Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Sammlung, Expansion und Differenzierung primärer humaner Nasensepithelzellmodelle zur Quantifizierung der Zilienschlagfrequenz
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die Entnahme, Expansion und Differenzierung nasaler Epithelzellen zu organotypischen Atemwegsepithelzellmodellen und die Quantifizierung der Zilienschlagfrequenz mittels Live-Cell-Bildgebung und maßgeschneiderten Skripten.
Zusammenfassung
Messungen der Zilienfunktion (Schlagfrequenz, Muster) haben sich als diagnostische Instrumente für Atemwegserkrankungen wie primäre Ziliardyskinesie etabliert. Die breitere Anwendung dieser Techniken ist jedoch durch die extreme Anfälligkeit der Ziliarfunktion gegenüber Änderungen der Umweltfaktoren wie Temperatur, Feuchtigkeit und pH-Wert begrenzt. In den Atemwegen von Patienten mit Mukoviszidose (CF) behindert die Schleimansammlung das Schlagen von Zilien. Die Zilienfunktion wurde in primären Atemwegszellmodellen als Indikator für die Kanalaktivität des CF Transmembrane Conductance Regulator (CFTR) untersucht. Es wurde jedoch eine beträchtliche Variabilität der Zilienschlagfrequenz als Reaktion auf CFTR-modulierende Medikamente festgestellt, selbst bei Patienten mit den gleichen CFTR-Mutationen . Darüber hinaus ist der Einfluss einer dysfunktionalen CFTR-regulierten Chloridsekretion auf die Ziliarfunktion kaum verstanden. Derzeit gibt es kein umfassendes Protokoll, das die Probenvorbereitung von In-vitro-Atemwegsmodellen, die Bildaufnahme und die Analyse der Zilienschlagfrequenz (CBF) demonstriert. Standardisierte Kulturbedingungen und Bildaufnahmen, die unter einem umweltkontrollierten Zustand durchgeführt werden, würden eine konsistente, reproduzierbare Quantifizierung von CBF zwischen Individuen und als Reaktion auf CFTR-modulierende Medikamente ermöglichen. Dieses Protokoll beschreibt die Quantifizierung von CBF in drei verschiedenen Atemwegsepithelzellmodellsystemen: 1) native Epithelschichten, 2) Luft-Flüssigkeits-Grenzflächenmodelle, die auf permeablen Stützeinsätzen abgebildet sind, und 3) extrazelluläre, in Matrix eingebettete dreidimensionale Organoide. Die beiden letzteren replizieren in vivo die Lungenphysiologie, mit schlagenden Zilien und Schleimproduktion. Die Ziliarfunktion wird mit einer Hochgeschwindigkeits-Videokamera in einer umgebungskontrollierten Kammer erfasst. Für die Analyse von CBF werden benutzerdefinierte Skripte verwendet. Die Übertragung von CBF-Messungen in die Klinik soll ein wichtiges klinisches Instrument zur Vorhersage des Ansprechens auf CFTR-modulierende Medikamente pro Patient sein.
Einleitung
Messungen der Zilienschlagfrequenz (CBF) und des Fasermusters haben sich als diagnostische Instrumente für Atemwegserkrankungen wie die primäre Ziliardyskinesie (PCD)1 etabliert. Bei Mukoviszidose (CF) führt eine Dysfunktion des CF-Transmembran-Leitfähigkeitsreglers (CFTR) zu einer Dehydratisierung der Atemwegsoberflächenflüssigkeit und einer gestörten mukoziliären Clearance2. Die Ziliarfunktion wurde in vitro in primären Atemwegszellmodellen als Indikator für die CFTR-Kanalaktivitätuntersucht 3. Allerdings besteht bei CBF eine beträchtliche Variabilität von Patient zu Patient als Reaktion auf CFTR-modulierende Medikamente, selbst bei Patienten mit den gleichen CFTR-Mutationen 3. Darüber hinaus ist der Einfluss einer dysfunktionalen CFTR-regulierten Chloridsekretion auf die Ziliarfunktion kaum verstanden. Derzeit gibt es kein umfassendes Protokoll, das die Probenvorbereitung von In-vitro-Atemwegsmodellen, die Bildaufnahme und die Analyse von CBF demonstriert.
Nasenepithelblätter, die aus Nasenschleimhautbürsten isoliert wurden, werden direkt für Messungen der Ziliarfunktion für die PKD-Diagnoseverwendet 4. Obwohl es keine Kontrolle über die Größe oder Qualität der erhaltenen Nasensepithelblätter gibt, variiert CBF je nachdem, ob es an einzelnen Zellen oder Zellblättern und an epithelialen Blattflanelländern gemessen wird, die gestört oder ungestört sind5. Daher können sekundäre Dyskinesien, die durch Zellschäden während der Entnahme von Nasenschleimhautbürsten verursacht werden, die CBF beeinflussen. Die primäre Zellkultur von Nasensepithelzellen und ihre Differenzierung an der Luft-Flüssigkeits-Grenzfläche (ALI) oder in dreidimensionaler Basalmembranmatrix in Flimmereitelepithelorganoide führen zu Zilien, die frei von sekundären Dyskinesiensind 4,6,7,8. Atemwegepithelzellen, die bei ALI differenziert wurden (im Folgenden als ALI-Modelle bezeichnet), gelten als wichtiges sekundäres diagnostisches Hilfsmittel, das die Ziliarschlagmuster und die Häufigkeit von Ex-vivo-Nasenschleimhautbürsten repliziert6 und die Analyse der Ziliarultrastruktur, des Schlagmusters und der Schlagfrequenz unter Beibehaltung patientenspezifischer Defekte ermöglicht 9 . Es gibt jedoch Diskrepanzen in den Methoden, die zur Erstellung dieser pseudogeschichteten, mukoziliären differenzierten Zellmodelle verwendet werden. Unterschiedliche Kulturexpansions- oder Differenzierungsprotokolle könnten unterschiedliche epitheliale Phänotypen (flimmerig oder sekretorisch)10 induzieren und zu signifikanten Unterschieden in CBF11 führen. CBF wurde quantifiziert in Nasepithelbürsten 4,6,12,13,14,15,16, Atemwegsepithelorganoiden 14,17,18 und ALI-Modellen 3,4,6,13,19,20, 21. Unter diesen Protokollen gibt es jedoch große Variabilitäten, und oft werden viele Parameter nicht kontrolliert. Zum Beispiel wird in einigen Studien CBF in situ abgebildet, während die Zellen des ALI-Modells auf dem durchlässigen Stützeinsatz 3,19,20,21 verbleiben, wieder andere kratzen die Zellen aus dem durchlässigen Stützeinsatz und bilden sie in den Medien 4,6,13 ab.
Darüber hinaus ist die breitere Anwendung von Techniken zur Messung der Ziliarfunktion durch die extreme Anfälligkeit der Ziliarfunktion gegenüber Veränderungen der Umweltfaktoren begrenzt. Umweltfaktoren wie Temperatur 22, Luftfeuchtigkeit23,24 und pH25,26 beeinflussen die Ziliarfunktion und müssen reguliert werden, um CBF genau zu quantifizieren. Die verschiedenen physiologischen Parameter, die in verschiedenen Laboratorien verwendet werden, und wie sie die CBF beeinflussen, wurden bereitsüberprüft 27.
Verschiedene Bildgebungstechnologien und Ansätze zur CBF-Messung werden in der Literatur beschrieben. Für die PCD-Diagnostik wird die Videomikroskopie zur Messung der Ziliarfunktion28,29 eingesetzt. Vor kurzem wurde ein Videoanalysealgorithmus, der auf differentieller dynamischer Mikroskopie basiert, verwendet, um sowohl die CBF- als auch die Zilienkoordination in den ALI-Modellen für Atemwegepithelzellenzu quantifizieren 3,30. Diese Methode ermöglicht die Charakterisierung von Ziliarschlägen in Atemwegsepithelzellen schnell und vollautomatisch, ohne dass Regionen segmentiert oder ausgewählt werden müssen. Verschiedene Methoden zur Bildgebung und Quantifizierung von CBF können zu den Unterschieden beitragen, die in der Literatur in CBF berichtet werden (Supplementary File 1).
Ein Protokoll von der Kultur bis zur Quantifizierung zur Rationalisierung bestehender Methoden, zur Standardisierung der Kulturbedingungen und zur Bildaufnahme, die unter streng umweltkontrollierten Bedingungen durchgeführt werden, würde eine konsistente, reproduzierbare Quantifizierung von CBF innerhalb und zwischen Individuen ermöglichen.
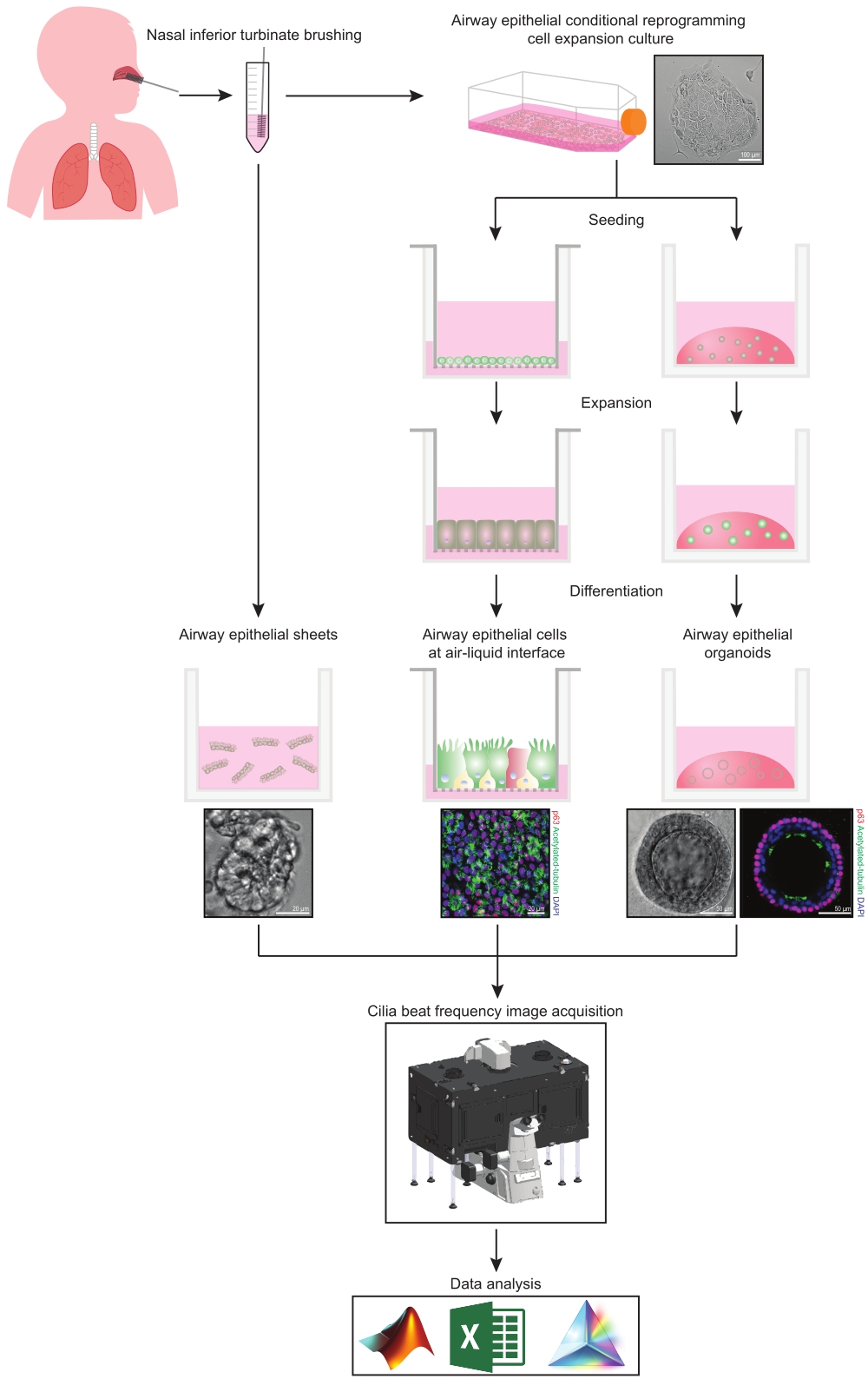
Dieses Protokoll bietet eine vollständige Beschreibung der Sammlung von Epithelzellen, der Expansions- und Differenzierungskulturbedingungen und der Quantifizierung von CBF in drei verschiedenen Atemwegepithelzellmodellsystemen nasalen Ursprungs: 1) native Epithelblätter, 2) ALI-Modelle, die auf permeablen Stützeinsätzen abgebildet wurden, und 3) extrazelluläre Matrix (ECM)-eingebettete dreidimensionale Organoide (Abbildung 1 ). Nasenepithelzellen, die aus Nasenbürsten der unteren Nasenmuscheln gewonnen werden, werden als Vertreter des Atemwegsepithels verwendet, da sie ein wirksamer Ersatz für Bronchialepithelzellen31 sind, während sie das invasive Verfahren überwinden, das mit dem Sammeln von Bronchialbürsten verbunden ist. Die Conditional Reprogramming Cell (CRC) Methode wird verwendet, um primäre Atemwegsepithelzellen für die Erstellung von ALI-Modellen und dreidimensionalen Organoiden zu erweitern. Die bedingte Reprogrammierung von Atemwegepithelzellen in einen stammzellähnlichen Zustand wird durch Kokultur mit wachstumshemmendem Fibroblasten-Feederzellsystem und Rho-assoziierter Kinase (ROCK)-Inhibitor32 induziert. Wichtig ist, dass die CRC-Methode die Verdopplung der Population in Atemwegepithelzellen erhöht und gleichzeitig ihr gewebespezifisches Differenzierungspotenzial beibehält33,34. In allen Epithelzellmodellen der Atemwege wird die Ziliarfunktion in einer temperaturgesteuerten Kammer mit einer Hochgeschwindigkeits-Videokamera mit standardisierten Bildaufnahmeeinstellungen erfasst. Für die Quantifizierung von CBF werden maßgeschneiderte Skripte verwendet.

Abbildung 1: Schematische Darstellung des Workflows. Nach dem Bürsten des Nasenmuschels werden die Atemwegsepithelzellen auf zwei Arten verwendet. Entweder werden Atemwegsepithelschichten isoliert und die Zilienschlagfrequenz wird sofort abgebildet, oder Atemwegsepithelzellen werden über die bedingte Reprogrammierungszellmethode erweitert. CRC-expandierte Atemwegsepithelzellen werden differenziert, um Atemwegepithelzellen an einer Luft-Flüssigkeits-Grenzfläche oder Atemwegepithel-Organoidkulturen zu etablieren. Die Bildgebung der Ziliarschlagfrequenz wird mit einem Lebendzell-Bildgebungsmikroskop mit einer Heiz- und Feuchtigkeits-Umgebungskammer und einer wissenschaftlichen Kamera mit schneller Bildrate (>100 Hz) aufgenommen. Die Datenanalyse wird mit benutzerdefinierten Skripten durchgeführt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Protokoll
Die Genehmigung für die Studie wurde vom Sydney Children's Hospital Network Ethics Review Board (HREC/16/SCHN/120) erteilt. Vor der Entnahme von Bioproben wurde die schriftliche Zustimmung aller Teilnehmer (oder des Vormunds der Teilnehmer) eingeholt.
1. Vorbereitungen zur Erstellung von Atemwegepithelzellmodellen
- Bereiten Sie Nasenzellentnahmemedien vor, indem Sie 80% Dulbeccos modifiziertes Adlermedium und 20% fötales Rinderserum kombinieren. Ergänzung mit 1 μL / ml Penicillin / Streptomycin. Bei 4 °C bis zu 3 Monate lagern.
- Beschichten Sie die Kolben oder durchlässigen Stützeinsätze je nach Bedarf mit Kollagenlösung gemäß den Schritten 1.2.1-1.2.4. Lagern Sie keine kollagenbeschichteten Gefäße langfristig.
- Eine 1:100-Verdünnung der Kollagenlösung Typ I (3 mg/ml Brühe) mit phosphatgepufferter Kochsalzlösung (PBS) auf eine Endkonzentration von 0,03 mg/ml herstellen. Gut mischen.
- Beschichten Sie die Zellkulturkolben (Abschnitt 4) mit 160 μL/cm 2 (d. h. 4 ml pro T25-Kolben) und durchlässigen Trägereinsätzen (Abschnitt 5) mit 455 μL/cm2 (d. h. 150 μL pro 6,5 mm Einsatz) der vorbereiteten Kollagenlösung.
- Bei 37 °C 2-24 h inkubieren.
- Entfernen Sie die Kollagenlösung vor der Aussaat der Zellen durch Pipette oder Vakuumsauger. Waschen Sie das Gefäß nicht, bevor Sie Zellen säen.
- Bereiten Sie CRC-Medien (Conditional Reprogramming Cell) vor, indem Sie die in Tabelle 1 aufgeführten Komponenten32 kombinieren. Filter sterilisieren mit einem Flaschen-Top-Vakuumfiltersystem. Bei 4 °C bis zu 2 Monate lagern.
- Fügen Sie am Tag der Anwendung den humanen epidermalen Wachstumsfaktor, den ROCK-Inhibitor und Antibiotika hinzu, wie in Tabelle 1 angegeben.
| Bestandteil | Volumen |
| DMEM, hohe Glukose | 156,7 ml |
| DMEM/F-12, HEPES | 313,3 ml |
| Hydrocortison | 55,6 μL |
| Insulin | 1,25 ml |
| Cholera-Toxin | 21 μL |
| Adenin | 1,2 ml |
| HI-FBS | 25 ml |
| Penicillin-Streptomycin | 5 ml |
| Humaner epidermaler Wachstumsfaktor | 1 μL/ml |
| ROCK-Inhibitor | 1 μL/ml |
| Fungizone | 2 μl/ml |
| Tobramycin | 2 μL/ml |
| Ceftazidim-Hydrat | 4 μL/ml |
| Gentamicin-Lösung | 1 μL/ml |
Tabelle 1: Komponenten für 500 mL bedingt reprogrammierendes Zellmedium
2. Sammlung von Nasenbürsten der unteren Nasenmuscheln
HINWEIS: Dieser Abschnitt des Protokolls erfordert ein Sammelröhrchen (50 ml) mit Nasenzellensammelmedien, Zytologiebürsten, Taschentüchern und geeigneter persönlicher Schutzausrüstung. Vermeiden Sie das Bürsten während einer Infektion der oberen Atemwege. Es besteht ein geringes Blutungsrisiko, das erhöht ist, wenn eine Entzündung vorliegt. Wenn der Zweck des Bürstens darin besteht, Atemwegsepithelblätter für Ex-vivo-CBF-Messungen zu erhalten, sollte das Bürsten mindestens 6 Wochen nach einer Infektion der oberen Atemwege erfolgen; Idealerweise mehr als 10 Wochen nach der Infektion35.
- Bereiten Sie das Nasenzellentnahmemedium vor (Abschnitt 1) und halten Sie das Röhrchen auf Eis.
- Beschreiben Sie dem Teilnehmer das Prozedere als unangenehm. Erklären Sie, dass während des Bürstens ein vollständiges Gefühl im Nasenloch zu spüren ist, ähnlich wie beim Springen in den Ozean / Pool und beim Wasser, das in den Nasengang strömt. Weisen Sie die Teilnehmer darauf hin, dass das Verfahren die Produktion von Tränen als Reflex induziert.
- Beurteilen Sie, welche Positionierung für den Teilnehmer geeignet ist. Legen Sie den Teilnehmer in Rückenlage, wenn eine Untersuchungsliege vorhanden ist, da die Rückenlage eine Bewegung des Kopfes des Teilnehmers von der Bürste während des Eingriffs verhindert. Alternativ setzen Sie den Teilnehmer neben eine Wand, gegen die er den Kopf zurückdrücken kann.
- Inspizieren Sie den Nasengang. Beachten Sie Septumabweichung, Polypen und andere anatomische Anomalien, die den Durchgang der Bürste im Nasengang beeinflussen und das Blutungsrisiko erhöhen können.
- Reinigen Sie die Nase von überschüssigem Schleim, indem Sie die Teilnehmer bitten, ihre Nase in ein Taschentuch zu blasen.
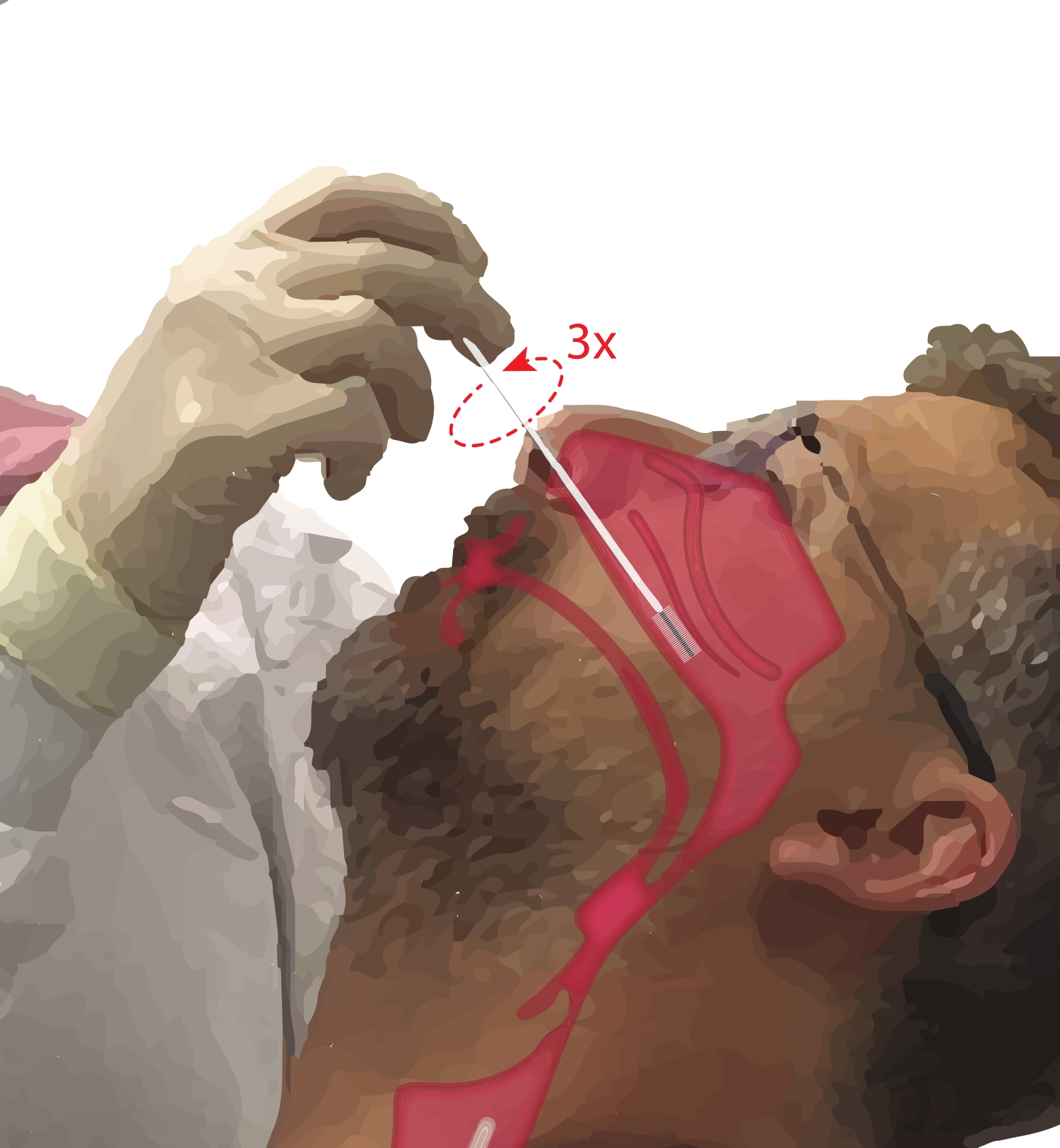
- Bitten Sie den Teilnehmer, durch den Mund zu atmen. Nehmen Sie einen Zytologiepinsel in die dominante Hand. Während Sie die fünfte Ziffer auf das Kinn des Teilnehmers legen, um die Hand zu verankern, führen Sie den Zytologiepinsel in den Nasengang des Teilnehmers ein (Abbildung 2). Führen Sie die Bürste bei ~ 45 ° in das Gesicht des Teilnehmers ein, um durch den Nasengang zu gelangen.
- Drehen Sie die Bürste aufrecht, so dass sie senkrecht zum Gesicht des Teilnehmers steht. Schieben Sie die Bürste vorsichtig, aber fest gegen die seitliche Wand der Nase unter dem unteren Nasenmuscheln, bis sie sich im mittleren bis hinteren Teil der unteren Nasenmuschel befindet.
HINWEIS: Vermeiden Sie übermäßiges Einfügen; Wenn ein plötzlicher Abfall des Widerstands zu spüren ist, wurde der Nasenpharynx betreten, und die Bürste sollte zurückgezogen werden, bis der Widerstand wieder vom Proceduralisten gefühlt wird. - Drehen Sie die Bürste bis zu dreimal um 360°. Entfernen Sie die Bürste vorsichtig in umgekehrter Reihenfolge des Einführmanövers, damit sich die Zellen nicht von der Bürste lösen.
- Legen Sie die Bürste in das vorbereitete Auffangröhrchen mit Nasenzellensammelmedien. Legen Sie das Auffangrohr auf Eis.
- Wiederholen Sie das Zähneputzen im zweiten Nasenloch, wenn der Teilnehmer einverstanden ist / eine große Anzahl von Zellen benötigt wird (z. B. um eine Zellkultur zu initiieren).
HINWEIS: Das gleiche Nasenloch kann erneut gebürstet werden, wenn sich keine sichtbaren Blutzellen auf der Bürste befinden, wobei jedoch zu beachten ist, dass das Blutungsrisiko mit einem zweiten Bürsten im selben Nasenloch leicht erhöht ist.

Abbildung 2: Sammlung von Nasepithelzellen. Illustration der Lage des Zytologiepinsels im mittleren bis hinteren Teil des unteren Nasenmuschels. Diese Position wird erreicht, indem die Bürste durch die Nasenlöcher eingeführt, die Bürste in einen 90°-Winkel zum Gesicht gedreht und die Bürste entlang des Nasengangs unterhalb des unteren Nasenmuschels geführt wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Vorbereitung von Atemwegsepithelblättern
HINWEIS: Dieser Abschnitt des Protokolls erfordert ein Sammelröhrchen (Zytologiebürste(n) + 1 ml Nasenzellsammelmedium) (Abschnitt 2) und eine 96-Well-Platte mit flachem Boden. Wenn Sie Nasenmuschelbürsten zum Zwecke der Bildgebung von Atemwegsepithelblättern sammeln, verwenden Sie nur 1 ml antibiotikafreie Nasenzellentnahmemedien. Andernfalls sind Epithelblätter für die Bildgebung zu verteilt.
- Schwenken Sie vorsichtig das Auffangröhrchen mit der (den) Zytologiebürste(n), um die Atemwegsepithelblätter von der/den Bürste(n) zu entfernen.
- Sammeln Sie alle Medien und Zellen mit einer P1000-Pipette. Geben Sie 5-6 Tropfen in eine Vertiefung einer 96-Well-Platte mit flachem Boden. Wiederholen Sie diesen Vorgang für ungefähr sieben Bohrungen.
- Übertragen Sie die Platte gemäß Schritt 7.1.4 auf das Mikroskop und folgen Sie dem Rest von Abschnitt 7, um die Zilienschlagfrequenz abzubilden.
- Bild Epithelblätter (Abbildung 1) und nicht einzelne ungebundene Zellen, da nachgewiesen wurde, dass sich die Ziliarfunktion zwischen Epithelblättern und einzelnen nicht angeschlossenen Zellen unterscheidet5.
4. Expansion und Aufrechterhaltung der Epithelzellen der Atemwege
- Atemwegepitheliale bedingte Reprogrammierung Zellexpansionskultur
HINWEIS: Kollagenlösungsbeschichtetes Gefäß (Abschnitt 1), bestrahlte embryonale Futterzellen der Maus (NIH-3T3), Conditional Reprogramming Cell (CRC) Medien (Abschnitt 1), zytologische Bürste(n) in Nasenzellentnahmemedien (Abschnitt 2).- Plattenbestrahlte Feederzellen in vorbereitete(r) mit Kollagenlösung beschichtete Kulturgefäße mit einer Aussaatdichte von 8.000 Zellen/cm2 mindestens 2 h und höchstens72 h vor der Kokultur mit Atemwegepithelzellen (siehe36 für Feederzellkultur und Bestrahlung).
- Die gebürsteten Zellen im Sammelröhrchen (Zytologiebürste(n) + Nasenzellsammelmedium) in den Wirbel auf Eis überführen. Bei niedriger Geschwindigkeit Wirbelrohr 10 s an, 10 s aus (zwischendurch auf Eis halten), um Zellen von der Bürste(n) zu entfernen. Kräftiges Wirbeln kann die Lebensfähigkeit der Zellen verringern. Untersuchen Sie die Bürste(n), um zu überprüfen, ob der Schleim noch haftet. Wenn ja, wiederholen Sie den Wirbel.
- Bringen Sie das/die Röhrchen(s) auf Eis zurück in die Biosicherheitswerkbank. Verwenden Sie eine serologische Pipette, um die Medien aus dem Sammelröhrchen in ein neues Röhrchen (Röhrchen B) zu überführen, wobei die zytologische Bürste zurückbleiben. Zentrifugenröhrchen B bei 300 × g für 7 min bei 4 °C.
- Röhrchen B aus der Zentrifuge nehmen, Überstand entsorgen. Wenn der Schleim sichtbar ist, waschen Sie das Pellet mit weiteren 5 ml Nasenzellsammelmedium und zentrifugieren Sie erneut.
- Fügen Sie 1 ml CRC-Medien hinzu, um das Zellpellet in Röhrchen B zu resuspendieren. Führen Sie die Zellen mit einer 5-ml-serologischen Pipette in kreisförmiger Bewegung durch ein Zellsieb, das auf einem 50-ml-Röhrchen (Röhrchen C) platziert ist.
- Wiederholen Sie mehrmals, um eine einzellige Suspension zu bilden. Sammeln Sie die Restmedien vom Boden des Siebs und integrieren Sie sie in das Medium. Entsorgen Sie das Zellsieb.
- Nehmen Sie mit einer serologischen 5-ml-Pipette 1 ml Medien aus Röhrchen C und geben Sie es in ein Mikrozentrifugenröhrchen.
- Nehmen Sie 10 μL dieser Zellsuspension und geben Sie sie in das Mikrozentrifugenröhrchen, das mit 10 μL Trypanblau voraliquotiert ist. Mischen Sie gut und verwenden Sie sofort einen automatisierten Zellzähler, um die Zellzahl und Lebensfähigkeit aufzuzeichnen.
- Die Epithelzellen der Atemwege werden in den mit bestrahlten Futterzellen vorbestrahlten T25-Kolben ausgesät.
- Erhaltung und Dissoziation von Epithelzellen der Atemwege
HINWEIS: CRC-Medien müssen auf 37 °C erwärmt werden, indem sie in ein temperaturgesteuertes Laborwasserbad oder ein Perlenbadgerät gegeben werden, bevor sie in die Zellen gegeben werden.- Überprüfen Sie die Zellen regelmäßig unter dem Zellkulturmikroskop (4× Objektivlinse) auf Anhaftung, Kontamination, Morphologie und Konfluenz.
- Wechseln Sie CRC-Medien jeden zweiten Tag. Wenn reprogrammierte Zellen beobachtet werden (Abbildung 1) und keine Kontamination vorhanden ist, reduzieren oder entfernen Sie Antibiotika.
- Wenn Zellen eine Konfluenz von 90% erreichen, verwenden Sie eine Doppeltrypsinmethode32 , um die Zellen zu dissoziieren und eine Zellzählung durchzuführen, wie in Schritt 4.1.8 beschrieben (siehe Zusatzdatei 2 für Zelldissoziation und Einfrieren).
5. Aussaat und Differenzierung von Atemwegsepithelzellen und Pflege differenzierter ALI-Modelle
- Aussaat von Atemwegsepithelzellen zu durchlässigen Stützeinsätzen
- Die mit Kollagenlösung beschichteten permeablen Stützeinsätze (Abschnitt 1) werden aus dem CO2-Inkubator in die Biosicherheitswerkbank überführt. Die Kollagenlösung absaugen und verwerfen. 750 μL Expansionsmedium (antibiotikafrei) in das Basalkompartiment der durchlässigen Stützeinsätze geben.
- Die dissoziierten Zellen oder aufgetauten Zellen auf Eis werden in die Biosicherheitswerkbank überführt. Fügen Sie das Volumen des Expansionsmediums, das benötigt wird, um 200.000-250.000 Zellen in 150 μL zu säen, in das apikale Kompartiment jedes permeablen Stützeinsatzes ein.
- Achten Sie darauf, keine Blasen zu erzeugen; Mischen Sie gut, um sicherzustellen, dass die Zellen homogen und in Suspension sind. Geben Sie 150 μL der Zellsuspension auf die apikale Seite jedes durchlässigen Stützeinsatzes.
- Resuspendieren Sie die Zellen nach der Aussaat alle drei durchlässigen Stützeinsätze, um eine homogene Zellsuspension zu erhalten.
- Jeden zweiten Tag, bis sich eine konfluierende Zellmonoschicht gebildet hat (normalerweise am Tag 4 nach der Aussaat), verwerfen Sie das Medium und fügen Sie ein frisches Expansionsmedium hinzu, das auf Raumtemperatur (RT, 15-25 °C) erwärmt ist.
- Differenzierung von Atemwegsepithelzellen an der Luft-Flüssigkeits-Grenzfläche
- Warme ALI-Medien (antibiotikafrei) bis RT (15-25 °C).
- Entfernen Sie das Expansionsmedium und wechseln Sie zu Differenzierungsmedien (ALI) sowohl an apikalen als auch an basalen Kompartimenten.
- Nach 2 Tagen Kultur in untergetauchten ALI-Medien aspirieren und entsorgen Sie die Medien.
- Fügen Sie nur 750 μL ALI-Medien in das Basalfach hinzu, um eine Luft-Flüssigkeits-Schnittstelle zu erzeugen.
HINWEIS: Wenn nach 1 Woche Kultur die Monoschicht nicht konfluent ist und immer noch Löcher beobachtet werden, haben Zellen möglicherweise nicht mehr die Fähigkeit, sich in die Hohlraumregionen auszudehnen, erwägen Sie, die Atemwegsepithelzellen zu verwerfen.
- Aufrechterhaltung eines differenzierten ALI-Modells und Schleimentfernung
- Wechseln Sie das apikale und basale Medium jeden zweiten Tag bis zur vollständigen Differenzierung (Tag 21-25 nach der Etablierung der Luft-Flüssigkeits-Grenzfläche).
- Einmal pro Woche Schleim von der apikalen Seite nach den Schritten 5.3.3-5.3.4 waschen.
- Warme PBS auf RT (15-25 °C).
- Geben Sie 200 μL PBS in das apikale Fach. Inkubieren imCO2-Inkubator für 10 min. Verwenden Sie eine Aspirationsvorrichtung oder Pipette, um das PBS zu entfernen.
6. Dreidimensionale Atemwegsepithelorganoide
- Vorbereitungen für die atepitheliale Organoidkultur der Atemwege
- 24-Well-Platten(n) in einenCO2-Inkubator geben, um sich über Nacht auf 37 °C zu erwärmen.
- Eine 10-ml-Durchstechflasche ECM (Table of Materials) gemäß den Anweisungen des Herstellers auf Eis auftauen. Bereiten Sie 500 μL Aliquots (einmalige Anwendung) vor, um die Anzahl der Gefrier-Tau-Zyklen zu minimieren.
HINWEIS: Es wird empfohlen, ECM mit einer Proteinkonzentration >10,5 mg / ml für die besten Kulturergebnisse zu verwenden. Eine geringere Konzentration beschleunigt den Zerfall der ECM-Kuppel und erhöht das Auftreten von apikal nach außen gerichteten Organoiden. - Verwenden Sie das Airway Organoid Kit (Table of Materials), um Atemwegs-Organoid-Seeding-Medien (AOSM) und Differenzierungsmedien (AODM) gemäß den Anweisungen des Herstellers vorzubereiten.
- Bereiten Sie die organoiden Basalmedien der Atemwege gemäß Tabelle 2 vor.
| Bestandteil | Volumen |
| Fortschrittliches DMEM/F-12 | 500 ml |
| HEPES | 5 ml |
| Alanyl-Glutamin | 5 ml |
| Penicillin-Streptomycin | 5 ml |
Tabelle 2: Komponenten der organoiden Basalmedien der Atemwege
- Verwenden Sie die Anzahl der in Abschnitt 4.2 dissoziierten lebenden Atemwegsepithelzellen, um zu berechnen, wie viele Vertiefungen bei einer Aussaatdichte von 10.000 Zellen ausgesät werden können (siehe Tabelle 3).
- Berechnen Sie das Gesamtvolumen von ECM und AOSM, das benötigt wird, um 1 x 50 μL 90% ECM-Dome (45 μL ECM und 5 μL AOSM) pro Vertiefung zu erzeugen.
HINWEIS: Die empfohlene Seeding-Dichte von 10.000 Zellen pro Vertiefung gilt für CRC-expandierte Nasepithelzellen bei Passage 1. Spätere Durchgangszellen benötigen möglicherweise eine höhere Seeding-Dichte, um die Bildung der gleichen Anzahl von Organoiden zu erreichen.
| Anzahl der Brunnen | Anzahl der Zellen | Anzahl der Kuppeln | Band von Matrigel ECM | Band von AOSM |
| 1 | 10.000 Zellen | 1 | 45 μL x 1,1 | 5 μL x 1,1 |
| 2 | 20.000 Zellen | 2 | 90 μL x 1,1 | 10 μL x 1,1 |
| 5 | 50.000 Zellen | 5 | 225 μL x 1,1 | 25 μL x 1,1 |
| ......... | ......... Zellen | ......... | .........μL x 1,1 | .........μL x 1,1 |
Tabelle 3: Berechnungen zur Aussaat von Atemwegsepithelzellen in ECM-Domen
- Aussaat von Atemwegsepithelzellen in ECM-Domen
HINWEIS: Halten Sie ECM immer auf Eis und führen Sie alle Schritte mit ECM auf Eis durch, da ECM bei Temperaturen >10 ° C zu erstarren beginnt.- Resuspendieren Sie die in Abschnitt 4.2 dissoziierten Atemwegepithelzellen mit dem berechneten Volumen von 90% ECM gemäß Tabelle 3.
- Halten Sie die Pipette in einem 90°-Winkel (vertikal) so nah wie möglich am Boden des Vertiefungs und dosieren Sie 50 μL (bis zum ersten Stopp, um Blasen zu vermeiden) der ECM-Zellsuspension in die Mitte der Vertiefung. Vermeiden Sie es, die Wand des Brunnens zu berühren.
- Die Platte bei 37 °C für 20 min inkubieren, bis das ECM erstarrt. Während sich das ECM verfestigt, erwärmen Sie AOSM auf RT (15-25 °C), um zu verhindern, dass es bei der Zugabe zu einer erneuten Verflüssigung und Desintegration der ECM-Kuppel kommt.
- Fügen Sie 500 μL erwärmtes AOSM zu jeder Vertiefung hinzu, indem Sie die Wand des Brunnens abgeben. Pipettieren Sie Medien nicht direkt auf die ECM-Kuppel.
- Wechseln Sie die Medien alle 2 Tage für 4-7 Tage. Um Medien abzusaugen, neigen Sie die Platte in einem Winkel von 45° und saugen Sie sie vom unteren Rand des Bohrlochs weg von der ECM-Kuppel ab.
- Nach 4-7 Tagen die Organoiddifferenzierung einleiten, indem Sie 500 μL AODM (15-25 °C) in jede Vertiefung geben und das Medium alle 2 Tage für 7 Tage wechseln.
- Replating von Atemwegepithelorganoiden am Tag 7 der Differenzierung
HINWEIS: Die Replattierung von Atemwegepithelorganoiden ist notwendig, da der Rand der ECM-Kuppeln während der 2-wöchigen Kulturperiode allmählich zerfällt. Epithelorganoide der Atemwege am Rand der Kuppel können verloren gehen (in die Medien verschoben) oder apikal nach außen gerichtet ausgerichtet sein, wenn sie nicht vollständig in ECM eingebettet sind. Der Replating-Schritt "reinigt" auch die ECM-Kuppel, indem Zellen / Trümmer entfernt werden, die Organoide nicht erfolgreich bilden.- Saugen Sie die Medien aus jedem Bohrloch ab. Fügen Sie 500 μL kaltes Atemwegs-Organoid-Basalmedium (im Folgenden Basalmedien genannt) in jede Vertiefung ein.
- Verwenden Sie die P1000-Pipette, da diese Pipettenspitze die größte Öffnung hat und die Wahrscheinlichkeit verringert, dass Organoide während des Pipettierens platzen. Stellen Sie die Pipette auf 350 μL ein, um Blasen zu vermeiden, und pipetten Sie dann vorsichtig auf und ab, um die ECM-Kuppel in jeder Vertiefung zu stören. Sammeln Sie alle ECM/Basalmedien in ein 15-ml-Zentrifugenröhrchen.
- Spülen Sie jede Vertiefung mit 500 μL kalten Basalmedien aus. Sammeln Sie die Basalmedien, die alle verbleibenden ECM und Organoide enthalten, in dasselbe 15-ml-Zentrifugenröhrchen wie oben.
- Bei 300 x g 5 min bei 4 °C zentrifugieren. Von den drei Schichten, die nach dem Zentrifugieren sichtbar sind - (1) Überstand, (2) ECM mit Zelltrümmern (flauschig) und (3) Pellet mit Organoiden - verwerfen Sie den Überstand und die ECM-Schicht und konservieren Sie das Organoidpellet.
- Fügen Sie 1 ml kaltes Basalmedium zum Organoidpellet hinzu und pipetten Sie vorsichtig auf und ab, um die verbleibende ECM zu trennen. 6 ml kalte Basalmedien in die Tube geben und vorsichtig mischen.
- Bei 300 × g 5 min bei 4 °C zentrifugieren. Verwerfen Sie den Überstand.
- Wenn noch überschüssige ECM sichtbar ist, wiederholen Sie die Schritte 6.3.5 bis 6.3.6, um eine weitere Wäsche durchzuführen.
- Resuspendieren Sie das Organoidpellet mit einem geeigneten Volumen von 90% ECM (verwenden Sie AODM anstelle von AOSM), um ~ 30 Organoide pro 50 μL der Kuppel zu platten.
- Überprüfen Sie die Dichte der Organoide unter dem Zellkulturmikroskop (4x Objektivlinse) nach der Beschichtung der ersten Kuppel. Wenn zu dicht, fügen Sie eine zusätzliche 90% ECM hinzu, um die gewünschte Dichte von ~ 30 Organoiden zu erreichen.
- Befolgen Sie die Schritte 6.2.3 bis 6.2.4, um die ECM zu verfestigen und die Zellen jeden zweiten Tag mit 500 μL erwärmtem AODM für weitere 14 Tage in jede Vertiefung zu leiten, bis sie die Reife erreichen (nach 21 Tagen der Differenzierung) mit Lumenbildung, umgeben von nach innen gerichteten pseudostratifizierten Epithel, das Basalzellen, Flimmerzellen und Becherzellen enthält.
HINWEIS: Die hier beschriebenen Atemwegepithelorganoide sind terminaldifferenziert und können nicht durchlässig oder kryokonserviert werden.
7. Bildgebende Zilienschlagfrequenz
HINWEIS: Dieser Abschnitt des Protokolls erfordert ein Lebendzell-Bildgebungsmikroskop mit einer Heiz- und Feuchtigkeits-Umgebungskammer, eine wissenschaftliche Kamera mit schneller Bildrate (>100 Hz), ein 20-faches Objektiv mit großem Arbeitsabstand und eine Bildgebungssoftware (siehe Materialtabelle für empfohlene Geräte in diesem Protokoll).
- Einrichtung des Mikroskops
- Stellen Sie sicher, dass das Mikroskopheizsystem eingeschaltet und auf 37 °C ausgeglichen ist. Schalten Sie das Mikroskop ein. Stellen Sie das Gas über den CO2/Luft-Gasmischer auf 5% CO2 ein.
- Füllen Sie die Flasche des Feuchtigkeitsmoduls, die das CO2 durchläuft, mit gereinigtem Wasser auf. Stellen Sie die relative Luftfeuchtigkeit über den Tischregler auf 85% ein, so dass das Wasser erwärmt und die Zellen mit befeuchteter Luft versorgt werden. Die Kammer für 30 min ins Gleichgewicht bringen.
- Setzen Sie den Einsatz der Mikroskopplatte in den Mikroskophalter ein.
- Übertragen Sie die Atemwegsepithelzellmodelle aus dem Inkubator in das Mikroskop auf einem Wärmeblock oder thermischen Kügelchen, die auf 37 °C äquilibriert sind, um die Probe auf einer physiologischen Temperatur zu halten.
- Legen Sie die Kulturplatte, die die Atemwegsepithelzellmodelle enthält, in den Einsatz der Mikroskopplatte. Schließen Sie die Mikroskop-Klimakammer.
- Lassen Sie die Probe in der vorgewärmten 37 °C, 5% CO2-gefüllten Mikroskopkammer für 30 min ausgleichen.
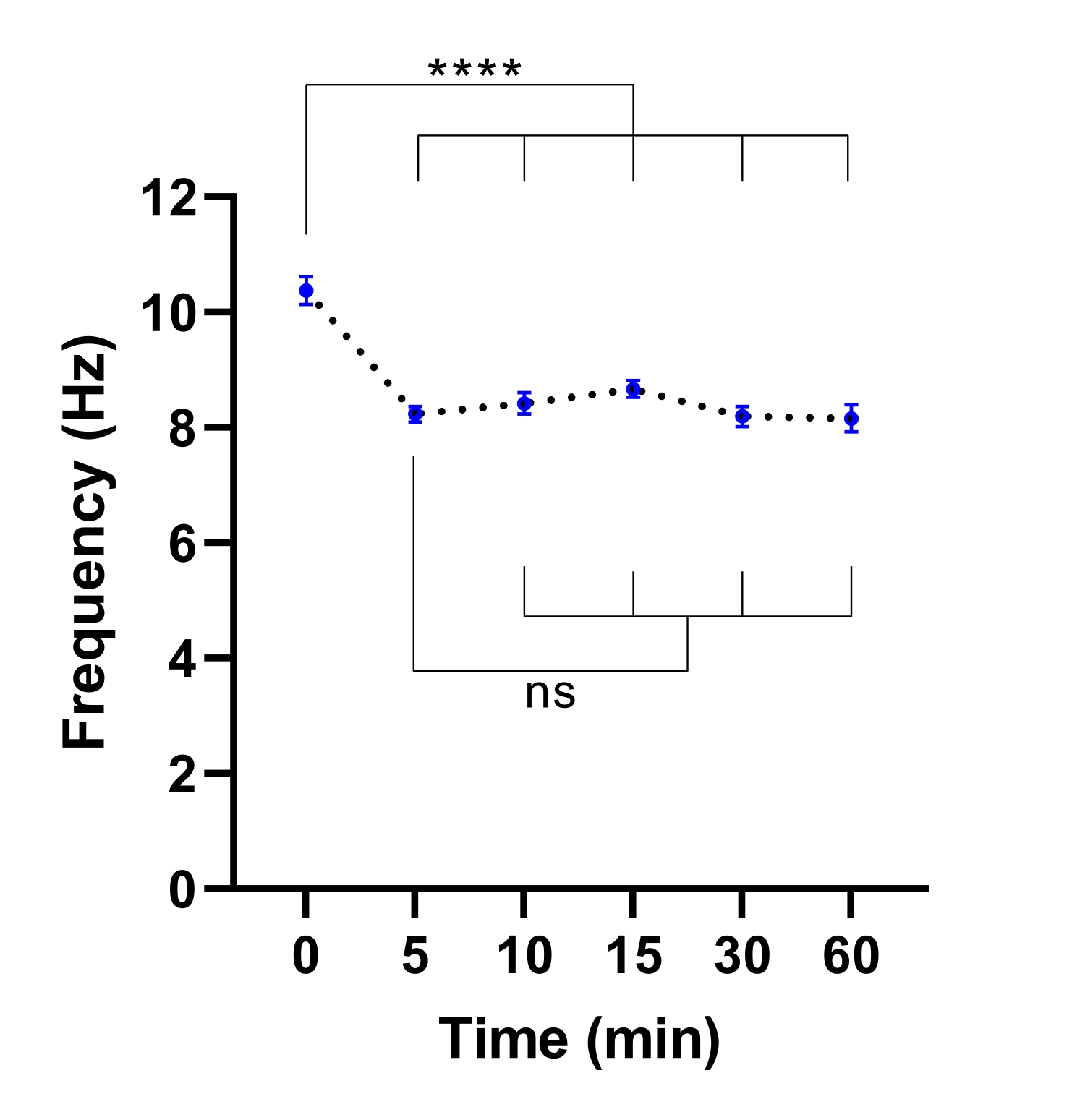
HINWEIS: Eine kürzere Äquilibrierungszeit kann ausreichend sein. Dies kann durch ein Experiment ermittelt werden, um die Zeit zu ermitteln, die für die Stabilisierung von CBF benötigt wird (siehe Abbildung 3).

Abbildung 3: Stabilisierung der Ziliarschlagfrequenz im Lebendzell-Bildgebungsmikroskop. Punktdiagramme der mittleren Zilienschlagfrequenz (CBF) in Atemwegsepithelzellen an der Luft-Flüssig-Grenzfläche (ALI-Modelle) nach Transfer in ein lebendes Zellbildgebungsmikroskop mit einer Klimakammer. Die Kammer wurde ausgeglichen und 30 min lang bei 37 °C, 5%CO2 und einer relativen Luftfeuchtigkeit von 85% gehalten, bevor die Kammertür geöffnet und die Kulturplatte in den Gitterplatteneinsatz eingesetzt wurde. Zellmodelle wurden für 60 Minuten in angegebenen Intervallen abgebildet. ALI-Modelle wurden von zwei Teilnehmern mit CF abgeleitet. Pro ALI-Modell wurden sechs Field of View (FOV)-Bilder aufgenommen. Jeder Punkt (blau) stellt den mittleren CBF in 12-36 FOV-Bildern dar. Die Daten werden als Mittelwert ± REM dargestellt, wobei der Mittelwert durch eine gepunktete Linie verbunden ist. Zur Bestimmung statistischer Unterschiede wurde die Einweg-Varianzanalyse (ANOVA) verwendet. P < 0,0001, ns: keine Signifikanz. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Öffnen Sie während der Äquilibrierungsphase am Computer die Erfassungssoftware. Wählen Sie das 20-fache Objektiv mit großem Arbeitsabstand.
- Am Mikroskopokular auf das Zellmodell fokussieren (~Z = 8000 μm).
- Stellen Sie sicher, dass das Mikroskop für Kohler-Beleuchtung so eingerichtet ist, dass die Glühfäden der Transmissionslichtquelle nicht auf die Probenebene fokussiert werden, um Artefakte in der Bildgebung zu vermeiden. Folgen Sie dazu den Schritten 7.1.10-7.1.13
- Schließen Sie die Feldblende über dem Kondensator vollständig. Öffnen Sie langsam die Halblichtblende und bewegen Sie den Kondensator nach oben/unten, bis eine Achteckform erscheint.
- Wenn die Feldblende nicht ausgerichtet ist (d. h. das Achteck befindet sich nicht in der Mitte des Sichtfelds (FOV)), richten Sie es mit Inbustasten an der Mitte aus.
- Sobald die Feldblende ausgerichtet ist, passen Sie den Kondensatorfokus an, um das Achteck scharf zu fokussieren.
- Öffnen Sie die Blendenblende des Feldes, bis sie im Sichtfeld nicht mehr sichtbar ist.
- Klicken Sie mit der Erfassungssoftware auf L100 , um den Lichtpfad zum Anschluss zu wechseln, an dem die Kamera montiert ist. Klicken Sie auf die grüne Play-Schaltfläche (Run), um das FOV des Mikroskops über die Software zu visualisieren. Überprüfen Sie, ob die Zilien scharf sind und passen Sie sie bei Bedarf an.
- Richten Sie das Mikroskop mithilfe der Erfassungssoftware mit den folgenden Einstellungen ein: Filter: leer; Kondensator: leer; Format: kein Binning; Belichtungszeit: 0,003 s; Auslesemodus: Rolling Shutter; ROI: 512 × 512 Pixel.
HINWEIS: Die Belichtungszeit basiert auf der höchsten Frequenz, die gemessen werden muss, da 1/Belichtungszeit mindestens doppelt so hoch sein muss. Wenn z.B. der maximale physiologische Bereich des Zilienschlagens = 30 Hz ist, dann 1/Belichtungszeit = 60, und die Belichtungszeit muss ≤ 0,016 s betragen. Wählen Sie einen ROI, der Bildraten >100 Hz erfasst.
- Bildaufnahme
- Um Zeitrafferbilder aus dem Menü zu erfassen, klicken Sie auf Erfassen und dann auf Schneller Zeitraffer. Wählen Sie im Popup-Fenster einen Speicherort und einen Dateinamen aus. Erwerben Sie 1000 Frames.
- Klicken Sie auf Übernehmen. Klicken Sie auf die grüne Play-Schaltfläche (Run), um eine Vorschau der Zilien im Mikroskop-Sichtfeld anzuzeigen und den Z-Fokus bei Bedarf anzupassen. Klicken Sie auf Jetzt ausführen , um den schnellen Zeitraffer aufzunehmen.
- Sobald der schnelle Zeitraffer erfasst wurde, klicken Sie auf die grüne Play-Schaltfläche (Run), um das Mikroskop-FOV zu visualisieren. Bewegen Sie sich mit dem Joystick des Mikroskops entlang der X/Y-Achse zu einem anderen Sichtfeld.
- Stellen Sie den Z-Fokus ein, um die Zilien in den Fokus zu bringen. Klicken Sie auf Jetzt ausführen , um einen weiteren schnellen Zeitraffer aufzunehmen.
- Wiederholen Sie die Schritte 7.2.3-7.2.4. Für ALI-Modelle und Atemwegsorganoide Bild 6x FOV in jeder der 3x Replikatproben. Bei Atemwegsepithelblättern sollten Sie mindestens 4x Bilder pro Teilnehmer replizieren.
8. Datenanalyse und Quantifizierung von CBF
- Vorbereitungen für die Datenanalyse
HINWEIS: Dieser Abschnitt des Protokolls erfordert benutzerdefinierte Analyseskripts (Zusatzdatei 3), Rohbilddateien (in Abschnitt 7.2 erfasst), eine Computersoftware und Analysesoftware.- Installieren Sie die Computersoftware, vorzugsweise die neueste Version, auf dem Analysecomputer. Stellen Sie sicher, dass Standard-Computing-Software-Toolboxes (elmat, ops, datafun, uitools, datatypes, iofun, iotools, audiovideo) und Bild- und Signalverarbeitungs-Toolboxen installiert sind.
- Kopieren Sie die benutzerdefinierten Analyseskripts "BeatingCiliaBatchOMEfiles_JOVE.m" und "LoadRawDataExportFilteredMovies_JOVE.m" sowie den Ordner "support scripts" auf das lokale Laufwerk des Computers.
- Klicken Sie in der Computersoftware auf die Registerkarte Start. Klicken Sie dann auf Set Path (Pfad festlegen) (Abbildung 4A-B).
- Klicken Sie im Popup-Fenster auf Add With Subfolders (Mit Unterordnern hinzufügen) (Abbildung 4C). Wählen Sie unter "MATLAB-Suchpfad" die in Abbildung 4D gezeigten Ordner aus und klicken Sie dann auf Speichern und schließen (Abbildung 4E-F).
- Vergewissern Sie sich, dass die Analyseskripts mit der Computersoftware verknüpft sind, indem Sie überprüfen, ob sie im linken Bereich angezeigt werden (Abbildung 4G).
- Übertragen Sie die in Abschnitt 7.2 erfassten Rohbilddateien (OME-Format (Open Microscopy Environment) auf das lokale Laufwerk des Computers.
HINWEIS: Auf Beispiel-RAW-Bilddateien kann zugegriffen werden unter: https://doi.org/10.6084/m9.figshare.16649878.v1.

Abbildung 4: Einrichten von Computersoftware für die Datenanalyse. (A) Öffnen Sie die Registerkarte Start . (B) Wählen Sie Pfad festlegen. (C) Wählen Sie Mit Unterordnern hinzufügen aus. (D) Wählen Sie Ordner aus, die die Analyseskripten enthalten. (E) Wählen Sie Speichern aus. (f) Wählen Sie Schließen. (G) Die Analyseskripte werden im linken Bereich angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Quantifizierung von CBF durch Peak-Detektion des Intensitätsspektrums einzelner Pixel
- Öffnen Sie die Computersoftware. Klicken Sie auf die Analyseskriptdatei 'BeatingCiliaBatchOMEfiles_JOVE.m' (Abbildung 5A).
- Klicken Sie auf die Registerkarte Editor und dann auf die grüne Wiedergabeschaltfläche (Ausführen), um das Skript auszuführen (Abbildung 5B-C). Wählen Sie im Eingabeaufforderungsfenster die zu analysierenden Rohbilddateien aus (Abbildung 5D).
- Geben Sie die Belichtungszeit aus Schritt 7.1.15 in das Eingabefenster für die Aufnahmezeit pro Bild ein und klicken Sie dann auf OK (Abbildung 5E).
- Warten Sie ~15 Minuten pro Datei, während das Skript die CBF berechnet und in der Datei 'AveSpectrum' (Supplementary File 4) ausgibt, die automatisch im selben Ordner wie die RAW-Bilddateien gespeichert wird. Visualisieren Sie den Fortschritt über den Fortschrittsbalken (Abbildung 5F).

Abbildung 5: Ausführen von Analyseskripten mit Computersoftware . (A) Öffnen Sie das Skript für die Analyse von CBF ('BeatingCiliaBatchOMEfiles_JOVE.m') oder die Erstellung eines Zilienschlagfilms ('LoadRawDataExportFilteredMovies_JOVE.m'). (B) Öffnen Sie die Registerkarte Editor . (C) Wählen Sie die grüne Schaltfläche Play (Ausführen), um das Analyseskript auszuführen. (D) Ein Eingabeaufforderungsfenster erfordert die Auswahl von Dateien für die Analyse oder Filmerstellung. (E) Während der Ausführung des Skripts 'BeatingCiliaBatchOMEfiles_JOVE.m' wird eine Eingabeaufforderung zur manuellen Eingabe der Erfassungszeit pro Frame(s) angezeigt, falls das Dateileseskript die Metadaten nicht richtig liest. (F) Fortschrittsbalken, der anzeigt, dass die Häufigkeit des Zilienschlags berechnet wird. (G) Während der Ausführung des Skripts "LoadRawDataExportFilteredMovies_JOVE.m" wird eine Eingabeaufforderung angezeigt, um den Typ des auszugebenden Films (mp4 oder avi), die Filmbildrate (fps), ob die immobile Komponente aus den Filmdaten entfernt wird ("y" oder "n"), die Bildzeit(en) und die Pixelgröße (Mikrometer) der in den Film exportierten Daten manuell einzugeben. Es wird empfohlen, "y" für die unbewegliche Filterung zu verwenden, da dadurch Schleim oder andere behindernde immobile Schichten in den Daten entfernt werden. (H) Fortschrittsbalken zur Anzeige des exportierten Films. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Führen Sie das Skript 'GetFirstAmplitude.m' für den Ordner aus, der die 'AveSpectrum'-Dateien enthält, indem Sie den Prozess in den Schritten 8.2.1-8.2.2 verwenden. Warten Sie, bis das Skript die Datei "FirstAmplitudeStacked.xlsx" ausgegeben hat, die die Frequenz mit der höchsten Amplitude enthält und im physiologischen Bereich der Atemwegepithelzilienschläge liegt, ≥3 und <30 Hz.
- Kopieren Sie die Frequenzwerte aus der Datei "FirstAmplitudeStacked.xlsx" und zeichnen Sie sie mit einer wissenschaftlichen Analysesoftware.
HINWEIS: Eine Erklärung, wie das benutzerdefinierte Analyseskript CBF quantifiziert, finden Sie in der Zusatzdatei 5. Beispiele für analysierte Datensätze finden Sie unter: https://doi.org/10.6084/m9.figshare.16649815.
- Exportieren eines Videos über das Schlagen von Zilien
- Öffnen Sie die Computersoftware. Klicken Sie auf die Skriptdatei "LoadRawDataExportFilteredMovies_JOVE.m" (Abbildung 5A), um das Skript zu laden.
- Klicken Sie auf die Registerkarte Editor , und klicken Sie dann auf die grüne Wiedergabeschaltfläche (Ausführen), um das Skript auszuführen (Abbildung 5C). Wählen Sie im Eingabeaufforderungsfenster die RAW-Bilddateien aus, die in Filmdateien exportiert werden sollen (Abbildung 5D).
- Geben Sie die in Tabelle 4 aufgeführten Einstellungen in das Popupfenster "Film erstellen" ein (Abbildung 5G).
- Warten Sie ~8 Minuten pro Datei, während das Skript die Filmdateien erstellt und sie an den Speicherort der RAW-Bilddateien ausgibt. Visualisieren Sie den Fortschritt über den Fortschrittsbalken (Abbildung 5H).
| Filmeingänge | Beschreibung |
| Dateityp | Geben Sie den Dateityp ein, den Sie exportieren möchten (mp4 oder avi). |
| Bildrate | Geben Sie die Bildrate ein, mit der der Film exportiert werden soll. Wenn Sie ~ 1000 Bilder pro Zeitreihe erfasst haben, wird empfohlen, die Bildrate ~ 30 fps einzustellen. |
| Immobile Filterung | Optionen sind 'y' oder 'n'. Der Standardwert ist 'y', und das Zeitfilterskript entfernt unter Verwendung von Fourierraum alle unbeweglichen Komponenten aus Filmdaten. Typischerweise tragen alle Zellschichten unter Zilien oder unbeweglichem Schleim eine Nullfrequenz-Offset-Komponente oder eine zeitinvariante Komponente in das Signal bei, die herausgefiltert werden kann. |
| Erfassungszeit pro Frame | Die Erfassungszeit pro Frame der erfassten Daten. Es wird verwendet, um einen Zeitstempel im Film in Sekunden anzuzeigen. |
| Pixelgröße | Die Pixelgröße in Mikrometern wird verwendet, um einen Maßstabsbalken im Film in Mikrometern anzuzeigen. |
Tabelle 4: Eingabeeinstellungen für die Filmerstellung
Ergebnisse
Um die Effizienz dieses Protokolls bei der Quantifizierung von CBF zu demonstrieren, werden die Ergebnisse von CBF vorgestellt, die in Atemwegsepithelzell-ALI-Modellen von drei Teilnehmern mit CF und drei gesunden Kontrollteilnehmern abgeleitet wurden. An Tag 14 der Kulturdifferenzierung waren schlagende Zilien vorhanden (Abbildung 6). Vom Tag 14 bis 21 der Kulturdifferenzierung wurde innerhalb beider Kohorten ein statistisch signifikanter (P < 0,0345) Anstieg der CBF beobachtet. Am Tag 21 d...
Diskussion
Es gibt mehrere Faktoren, die die Quantifizierung von CBF in Nasepithelblättern verschleiern könnten. Epithelblätter sollten innerhalb von 3-9 Stunden nach der Probenentnahme abgebildet werden, da die Ziliarfunktion während dieser Zeit am stabilsten ist37. Weniger rote Blutkörperchen und Ablagerungen sind am besten für die Bildgebung geeignet, da diese die Datenerfassung stören. Bei der Auswahl eines ROI für die Bildgebung ist es wichtig, eine Epithelschicht auszuwählen, deren Kante währ...
Offenlegungen
Die Autoren erklären, dass sie nichts offenzulegen haben.
Danksagungen
Wir danken den Studienteilnehmern und ihren Familien für ihre Beiträge. Wir schätzen die Unterstützung der Randwick Atemwegsabteilung des Sydney Children's Hospitals (SCH) bei der Organisation und Sammlung von Patientenbioproben - besonderer Dank gilt Dr. John Widger, Dr. Yvonne Belessis, Leanne Plüsch, Amanda Thompson und Rhonda Bell. Wir danken Iveta Slapetova und Renee Whan von der Katharina Gaus Light Microscopy Facility innerhalb des Mark Wainwright Analytical Centre an der UNSW Sydney für ihre Unterstützung. Diese Arbeit wird vom National Health and Medical Research Council (NHMRC) Australia (GNT1188987), der CF Foundation Australia und der Sydney Children's Hospital Foundation unterstützt. Die Autoren möchten Luminesce Alliance - Innovation for Children's Health für ihren Beitrag und ihre Unterstützung danken. Luminesce Alliance - Innovation for Children's Health ist ein gemeinnütziges Kooperationsunternehmen zwischen dem Sydney Children's Hospitals Network, dem Children's Medical Research Institute und dem Children's Cancer Institute. Es wurde mit Unterstützung der Regierung von NSW gegründet, um die pädiatrische Forschung zu koordinieren und zu integrieren. Luminesce Alliance ist auch mit der University of Sydney und der University of New South Wales Sydney verbunden. KMA wird durch ein Stipendium des Australian Government Research Training Program unterstützt. LKF wird vom Rotary Club Sydney Cove/Sydney Children's Hospital Foundation und Postgraduate Award Stipendien der UNSW University unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Adenine | Sigma-Aldrich | A2786 | 10 mg/mL |
| Advanced DMEM/F-12 | Thermo Fisher Scientific | 12634-010 | |
| Alanyl-glutamine | Sigma-Aldrich | G8541 | 200 mM |
| Andor Zyla 4.2 sCMOS | Oxford Instruments | Fast frame rate (>100 Hz) scientific camera | |
| Bottle-top vacuum filter system | Sigma-Aldrich | CLS431098 | |
| Ceftazidime hydrate | Sigma-Aldrich | A6987 | 50 mg/mL |
| Cell Culture Microscope | Olympus | CKX53 | |
| CFI S Plan Fluor ELWD 20XC | Nikon Instruments Inc. | MRH08230 | Long working distance objective lens. NA0.45 WD 8.2-6.9 |
| Cholera toxin | Sigma-Aldrich | C8052-1MG | 200 µg/mL |
| Corning Gel Strainer 40 UM | Sigma-Aldrich | CLS431750 | Pore size 40 μm |
| Corning Matrigel Matrix (Phenol red-free) | Corning | 356231 | Extracellular matrix (ECM) |
| Corning bottle-top vacuum filter system | Sigma-Aldrich | CLS431098 | |
| Corning CoolCell LX Cell Freezing Container | Sigma-Aldrich | CLS432002 | |
| Corning Transwell polyester membrane cell culture inserts | Sigma-Aldrich | CLS3470 | Permeable support inserts. 6.5 mm Transwell with 0.4 μm pore polyester membrane insert. |
| Countess Cell Counting Chamber Slides | Thermo Fisher Scientific | C10228 | |
| Countess II Automated Cell Counter | ThermoFisher Scientific | AMQAX1000 | Automated cell counter |
| Cytology brushes | McFarlane Medical | 33009 | |
| DMEM/F12-Ham | Thermo Fisher Scientific | 11330032 | |
| DMEM/F12-Ham | Thermo Fisher Scientific | 11330032 | |
| DMEM-High Glucose | Thermo Fisher Scientific | 11965-092 | |
| Dulbecco′s Phosphate Buffered Saline (PBS) | Sigma-Aldrich | D8537 | |
| Eclipse Ti2-E | Nikon | Live-cell imaging microscope. | |
| Fetal Bovine Serum, certified, heat inactivated, United States | Thermo Fisher Scientific | 10082147 | |
| Fungizone (Amphotericin B) | Thermo Fisher Scientific | 15290018 | 250 µg/mL |
| Gentamicin solution | Sigma-Aldrich | G1397 | 50 mg/mL |
| Graphpad Prism | Graphpad | Scientific analysis software | |
| Greiner Cryo.s vials | Sigma-Aldrich | V3135 | Cryogenic vials |
| HEPES solution | Sigma-Aldrich | H0887 | 1 M |
| HI-FBS | Thermo Fisher Scientific | 10082-147 | |
| Hydrocortisone | Sigma-Aldrich | H0888 | 3.6 mg/mL |
| Incubator NL Ti2 BLACK 2000 | PeCon | Microscope environmental chamber. Allows warm air incubation and local CO2 and O2 gassing | |
| Insulin | Sigma-Aldrich | I2643 | 2 mg/mL |
| Lab Armor 74220 706 Waterless Bead Bath 6L | John Morris Group | 74220 706 | Bead bath |
| Lab Armor Beads | Thermo Fisher Scientific | A1254302 | Thermal beads |
| MATLAB | MathWorks | Computing software | |
| Microsoft Excel | Microscoft | Spreadsheet software | |
| NIH/3T3 | American Type Culture Collection | CRL-1658 | Irradiated NIH-3T3 mouse embryonic feeder cells |
| NIS-Elements AR | Nikon Instruments Inc. | Image acquisition software | |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 | 10,000 units penicillin and 10 mg streptomycin/mL |
| Dulbecco′s Phosphate Buffered Saline (PBS) | Sigma-Aldrich | D8537 | |
| PneumaCult Airway Organoid Kit | StemCell Technologies | 5060 | Airway Organoid Kit |
| PneumaCult-ALI Medium | StemCell Technologies | 5001 | |
| PneumaCult-Ex Plus Medium | StemCell Technologies | 5040 | |
| PureCol-S | Advanced BioMatrix | 5015 | Type I Collagen solution |
| ReagentPack Subculture Reagents | Lonza | CC-5034 | |
| rhEGF (Epidermal Growth Factor, human) | Sigma-Aldrich | E9644 | 25 µg/mL |
| Y-27632 2HCl (ROCK inhibitor) | Selleckchem | S1049 | 10 mM |
| Tobramycin | Sigma-Aldrich | T4014 | 100 mg/mL |
| Trypan blue solution | Sigma-Aldrich | T8154 | 0.4% |
| UNO Stage Top Incubator | Okolab | Microscope incubator. Allows temperature, humidity and CO2 conditioning |
Referenzen
- Barbato, A., et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. European Respiratory Journal. 34 (6), 1264-1276 (2009).
- Cutting, G. R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nature Reviews Genetics. 16 (1), 45-56 (2015).
- Chioccioli, M., Feriani, L., Kotar, J., Bratcher, P. E., Cicuta, P. Phenotyping ciliary dynamics and coordination in response to CFTR-modulators in Cystic Fibrosis respiratory epithelial cells. Nature Communications. 10 (1), 1763 (2019).
- Hirst, R. A., Rutman, A., Williams, G., O'Callaghan, C. Ciliated air-liquid cultures as an aid to diagnostic testing of primary ciliary dyskinesia. Chest. 138 (6), 1441-1447 (2010).
- Thomas, B., Rutman, A., O'Callaghan, C. Disrupted ciliated epithelium shows slower ciliary beat frequency and increased dyskinesia. European Respiratory Journal. 34 (2), 401-404 (2009).
- Coles, J. L., et al. A revised protocol for culture of airway epithelial cells as a diagnostic tool for primary ciliary dyskinesia. Journal of Clinical Medicine. 9 (11), (2020).
- Pifferi, M., et al. Simplified cell culture method for the diagnosis of atypical primary ciliary dyskinesia. Thorax. 64 (12), 1077-1081 (2009).
- Pifferi, M., et al. Rapid diagnosis of primary ciliary dyskinesia: cell culture and soft computing analysis. European Respiratory Journal. 41 (4), 960-965 (2013).
- Lee, D. D. H., et al. Higher throughput drug screening for rare respiratory diseases: Readthrough therapy in primary ciliary dyskinesia. European Respiratory Journal. 58 (4), 2000455 (2021).
- Saint-Criq, V., et al. Choice of differentiation media significantly impacts cell lineage and response to CFTR modulators in fully differentiated primary cultures of cystic fibrosis human airway epithelial cells. Cells. 9 (9), (2020).
- Awatade, N. T., et al. Significant functional differences in differentiated Conditionally Reprogrammed (CRC)- and Feeder-free Dual SMAD inhibited-expanded human nasal epithelial cells. Journal of Cystic Fibrosis. 20 (2), 364-371 (2021).
- Dabrowski, M., Bukowy-Bieryllo, Z., Jackson, C. L., Zietkiewicz, E. Properties of non-aminoglycoside compounds used to stimulate translational readthrough of PTC mutations in primary ciliary dyskinesia. International Journal of Molecular Sciences. 22 (9), (2021).
- Hirst, R. A., et al. Culture of primary ciliary dyskinesia epithelial cells at air-liquid interface can alter ciliary phenotype but remains a robust and informative diagnostic aid. PloS One. 9 (2), 89675 (2014).
- Marthin, J. K., Stevens, E. M., Larsen, L. A., Christensen, S. T., Nielsen, K. G. Patient-specific three-dimensional explant spheroids derived from human nasal airway epithelium: a simple methodological approach for ex vivo studies of primary ciliary dyskinesia. Cilia. 6, 3 (2017).
- Chilvers, M. A., O'Callaghan, C. Analysis of ciliary beat pattern and beat frequency using digital high speed imaging: comparison with the photomultiplier and photodiode methods. Thorax. 55 (4), 314-317 (2000).
- Chilvers, M. A., Rutman, A., O'Callaghan, C. Functional analysis of cilia and ciliated epithelial ultrastructure in healthy children and young adults. Thorax. 58 (4), 333-338 (2003).
- Castillon, N., et al. Polarized expression of cystic fibrosis transmembrane conductance regulator and associated epithelial proteins during the regeneration of human airway surface epithelium in three-dimensional culture. Laboratory Investigation. 82 (8), 989-998 (2002).
- Jorissen, M., Bessems, A. Normal ciliary beat frequency after ciliogenesis in nasal epithelial cells cultured sequentially as monolayer and in suspension. Acta Oto-Laryngologica. 115 (1), 66-70 (1995).
- Conger, B. T., et al. Comparison of cystic fibrosis transmembrane conductance regulator (CFTR) and ciliary beat frequency activation by the CFTR Modulators Genistein, VRT-532, and UCCF-152 in primary sinonasal epithelial cultures. JAMA Otolaryngology-Head & Neck Surgery. 139 (8), 822-827 (2013).
- Pique, N., De Servi, B. Rhinosectan((R)) spray (containing xyloglucan) on the ciliary function of the nasal respiratory epithelium; results of an in vitro study. Allergy, Asthma & Clinical Immunology. 14, 41 (2018).
- Chen, Q., et al. Host antiviral response suppresses ciliogenesis and motile ciliary functions in the nasal epithelium. Frontiers in Cell and Developmental Biology. 8, 581340 (2020).
- Clary-Meinesz, C. F., Cosson, J., Huitorel, P., Blaive, B. Temperature effect on the ciliary beat frequency of human nasal and tracheal ciliated cells. Biology of the Cell. 76 (3), 335-338 (1992).
- Ballenger, J. J., Orr, M. F. Quantitative measurement of human ciliary activity. Annals of Otology, Rhinology and Laryngology. 72, 31-39 (1963).
- Mercke, U. The influence of varying air humidity on mucociliary activity. Acta Oto-Laryngologica. 79 (1-2), 133-139 (1975).
- Sutto, Z., Conner, G. E., Salathe, M. Regulation of human airway ciliary beat frequency by intracellular pH. Journal of Physiology. 560, 519-532 (2004).
- Salathe, M. Regulation of mammalian ciliary beating. Annual Review of Physiology. 69, 401-422 (2007).
- Kempeneers, C., Seaton, C., Garcia Espinosa, B., Chilvers, M. A. Ciliary functional analysis: Beating a path towards standardization. Pediatric Pulmonology. 54 (10), 1627-1638 (2019).
- Kempeneers, C., Seaton, C., Chilvers, M. A. Variation of ciliary beat pattern in three different beating planes in healthy subjects. Chest. 151 (5), 993-1001 (2017).
- Jackson, C. L., et al. Accuracy of diagnostic testing in primary ciliary dyskinesia. European Respiratory Journal. 47 (3), 837-848 (2016).
- Feriani, L., et al. Assessing the collective dynamics of motile cilia in cultures of human airway cells by multiscale DDM. Biophysical Journal. 113 (1), 109-119 (2017).
- Brewington, J. J., et al. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight. 3 (13), (2018).
- Liu, X., et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. The American Journal of Pathology. 180 (2), 599-607 (2012).
- Suprynowicz, F. A., et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 109 (49), 20035-20040 (2012).
- Martinovich, K. M., et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Scientific Reports. 7 (1), 17971 (2017).
- Wong, J. Y., Rutman, A., O'Callaghan, C. Recovery of the ciliated epithelium following acute bronchiolitis in infancy. Thorax. 60 (7), 582-587 (2005).
- Gentzsch, M., et al. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 56 (5), 568-574 (2017).
- Sommer, J. U., Gross, S., Hormann, K., Stuck, B. A. Time-dependent changes in nasal ciliary beat frequency. European Archives of Oto-Rhino-Laryngology. 267 (9), 1383-1387 (2010).
- Ratjen, F., et al. Cystic fibrosis. Nature Reviews Disease Primers. 1, 15010 (2015).
- Delmotte, P., Sanderson, M. J. Ciliary beat frequency is maintained at a maximal rate in the small airways of mouse lung slices. American Journal of Respiratory Cell and Molecular Biology. 35 (1), 110-117 (2006).
- Smith, C. M., et al. Cooling of cilia allows functional analysis of the beat pattern for diagnostic testing. Chest. 140 (1), 186-190 (2011).
- Raidt, J., et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. European Respiratory Journal. 44 (6), 1579-1588 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten