasst werden kann. In diesem Protokoll ist entweder ex utero Elektroporation (EUE; Abbildung 2A, 2C-I) oder in utero Elektroporation (IUE; Abbildung 2B, C und 2J-Q) wurden verwendet, um Plasmide in die Vorläuferneuronen zu bringen, die die lateralen Ventrikel auskleiden. Diese Vorläufer sind die Quelle zukünftiger kortikaler projizierender Neuronen (CPN)15,16. Plasmidmischungen wurden hergestellt, um eine spärliche neuronenspezifische Expression von entweder membrangerichtetem (Lyn)-mNeonGreen (Abbildung 1A) oder LifeAct-verstärktem (E)GFP (Abbildung 1B) anzutreiben, um das Gesamtverhalten bzw. die Aktindynamik in Wachstumskegeln zu bewerten. Darüber hinaus wurde eine Plasmidmischung eingeschlossen, die darauf abzielte, einzelne Neuronen entweder mit Turbo(t)-RFP oder Zoanthus sp. (Zs) grün fluoreszierendem Protein (ZsGreen) zu markieren (Abbildung 1C). Dies erleichtert die Überwachung des Wachstumskegelverhaltens von unabhängigen benachbarten Neuronen.
Die Hirndissektion aus elektropoierten Embryonen ist ein entscheidender Schritt, der sorgfältig durchgeführt werden muss, um qualitativ hochwertige Scheiben zu erhalten und die native Gehirnstruktur zu erhalten. Dissektionsinstrumente und Vibratom wurden vorher vorbereitet und sorgfältig mit Ethanol sterilisiert (Abbildung 3A,B). Als nächstes wurden die Köpfe der elektropolierten Embryonen sorgfältig seziert und die Gehirne extrahiert. Hier wird eine repräsentative Dissektion von Gehirnen aus den Embryonen gezeigt, die bei E15 (Abbildung 3C-F) und E12.5 (Abbildung 3G-J) einer EUE unterzogen wurden. Gehirne werden sofort in eine Agarosematrix eingeschlossen, geschnitten und auf PTFE-Membraneinsätze in einer unteren Glasschale für die Inkubation gelegt (Abbildung 3K-M).
Der Gesundheitszustand von Gehirnschnitten ist ein wichtiger Punkt für die Kontrolle, um zuverlässige Ergebnisse zu gewährleisten. Eine Sichtprüfung auf Verunreinigungen wurde täglich durchgeführt. Sobald die Kultur fertiggestellt ist, werden die Gehirnschnitte fixiert und der Immunhistochemie unterzogen. Hier wurde 4′,6-Diamidino-2-phenylindol (DAPI) verwendet, um die gesamte zelluläre Organisation und die Vimentinfärbung zu kontrollieren, um die Glia-Organisation aufzudecken; insbesondere Radialglia (RG) Gerüst. Typischerweise zeigen erfolgreich kultivierte Hirnschnitte, die entweder aus IUE oder EUE stammen, eine normale zelluläre Verteilung, wie DAPI und eine etwas organisierte Anordnung von RG mit apikal orientierten Pial-Kontaktierungsprozessen17 zeigen (Abbildung 4A, B). Gelegentlich werden deutliche Störungen im RG-Gerüst in kultivierten Hirnschnitten beobachtet, insbesondere bei solchen, die aus der EUE-Elektroporation stammen (Abbildung 4C). Hirnschnitte mit extrem unorganisiertem RG-Gerüst zeigen eine gestörte neuronale Migration und ein defektes Axonwachstum (nicht abgebildet). Daher ist die Steuerung des RG-Gerüsts eine einfache postkulturelle Methode, um die aus zuverlässigen Gehirnschnitten gewonnenen Daten zu sortieren.
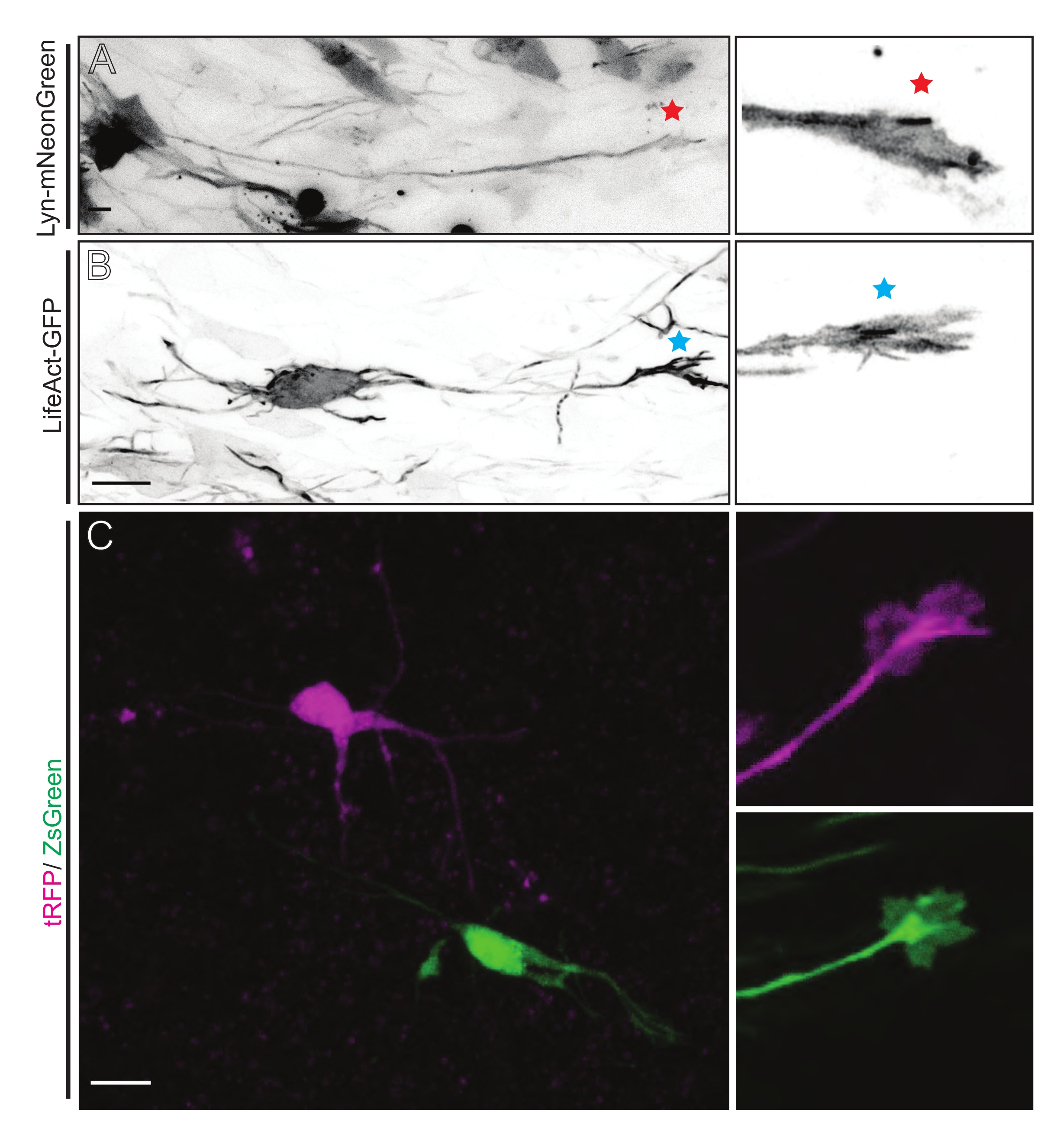
Gehirnschnitte, die entweder aus IUE oder EUE mit Lyn-mNeonGreen-exprimierender Plasmidmischung gewonnen werden, führen zu einer ähnlich spärlichen Neuronenmarkierung. Ein repräsentatives pyramidenförmiges CPN, das Lyn-mNeonGreen und das dynamische Verhalten seines Wachstumskegels exprimiert, ist als Beispiel gezeigt (Abbildung 5A und ergänzendes Video 1, oben links). Darüber hinaus wurden Neuronen mit einem Plasmid markiert, das eine Aktinsonde exprimierte, um die Aktindynamik von axonalen Wachstumskegeln in situ zu analysieren (Abbildung 5B und ergänzendes Video 1, unten links). In-situ-Experimente wurden auch mit einem Dual-Cre/Dre-Fluorophor-exprimierenden Plasmid-Design durchgeführt (Abbildung 1C und ergänzendes Video 1, rechts). tRFP- oder ZsGreen-Fluorophore in diesem Plasmid könnten spezifisch und individuell entweder durch Dre- bzw. Cre-Rekombinasen in benachbarten Neuronen aktiviert werden (Abbildung 5C). Diese experimentelle Aufstellung ermöglicht die parallele Analyse von Wachstumskegeln von Kontrollneuronen mit benachbarten modifizierten Neuronen (bei gegebenem Funktionsverlust oder -gewinn). Dadurch wird die Variabilität umgangen, die sich aus der Verwendung verschiedener Schichten zum Testen von Kontroll- und Versuchsbedingungen ergibt.
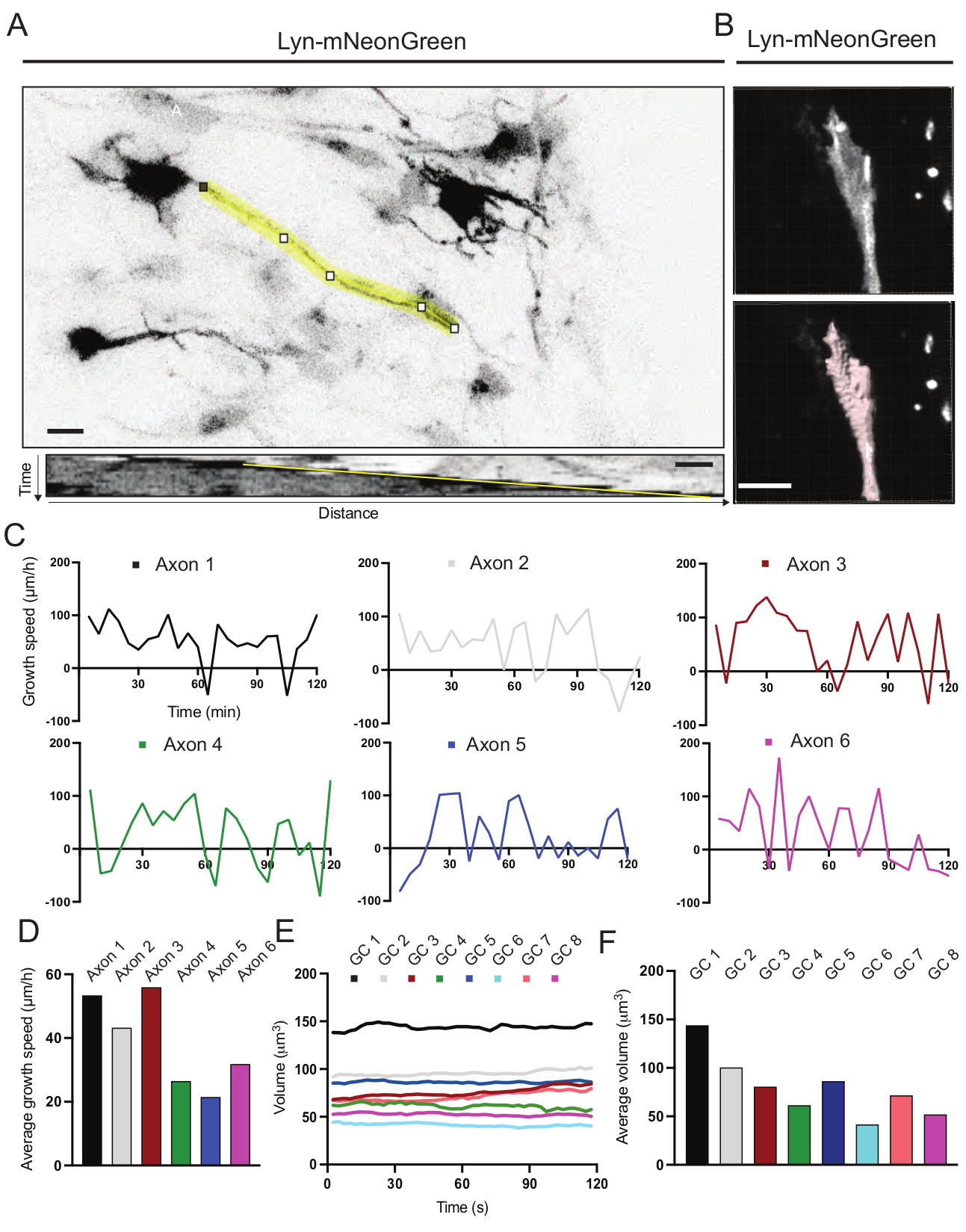
Aus dem aufgezeichneten Film generierte Kymographen wurden analysiert, aus denen dynamische Wachstumsparameter wie protrusive Aktivität über die Zeit und Wachstumslänge leicht gewonnen werden können (Abbildung 6A). Beachten Sie, dass eine einfache Anpassung der zeitlichen Auflösung des Zeitraffers die Messung der Axondehnungsgeschwindigkeit für 2 h ermöglicht (Abbildung 6A). Darüber hinaus kann die Variation des Wachstumskegelvolumens im Laufe der Zeit - ein Maß für die allgemeine dynamische Aktivität des Wachstumskegels - leicht erhalten werden, in diesem Fall mit lizenzierter Software (Abbildung 6B und Abbildung 6E, F). Dies kann verwendet werden, um die Geschwindigkeit des Aktinlaufbandes und das Gleichgewicht von Filopodien / Lamellipodien während der Wachstumskegelerkundungsaktivität zu bewerten.

Abbildung 1: Schemata der im Protokoll verwendeten Plasmide . (A) pCAG-lox-STOP-lox-Lyn-mNeonGreen. (B) p-Tub-alpha-1-LifeAct-GFP. (C) pCAG-lox-rox-STOP-rox-tRFP-pA-lox-ZsGreen-pA. Relevante Informationen über Plasmidkomponenten und die Herkunft von Fluorophor finden Sie in den Boxen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Arbeitsablauf von ex utero und in utero Elektroporation von E15.5-Mäusen. (A) Einrichtung einer Operationsstation für die Elektroporation ex utero. (B) Einrichtung einer Operationsstation für die In-utero-Elektroporation. (C) Uterushörner, die außerhalb der Bauchhöhle der betäubten Maus gezogen werden. (D) Entnahme eines Embryos aus dem Gebärmuttersack. (E) Embryonenopfer durch vollständige Rückenmarkstransektion über einen diagonalen Einschnitt; Beachten Sie, dass eine Enthauptung vermieden wurde. (F) Einsetzen des Embryos in den Halter und Injektion mit DNA/Fast Green-Gemisch in den linken seitlichen Ventrikel. (G,H) Positionieren Sie den Kopf des Embryos zwischen Platinpinzettenelektroden mit der Kathode (roter Pfeil) über dem Kortex in einem Winkel von 60°. (I) Platzierung der Arme des Embryos (schwarze Pfeile) außerhalb des Halters, um ein Abrutschen des Embryos während des Eingriffs zu verhindern. (J) Rotation des Embryos im Gebärmuttersack, um den Kopf freizulegen. (K,L) Injektion der DNA / Fast Green-Mischung in die lateralen Ventrikel des Embryos durch die Gebärmutterwand. (M) Positionieren Sie den Kopf des Embryos zwischen Platinpinzettenelektroden mit einer Kathode (roter Pfeil) über dem Kortex in einem Winkel von 60°. (N) Vernähter Muskelschnitt durch laufende Verriegelungsnaht. (O) Vernähter Hautschnitt über eine unterbrochene Naht. (P) Sicherung der Wunde mit chirurgischen Wundclips und Desinfektion mit Betadin. (Q) Platzierung der Maus im Auffangkäfig mit Ferninfrarot-Wärmelicht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Extraktion von E15.5 und E12.5 Gehirnen und organotypisches Schnittkulturverfahren. (A) Werkzeuge, die für das Verfahren zur Gehirnextraktion verwendet werden. (B) Einrichtung einer organotypischen Kulturstation. (C-F) Extraktion von E15.5 Gehirn. (G-J) Extraktion von E12.5 Gehirn. Gepunktete Linien markieren die Position der Einschnitte. Rote Pfeile weisen auf die Richtung des Ziehens durch eine Pinzette hin. (K) Einbettung des Gehirns in eine 3-cm-Schale mit 3% schmelzarmer Agarose, wobei ein Abstand von 1-2 mm Agarose unter dem Gehirn verbleibt. (L) Sammlung von 150 μm Hirnschnitt. (M) Platzierung von Hirnschnitten auf PTFE-Membraneinsätzen, die in einer 35-mm-Schale mit Paraffinfilm immobilisiert wurden (blauer Pfeil). Die Markierung des roten Sterns zeigt eine bestimmte Gehirnschnittsammlung aus Vibratom (L) und deren Übertragung auf die PTFE-Membran (M) an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Erhaltene radiale Gliazellstruktur in gesunden organotypischen Schnitten. Konfokale Bilder von E17.5-Gehirnschnitten, die das RG-Array (Vimentin; grün) und die Gesamtzellorganisation (DAPI; Magenta) nach IUE (A) und EUE (B, C) zeigen. Beachten Sie die starken Störungen im RG-Array, die gelegentlich durch EUE (C) entstehen können. Die Vergrößerungen entsprechen den rot gepunkteten Rahmen in der Hauptfigur: Maßstabsbalken, 10 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: In-situ-Visualisierung der Wachstumskegeldynamik in akuten organotypischen Schnitten. (A,B) Neuronen und ihre entsprechenden Wachstumskegel, die mit Lyn-mNeonGreen bzw. LifeAct-GFP gekennzeichnet sind. Roter Stern markiert Wachstumskegel von Lyn-mNeonGreen exprimierendem Neuron. Blaues Sternchen, das den Wachstumskegel des LifeAct-GFP-exprimierenden Neurons markiert. (C) Benachbarte Neuronen, die mit dem dualen Plasmidsystem markiert sind, das tRFP (Magenta) und ZsGreen (grün) und ihre entsprechenden Wachstumskegel enthält. Die abgebildeten Wachstumskegel (rechts) befanden sich außerhalb des erfassten Rahmens (links), der kurz nach dem Erwerb des Wachstumskegel-Zeitraffers aufgenommen wurde; Maßstabsstäbe, 5 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 6: Analyse der Axonwachstumsgeschwindigkeit und des Wachstumskegelvolumens. (A) Axon-Tracing auf einem Neuron, das Lyn-mNeonGreen (oben) exprimiert, und seinem entsprechenden Kymographen (unten), der mit ImageJ erzeugt wurde. (B) Rekonstruktion des Z-Stack-Videos des Wachstumskegels mit der Bildanalysesoftware (oben) und dem gleichen Wachstumskegel, der mit dem Oberflächenmesswerkzeug (unten) hervorgehoben wurde. (C) Graphen, die Veränderungen der Wachstumsgeschwindigkeit im Laufe der Zeit für mehrere Axone zeigen. (D) Die durchschnittliche Wachstumsgeschwindigkeit von Axonen wird in (C) quantifiziert. (E) Schaubild der Veränderungen des Wachstumskegelvolumens im Zeitablauf. (F) Das durchschnittliche Volumen der Wachstumskegel wird in (E) quantifiziert; Maßstabsstab, 5 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

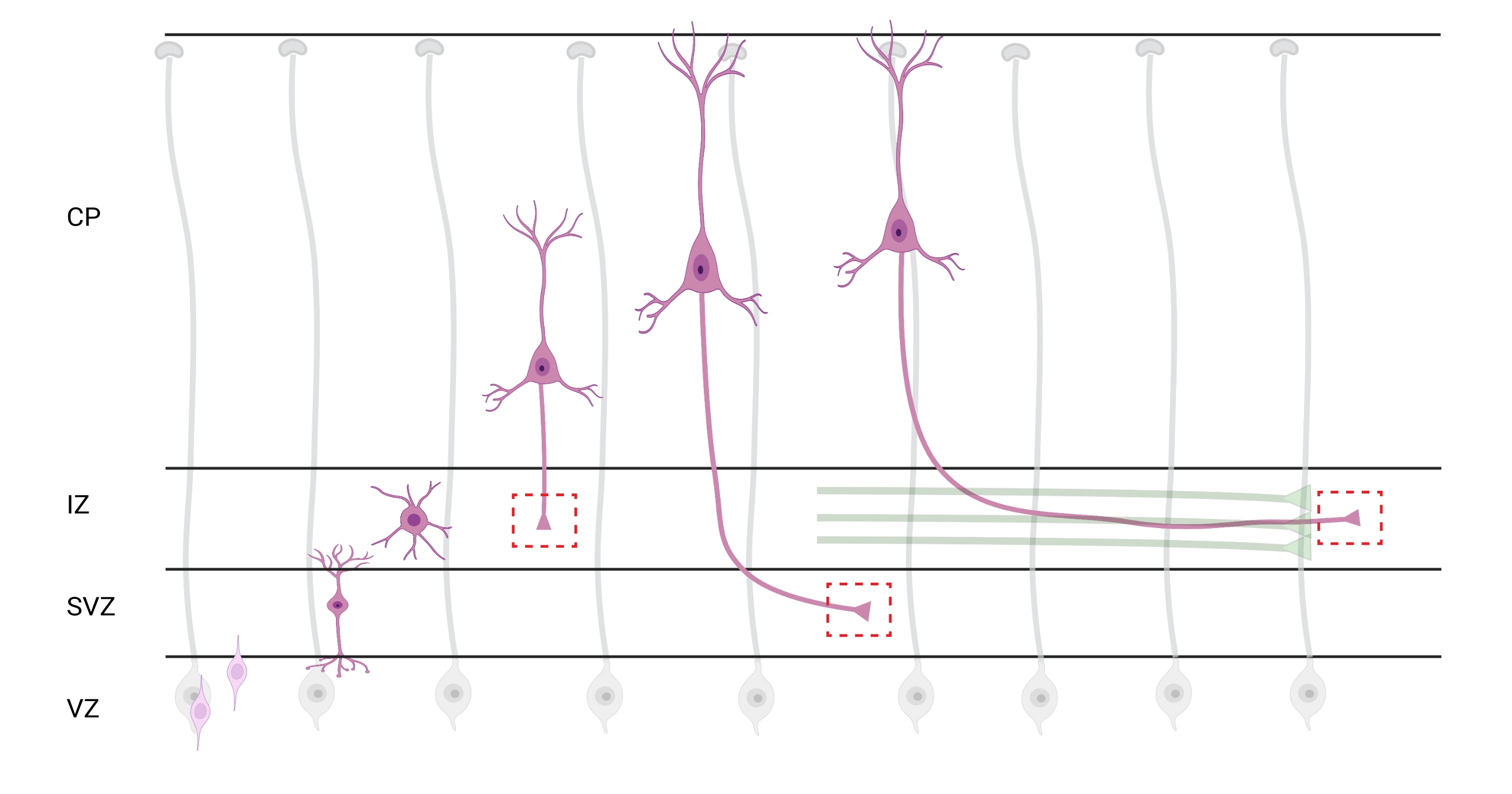
Abbildung 7: Radiale Migration und neuronale Polarisation pyramidaler kortikaler Neuronen. Diagramm, das die Entwicklung pyramidenförmiger kortikaler Neuronen (rosa) veranschaulicht, die radial von der keimventrikulären Zone (VZ) in Richtung der Pia-Oberfläche wandern. Geleitet von radialen Gliaprozessen (grau) etablieren wandernde polarisierte Neuronen einen führenden Prozess, den zukünftigen Dendrit, und einen nachlaufenden Prozess, das zukünftige Axon, die sich weiter nach unten in Richtung der Zwischenzone (IZ) erstrecken. Gestrichelte rote Kästchen stellen die kortikalen Bereiche dar, in denen Wachstumskegel abgebildet wurden. Speziell im IZ, in der subventrikulären Zone (SVZ) oder beim Verbinden von Axonbündeln (grün). Die Illustration wurde mit dem webbasierten Tool BioRender.com erstellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Plasmid | Konzentration (μg/μL) | Verwendungszweck |
| pCAG-lox-STOP-lox-Lyn-mNeonGreen | 0.25 | Markierung von membranzielgerichtetem Protein (Lyn) |
| + | + | |
| p-Wanne-alpha-1-iCre | 0.08 | |
| p-Wanne-alpha-1-LifeAct-GFP | 0.125 | Markierung von filamentösem Aktin (F-Aktin) in Wachstumskegeln |
| pCAG-lox-rox-STOP-rox-tRFP-lox-Lyn-ZsGreen | 1 | Unabhängige Markierung von zwei Populationen benachbarter Neuronen |
| + | + | |
| p-Wanne-alpha-1-iCre | 0.004 | |
| + | + | |
| p-Wanne-alpha-1-Dre | 0.2 |
Tabelle 1: Liste der im Protokoll verwendeten Plasmide. Name, Konzentration und Verwendungszweck jedes verwendeten Plasmids.
Ergänzendes Video 1: In situ Visualisierung der Wachstumskegeldynamik in akuten organotypischen Schnitten. Dynamik von Wachstumskegeln, die mit Lyn-mNeonGreen (oben links) und LifeAct-GFP (unten links) gekennzeichnet sind. Benachbarte Wachstumskegel werden mit dem dualen Plasmidsystem, das tRFP (Magenta; oben rechts) und ZsGreen (grün; unten rechts) enthält, unterschiedlich markiert. Bildgebungsintervall, 2,5 s. Maßstabsstäbe, 5 μm. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Wie die Wachstumskegel ihre Umgebung wahrnehmen und darauf reagieren, um die gleichzeitige Axonausdehnung und -führung zu koordinieren, ist immer noch Gegenstand der Debatte 3,18. Bahnbrechende Studien an 2D-Substraten gaben einen Einblick in die grundlegenden molekularen Mechanismen, die die Kräfte erzeugen, die die Wachstumskegeldynamik während der Axonbildung, des Auswachsens und der Navigation antreiben 2,10,11,12,19. In jüngerer Zeit zeigten Studien in 3D-Matrizen, wie viel Einfluss eine dritte Dimension auf das Verhalten des Wachstumskegels und damit auf das Axonwachstum hat 8,9. Dennoch müssen die komplizierten Mechanismen, die die Wachstumskegeldynamik in vivo anweisen, noch gründlich untersucht werden.
Die Herstellung von organotypischen Schnittkulturen aus IUE- oder EUE-Gehirnen ist weit verbreitet und gut dokumentiert. Es ist zu einem goldenen Standard geworden, der es Wissenschaftlern ermöglicht, Einblicke in die Entwicklung und das Verhalten von Neuronen im lebenden Hirngewebezu gewinnen 20,21. Tatsächlich wurde diese Technik in Kombination mit verschiedenen hochauflösenden bildgebenden Verfahren erfolgreich eingesetzt, um spezifische molekulare Prozesse und morphologische Ereignisse in situ zu visualisieren. Solche Studien umfassen, sind aber nicht beschränkt auf die Axonbildung und -erweiterung 19,22, die kortikale neuronale Migration 19,22,23,24, die Zentrosomendynamik 25,26, die Mikrotubulidynamik 27 sowie die funktionelle Dynamik prä- und postsynaptischer Kompartimente 28,29.
Dieses Protokoll befasst sich mit einer Lücke in der experimentellen Neurobiologie, indem es die Wachstumskegeldynamik der sich entwickelnden kortikalen Neuronen in situ, in ex vivo akuten Gehirnschnittkulturen und die Werkzeuge zur Analyse der erhaltenen Daten visualisiert.
Akute Gehirnschnittkulturen wurden verwendet, um dieses Protokoll zu etablieren, weil sie (1) mit etwas Übung leicht zu erzeugen sind; (2) ein zugängliches System zur Untersuchung von Wachstumskegeln zu präsentieren, die in eine quasi-vollständig physiologische Umgebung eingebettet sind, aber transparent genug sind, um eine hochauflösende Bildgebung lebender Zellen zu ermöglichen; (3) kann für seine Verwendung mit einer Vielzahl von transgenen Mauslinien erweitert werden; (4) bieten in Kombination mit IUE oder EUE praktisch unbegrenztes Potenzial, molekulare Werkzeuge zur Bewertung der Leistung von Wachstumskegeln und Axonen in vivo unter Verlust-/Gewinn-von-Funktionsregimen zusammen mit fluoreszierenden Reportern und Zytoskelettsonden bereitzustellen.
Diese Methodik wurde sowohl im Zusammenhang mit EUE als auch mit IUE beschrieben. Obwohl es sich immer noch um eine sehr zuverlässige Methode handelt, führte EUE zu einer erhöhten Inzidenz von Gehirnschnitten, die ein unorganisiertes RG-Netzwerk zeigten, verglichen mit denen, die mit IUE als Verabreichungsmethode erhalten wurden (Abbildung 4C). Störungen im RG-Array beeinflussen stark die neuronale Migration und das Muster der Axondehnung30,31. Dies sind Schlüsselparameter, die vorhersagen, wo Axone zu einem bestimmten Zeitpunkt zur Analyse zu finden sind und in welcher Art von Umgebung sie navigieren. Gehirnschnitte mit einem signifikant gestörten RG-Netzwerk haben typischerweise eine gestörte kortikale Neuronenschichtung. Dies wiederum erzeugt Axone mit chaotischen Flugbahnen. Daher wird dringend empfohlen, die strukturelle Integrität des RG-Netzwerks zu kontrollieren. Interessanterweise korreliert eine schlechte strukturelle Integrität mit einem erhöhten Alter des embryonalen Gehirns. Tatsächlich wurden solche Effekte bei jüngeren E12.5-E13.5-Embryonen typischerweise nicht beobachtet19.
Das vorliegende Protokoll ist gründlich und unkompliziert. Dennoch gibt es einige kritische Schritte, bei denen besondere Sorgfalt und Aufmerksamkeit auf sich genommen werden muss, um optimale Ergebnisse zu erzielen. Diese wurden ausdrücklich im Protokoll erwähnt und umfassen (1) die Abstimmung der DNA-Menge, die bei der Elektroporation verwendet wird, um eine spärliche Markierung zu erhalten; (2) Vermeidung von Schäden bei der Extraktion von Gehirnen; (3) Kontrolle der Temperatur der Agarose während der Hirnhülle; (4) Fehlerbehebung des idealen Prozentsatzes von Agarose für Gehirne eines bestimmten Alters; und (5) Auswahl von Fluorophoren, deren Erfahrung folgt. Während der Protokolloptimierung wurde die Leistung mehrerer Fluorophore in der Live-Cell-In-situ-Bildgebung getestet. Für dieses Protokoll wurden die monomeren GFP-Varianten EGFP und NeonGreen zur Herstellung der LifeAct- und Lyn-markierten Plasmide gewählt (Abbildung 5A,B). Zusätzlich wurde die RFP-Variante mScarlet getestet und für diesen Aufbau als sehr geeignet befunden (Daten nicht gezeigt). Multimere tRFP (Dimer) und ZsGreen (Tetramer) (Abbildung 5C und ergänzendes Video 1, rechts) wurden ebenfalls getestet. Diese schnell faltenden, superhellen Fluorophore werden empfohlen, wenn die Methode eine schnelle Fluoreszenzsignalerzeugung nach der DNA-Lieferung erfordert.
Eine gängige Praxis bei der Verwendung von Slice-Kulturen ist die Verwendung von Slices aus verschiedenen Gehirnen, um Kontroll- und experimentelle Bedingungen zu testen. Dies stellt eine inhärente Quelle unerwünschter Variabilität dar. Hier wurde ein Expressionssystem verwendet, das eine unabhängige Modifikation benachbarter Neuronen und die Expression von Reportern zur Identifizierung ermöglicht. Beachten Sie, dass es in dieser Demonstration (Abbildung 5C) keine Unterschiede zwischen Neuronen gab, die einen der Fluorophore exprimierten. Zum Beispiel wird eine solche Plasmidmischung in Kombination mit einer transgenen Mauslinie, die ein Cre-sensitives Gen beherbergt, mit tRFP-Neuronen (Dre-sensitive) markiert, die als Wildtyp erhalten geblieben sind. Im Gegensatz dazu markiert das ZsGreen (auch Cre-sensitive) die rekombinierten Neuronen. Daher könnten Wachstumskegel der beiden verschiedenen Genotypen und wahrscheinlich auch Phänotypen gleichzeitig in derselben Gehirnscheibe untersucht werden.
Die Lokalisierung von Axonen und Wachstumskegeln für die Analyse ist eine wichtige Überlegung. Kortikale Neuronen polarisieren, während sie radial von der ventrikulären Zone (VZ) in Richtung der kortikalen Platte (CP) wandern. Während dieses Prozesses bilden Neuronen einen führenden Prozess (einen zukünftigen Dendriten) und einen nachlaufenden Prozess, der zum Axon wird und sich schließlich mit Pionieraxonen in der Zwischenzone (IZ) verbindet und Axontrakte32 bildet. Um axonale Wachstumskegel zu erfassen, wurde daher eine Bildgebung an axonalen Fasern im IZ durchgeführt, einschließlich Axonen, die aus dem CP austreten, und früh erzeugten Axonen, die bereits mit axonalen Bündeln assoziiert sind; oder schließlich in Fasern, die den IZ durchqueren und darunter ausdehnen (Abbildung 7).
Dieses Protokoll macht es möglich, eine hochauflösende Bildgebung von Neuronen in organotypischen Schnitten durchzuführen. In der Vergangenheit war die Lichtstreuung ein erhebliches Problem bei der Abbildung dicker Proben. In den letzten zwei Jahrzehnten ermöglichten umfangreiche Fortschritte in der optischen Technologie die Abbildung dicker Proben. Hier wurde ein Fernabstandsobjektiv verwendet, um kleinere Strukturen, wie z.B. Wachstumskegel, besser sichtbar zu machen. Es ist unvermeidlich, dass dieses Protokoll detailliertere Ereignisse wie den retrograden Aktinfluss oder die Mikrotubulidynamik nicht erfasst. Das Objektiv mit langem Arbeitsabstand, das eine niedrigere numerische Apertur (NA) erfordert, bewahrt Informationen aus dicken Scheiben. Es war jedoch auch möglich, dieses Protokoll an die Verwendung mit Zielen der kürzeren Arbeitsentfernung anzupassen. Dies erforderte einen reibungslosen Transfer der Scheiben in eine Glasbodenschale, um die strukturelle Integrität zu erhalten. Die Verwendung dieser Methode führte jedoch zu einem kürzeren Überleben - ~ 15 h - aufgrund des Verlustes des Gasaustauschs (Daten nicht gezeigt). Im Gegensatz zu 2D-Kulturen nehmen Wachstumskegel in 3D ein größeres Volumen ein und erfordern eine Kompensation von Bewegungsartefakten in der z-Achse. Um die Fähigkeit zur Abbildung detaillierter Ereignisse zu erhöhen, muss moderne konfokale Technologie eingesetzt werden. Daher wird empfohlen, einen schnell scannenden Z-Stack-Motor zu verwenden, wie z-Galvo, der für hochempfindliche konfokale Mikroskope33 verfügbar ist.
Bemerkenswert ist, dass dieses Protokoll drei Haupteinschränkungen aufweist. Erstens ist es oft eine Herausforderung, das Niveau der Expression / Anzahl der exprimierenden Zellen eines bestimmten Plasmids in vivo zu kontrollieren. Dies führt zu einer Variabilität zwischen allen Schichten, selbst wenn die gleiche Plasmidkonzentration beibehalten wird. Daher muss die Auswahl der regulatorischen Elemente in den verwendeten Ausdrucksvektoren sorgfältig vorgegeben werden. Zweitens ist die Abbildung detaillierter Ereignisse mit Membraneinsätzen derzeit nicht möglich. Diese zweite Einschränkung kann mit den im vorhergehenden Absatz vorgeschlagenen methodischen Aktualisierungen überwunden werden. Schließlich sind Wachstumskegel sehr lichtempfindlich und können schnell photogebleicht werden. Daher kann eine häufige Abbildung der Wachstumskegel für nur 5 Minuten mit Laserscanning-Mikroskopen oft die Wachstumskegel kollabieren lassen. In dieser Hinsicht können neue Fortschritte bei der Lichtblattmikroskopie erzeugten Geräten für die Langzeitbildgebung der Gehirnschnitteangepasst werden 34.
Solche Protokolle sollen neue Forschungswege eröffnen, die ein besseres Verständnis dafür ermöglichen, was es braucht, damit ein Wachstumskegel eine komplexe In-vivo-Umgebung liest und darauf reagiert und, was noch wichtiger ist, die Mechanik dieses ausgeklügelten Zusammenspiels entwirrt.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Wir danken Maria Eugenia Bernis für das Fotografieren der Verfahren. Wir danken auch Emily Burnside, Emily Handley, Thorben Pietralla, Max Schelski und Sina Stern für das Lesen und Diskutieren des Manuskripts. Wir danken unseren hervorragenden technischen Assistentinnen Jessica Gonyer, Blanca Randel und Anh-Tuan Pham. Wir würdigen die wertvolle Unterstützung der Lichtmikroskopanlage und der Tieranlage des DZNE. Diese Arbeit wurde von der Deutschen Forschungsgesellschaft (DFG), der International Foundation for Research in Paraplegia (IRP) und Wings for Life (bis F.B) unterstützt. F.B. ist Mitglied des Exzellenzclusters ImmunoSensation2, der SFBs 1089 und 1158 und ist Träger des Roger-De-Spoelberch-Preises.
Materialien
| Name | Company | Catalog Number | Comments |
| Adson Forceps | Fine Science Tools | 11006-12 | |
| Alexa Fluor 488 | Invitrogen | A21202 | Goat Anti-Mouse |
| Alexa Fluor 647 | Invitrogen | A21236 | Goat Anti-Mouse |
| Anti-Vimentin antibody | sigma-Aldrich | V2258-.2ML | Monoclonal mouse, clone LN-6, ascites fluid |
| B27 supplement | ThermoFisher Scientific | 17504044 | |
| Betadine | B. Braun | 3864154 | |
| Biozym Sieve GP Agarose | Biozyme | 850080 | |
| Braunol, Sprühflasche | B. Braun | 3864073 | |
| Buprenorphine (Temgesic) | GEHE Pharma | 345928 | |
| DAPI | sigma-Aldrich | D9542 | |
| DMZ unevirsal electrode puller | Zeitz | NA | |
| Electric razor | Andes | NA | ProClip UltraEdge Super 2-Speed model |
| Enrofloxacin (Baytril) | Bayer | 3543238 | 2,5% (wt/vol) |
| Eppendorf microloader pipette tips | FischerScientific | 10289651 | |
| Fast Green FCF | Sigma-Aldrich | F7252-5G | Dye content ≥ 85 % |
| Fetal Bovine Serum | ThermoFisher Scientific | 10500064 | |
| Fiji 2.1.0 | NIH | NA | https://imagej.net/software/fiji/downloads |
| Fine Scissors | Fine Science Tools | 14058-09 | ToughCut/Straight/9cm |
| FluoroDish Cell Culture Dish | World Precision Instruments | FD5040-100 | |
| Fluoromount Aqueous Mounting Medium | sigma-Aldrich | F4680-25ML | |
| Glucose | MedPex | 3705391 | 5% |
| GlutaMAX Supplement | ThermoFisher Scientific | 35050061 | |
| Glycine | Sigma-Aldrich | G8898 | |
| HBSS | Life Technologies | 14025092 | calcium, magnesium, no phenol red |
| Horse serum | Pan-Biotech | P30-0711 | |
| Imaris 9.7.2 | Bitplane | NA | https://imaris.oxinst.com/products/imaris-for-neuroscientists |
| Isoflurane | Virbac | NA | |
| Isotonic saline solution | B. Braun | 8609261 | 0.90% |
| Leica VT1200 S vibratome | Leica | 14048142066 | |
| LSM 880 with Airyscan | Zeiss | NA | |
| Metacam | Venusberg Apotheke | 8890217 | 5 mg/ml |
| Mice | Janvier Labs | NA | C57BL/6JRj |
| Micro-Adson Forceps | Fine Science Tools | 11018-12 | |
| Micropipette Storage Jar | World Precision Instruments | E210 | 16.16.27 |
| Microsoft Excel | Microsoft | NA | https://www.microsoft.com/en-us/microsoft-365/p/excel/cfq7ttc0k7dx?activetab=pivot:overviewtab |
| Millicell Cell Culture Insert | EMD Millipore | PICM0RG50 | 30 mm, hydrophilic PTFE, 0.4 µm |
| Moria Perforated Spoons | Fine Science Tools | 10370-18 | |
| Moria Spoon | Fine Science Tools | 10321-08 | |
| Neurobasal Medium, minus phenol red | ThermoFisher Scientific | 12348017 | |
| Neuropan-2 supplement | Pan-Biotech | P07-11010 | |
| Normal goat serum | Abcam | ab138478 | |
| Olsen-Hegar Needle Holder with Scissors | Fine Science Tools | 12002-12 | |
| p-Tub-alpha-1-Dre | Addgene | 133925 | |
| p-Tub-alpha-1-iCre | Addgene | 133924 | |
| p-Tub-alpha-1-LifeAct-GFP | Addgene | 175437 | |
| Parafilm | VWR | 52858-000 | |
| Paraformaldehyde | sigma-Aldrich | P6148 | |
| PBS | Sigma-Aldrich | P3813-10PAK | |
| pCAG-lox-rox-STOP-rox-tRFP-lox-Lyn-ZsGreen | Addgene | 175438 | |
| pCAG-lox-STOP-lox-Lyn-mNeonGreen | Addgene | 175257 | |
| Penicillin-Streptomycin | ThermoFisher Scientific | 15140122 | |
| PicoNozzle Kit v2 | World Precision Instruments | 5430-ALL | |
| Platinum Tweezertrodes | Harvard Apparatus | 45-0487 | 1 mm / 3 mm |
| QIAGEN Maxi kit | QIAGEN | 12162 | |
| Reflex wound closure Clip | World Precision Instruments | 500344-10 | 7 mm |
| Sekundenkleber Pattex Mini Trio | Lyreco | 4722659 | |
| Square wave electroporation system ECM830 | Harvard Apparatus | W3 45-0052 | |
| Sterile gauze | Braun Askina | 9031216 | |
| Sterile lubricant eye ointment | Bayer Vital | PZN1578675 | |
| Sterile surgical gloves | Sempermed | 14C0451 | |
| Sucrose | Roth | 4621.2 | |
| Supramid 5-0 surgical silk sutures | B. Braun | NA | |
| Thin-wall glass capillaries | World Precision Instruments | TW100-4 | |
| Triton X-100 | Sigma-Aldrich | X100 | |
| Vannas spring scissors | Fine Science Tools | 15000-03 | |
| µ-Slide 8 Well Glass Bottom | Ibidi | 80827 |
Referenzen
- Schelski, M., Bradke, F. Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Molecular and Cellular Neurosciences. 84, 11-28 (2017).
- Lowery, L. A., Van Vactor, D. The trip of the tip: understanding the growth cone machinery. Nature Reviews: Molecular Cell Biology. 10 (5), 332-343 (2009).
- Stoeckli, E. T. Understanding axon guidance: are we nearly there yet. Development. 145 (10), (2018).
- Bradke, F., Dotti, C. G. The role of local actin instability in axon formation. Science. 283 (5409), 1931-1934 (1999).
- Neukirchen, D., Bradke, F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. Journal of Neuroscience. 31 (4), 1528-1538 (2011).
- Witte, H., Bradke, F. The role of the cytoskeleton during neuronal polarization. Current Opinion in Neurobiology. 18 (5), 479-487 (2008).
- Witte, H., Neukirchen, D., Bradke, F. Microtubule stabilization specifies initial neuronal polarization. Journal of Cell Biology. 180 (3), 619-632 (2008).
- Nichol, R. H., Catlett, T. S., Onesto, M. M., Hollender, D., Gomez, T. M. Environmental elasticity regulates cell-type specific RHOA signaling and neuritogenesis of human neurons. Stem Cell Reports. 13 (6), 1006-1021 (2019).
- Santos, T. E., et al. Axon growth of CNS neurons in three dimensions is amoeboid and independent of adhesions. Cell Reports. 32 (3), 107907(2020).
- Mitchison, T., Kirschner, M. Cytoskeletal dynamics and nerve growth. Neuron. 1 (9), 761-772 (1988).
- Lin, C. H., Thompson, C. A., Forscher, P. Cytoskeletal reorganization underlying growth cone motility. Current Opinion in Neurobiology. 4 (5), 640-647 (1994).
- Myers, J. P., Gomez, T. M. Focal adhesion kinase promotes integrin adhesion dynamics necessary for chemotropic turning of nerve growth cones. Journal of Neuroscience. 31 (38), 13585-13595 (2011).
- Turney, S. G., et al. Nerve growth factor stimulates axon outgrowth through negative regulation of growth cone actomyosin restraint of microtubule advance. Molecular Biology of the Cell. 27 (3), 500-517 (2016).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S., Kriegstein, A. R. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 409 (6821), 714-720 (2001).
- Noctor, S. C., Martinez-Cerdeno, V., Ivic, L., Kriegstein, A. R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nature Neuroscience. 7 (2), 136-144 (2004).
- Ferent, J., Zaidi, D., Francis, F. Extracellular control of radial glia proliferation and scaffolding during cortical development and pathology. Frontiers in Cell and Developmental Biology. 8, 578341(2020).
- Dent, E. W., Gupton, S. L., Gertler, F. B. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harbor Perspectives in Biology. 3 (3), (2011).
- Dupraz, S., et al. RhoA controls axon extension independent of specification in the developing brain. Current Biology. 29 (22), 3874-3886 (2019).
- Azzarelli, R., Oleari, R., Lettieri, A., Andre, V., Cariboni, A. In vitro, ex vivo and in vivo techniques to study neuronal migration in the developing cerebral cortex. Brain Sciences. 7 (5), (2017).
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience. 305, 86-98 (2015).
- Namba, T., et al. Pioneering axons regulate neuronal polarization in the developing cerebral cortex. Neuron. 81 (4), 814-829 (2014).
- Shah, B., et al. Rap1 GTPases are master regulators of neural cell polarity in the developing neocortex. Cerebral Cortex. 27 (2), 1253-1269 (2017).
- Wiegreffe, C., Feldmann, S., Gaessler, S., Britsch, S. Time-lapse confocal imaging of migrating neurons in organotypic slice culture of embryonic mouse brain using in utero electroporation. Journal of Visualized Experiments: JoVE. (125), (2017).
- de Anda, F. C., Meletis, K., Ge, X., Rei, D., Tsai, L. H. Centrosome motility is essential for initial axon formation in the neocortex. Journal of Neuroscience. 30 (31), 10391-10406 (2010).
- Sakakibara, A., et al. Dynamics of centrosome translocation and microtubule organization in neocortical neurons during distinct modes of polarization. Cerebral Cortex. 24 (5), 1301-1310 (2014).
- Schatzle, P., Kapitein, L. C., Hoogenraad, C. C. Live imaging of microtubule dynamics in organotypic hippocampal slice cultures. Methods in Cell Biology. 131, 107-126 (2016).
- Qu, X., Kumar, A., Bartolini, F. Live imaging of microtubule dynamics at excitatory presynaptic boutons in primary hippocampal neurons and acute hippocampal slices. STAR Protocols. 2 (1), 100342(2021).
- Tonnesen, J., Katona, G., Rozsa, B., Nagerl, U. V. Spine neck plasticity regulates compartmentalization of synapses. Nature Neuroscience. 17 (5), 678-685 (2014).
- Buchsbaum, I. Y., Cappello, S. Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development. 146 (1), (2019).
- Rigby, M. J., Gomez, T. M., Puglielli, L. Glial cell-axonal growth cone interactions in neurodevelopment and regeneration. Frontiers in Neuroscience. 14, 203(2020).
- Barnes, A. P., Polleux, F. Establishment of axon-dendrite polarity in developing neurons. Annual Review of Neuroscience. 32, 347-381 (2009).
- Multiphoton Microscope Leica TCS SP8 MP. Microsystems. , Available from: http://www.leica-microsystems.com/products/confocal-microscopes/p/leica-tcs-sp8-sted-one/ (2021).
- ZEISS Lattice Lightsheet 7. Zeiss. , Available from: http://www.zeiss.com/microscopy/int/products/imaging-systems/lattice-lightsheet-7.html (2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten