
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Dreidimensionale Charakterisierung von interorganellen Kontaktstellen in Hepatozyten mittels serieller Schnittelektronenmikroskopie
In diesem Artikel
Zusammenfassung
Ein einfaches und umfassendes Protokoll, um dreidimensionale Details von Membrankontaktstellen zwischen Organellen in Hepatozyten aus der Leber oder Zellen in anderen Geweben zu erfassen.
Zusammenfassung
Die Transmissionselektronenmikroskopie gilt seit langem als Goldstandard für die Visualisierung zellulärer Ultrastrukturen. Die Analyse ist jedoch oft auf zwei Dimensionen beschränkt, was die Fähigkeit behindert, die dreidimensionale (3D) Ultrastruktur und die funktionelle Beziehung zwischen Organellen vollständig zu beschreiben. Die Volumenelektronenmikroskopie (vEM) beschreibt eine Sammlung von Techniken, die die Abfrage der zellulären Ultrastruktur in 3D bei mesoskaligen, mikroskaligen und nanoskaligen Auflösungen ermöglichen.
Dieses Protokoll bietet eine zugängliche und robuste Methode zur Erfassung von vEM-Daten mittels serieller Schnittübertragung EM (TEM) und deckt die technischen Aspekte der Probenbearbeitung bis hin zur digitalen 3D-Rekonstruktion in einem einzigen, unkomplizierten Workflow ab. Um die Nützlichkeit dieser Technik zu demonstrieren, wird die ultrastrukturelle 3D-Beziehung zwischen dem endoplasmatischen Retikulum und den Mitochondrien und ihren Kontaktstellen in Leberhepatozyten vorgestellt. Interorganelle Kontakte spielen eine wichtige Rolle bei der Übertragung von Ionen, Lipiden, Nährstoffen und anderen kleinen Molekülen zwischen Organellen. Trotz ihrer ersten Entdeckung in Hepatozyten gibt es jedoch noch viel über ihre physikalischen Eigenschaften, Dynamik und Funktionen zu lernen.
Interorganellenkontakte können eine Reihe von Morphologien aufweisen, die sich in der Nähe der beiden Organellen zueinander (typischerweise ~ 10-30 nm) und der Ausdehnung der Kontaktstelle (von punktuellen Kontakten bis zu größeren 3D-zisternenartigen Kontakten) unterscheiden. Die Untersuchung enger Kontakte erfordert eine hochauflösende Bildgebung, und der serielle Schnitt TEM eignet sich gut, um die ultrastrukturelle 3D-Struktur von interorganellen Kontakten während der Hepatozytendifferenzierung sowie Veränderungen in der Hepatozytenarchitektur im Zusammenhang mit Stoffwechselerkrankungen zu visualisieren.
Einleitung
Seit ihrer Erfindung in den 1930er Jahren haben Elektronenmikroskope es Forschern ermöglicht, die strukturellen Bestandteile von Zellen und Geweben zu visualisieren 1,2. Die meisten Untersuchungen haben 2D-Informationen geliefert, da das Erstellen von 3D-Modellen eine sorgfältige Sammlung serieller Abschnitte, manuelle Fotografie, Negativverarbeitung, manuelle Nachverfolgung und die Erstellung und Montage von 3D-Modellen aus Glas-, Kunststoff- oder Styropor 3,4 erfordert. Fast 70 Jahre später gab es erhebliche Fortschritte in zahlreichen Aspekten des Prozesses, von der Mikroskopleistung, der seriellen Schnittsammlung, der automatisierten digitalen Bildgebung, der ausgefeilten Software und Hardware für die 3D-Rekonstruktion, Visualisierung und Analyse bis hin zu alternativen Ansätzen für das, was heute allgemein als Volume EM (vEM) bezeichnet wird. Es wird allgemein angenommen, dass diese vEM-Techniken ultrastrukturelle 3D-Informationen bei Nanometerauflösungen über Mikrometerskalen liefern und Transmissionselektronenmikroskopie (TEM) und neuere Rasterelektronenmikroskopie- (REM) Techniken umfassen. Siehe Bewertungen 5,6,7,8.
Zum Beispiel verwendet der fokussierte Ionenstrahl SEM (FIB-SEM) einen fokussierten Ionenstrahl in einem REM, um die Oberfläche des Blocks zwischen sequentiellen REM-Bildaufnahmen der Blockoberfläche wegzufräsen, was das wiederholte automatisierte Fräsen / Abbilden einer Probe ermöglicht und einen 3D-Datensatz für die Rekonstruktion 9,10 erstellt. Im Gegensatz dazu verwendet serielles Blockgesicht SEM (SBF-SEM) ein Ultramikrotom im REM, um Material von der Blockfläche vor der Bildgebung zu entfernen 11,12, während die Array-Tomographie ein zerstörungsfreier Prozess ist, der die Sammlung von seriellen Abschnitten auf Deckgläsern, Wafern oder Bändern erfordert, bevor ein automatisierter Workflow der Bildgebung des interessierenden Bereichs in sequenziellen Abschnitten im REM eingerichtet wird, um den 3D-Datensatz zu generieren 13 . Ähnlich wie bei der Array-Tomographie erfordert die serielle Schnitt-TEM (ssTEM), dass vor der Bildgebung physikalische Schnitte gesammelt werden. Diese Abschnitte werden jedoch in TEM-Rastern gesammelt und in einem TEM14,15,16 abgebildet. ssTEM kann durch Tilttomographie17,18,19 erweitert werden. Die serielle Neigungstomographie bietet die beste Auflösung in x, y und z, und obwohl sie zur Rekonstruktion ganzer Zellen20 verwendet wurde, ist sie eine ziemliche Herausforderung. Dieses Protokoll konzentriert sich auf die praktischen Aspekte von ssTEM als der am besten zugänglichen vEM-Technik, die vielen EM-Labors zur Verfügung steht, die derzeit möglicherweise keinen Zugang zu spezialisierten Schnitt- oder vEM-Instrumenten haben, aber von der Generierung von 3D-vEM-Daten profitieren würden.
Die serielle Ultramikrotomie für die 3D-Rekonstruktion wurde bisher als Herausforderung angesehen. Es war schwierig, gerade Bänder mit gleichmäßiger Schnittstärke zu schneiden, in der Lage zu sein, Bänder der richtigen Größe in der richtigen Reihenfolge auf Gittern mit ausreichender Unterstützung anzuordnen und aufzunehmen, aber ohne Gitterbalken, die Bereiche von Interesse verdeckten, und vor allem, ohne Abschnitte zu verlieren, da eine unvollständige Serie eine vollständige 3D-Rekonstruktion verhindern kann21. Verbesserungen an kommerziellen Ultramikrotomen, Diamantschneid- und Trimmmessern 22,23, elektronenluzenten Stützfolien auf Gittern 21,24 und Klebstoffen zur Unterstützung der Schnitthaftung und Bandkonservierung 13,21 sind jedoch nur einige der inkrementellen Fortschritte im Laufe der Jahre, die die Technik in vielen Labors zur Routine gemacht haben. Sobald serielle Abschnitte gesammelt wurden, ist die serielle Bildgebung in TEM einfach und kann EM-Bilder mit Subnanometer-px-Größen in x und y liefern, was eine hochauflösende Abfrage der subzellulären Strukturen ermöglicht - eine potenzielle Anforderung für viele Forschungsfragen. Die hier vorgestellte Fallstudie zeigt die Verwendung von ssTEM und 3D-Rekonstruktion bei der Untersuchung von endoplasmatischen Retikulum (ER)-Organellenkontakten in Leberhepatozyten, wo ER-Organellen-Kontakte zuerst beobachtet wurden25,26.
Während die Notaufnahme an die Kernhülle angrenzt, nimmt sie auch enge Kontakte zu zahlreichen anderen Zellorganellen auf, darunter Lysosomen, Mitochondrien, Lipidtröpfchen und die Plasmamembran27. ER-Organellen-Kontakte wurden mit dem Fettstoffwechsel28, dem Phosphoinositid- und Calcium-Signalsystem29, der Autophagieregulation und der Stressreaktion30,31 in Verbindung gebracht. Die ER-Organellenkontakte und andere interorganelle Kontakte sind hochdynamische Strukturen, die auf zelluläre Stoffwechselbedürfnisse und extrazelluläre Hinweise reagieren. Es wurde gezeigt, dass sie morphologisch in ihrer Größe und Form und den Abständen zwischen Organellenmembranenvariieren 32,33. Es wird angenommen, dass diese ultrastrukturellen Unterschiede wahrscheinlich ihre unterschiedliche Protein-Lipid-Zusammensetzung und Funktionwiderspiegeln 34,35. Es ist jedoch immer noch eine herausfordernde Aufgabe, interorganelle Kontakte zu definieren und zu analysieren36. Daher ist ein zuverlässiges und dennoch einfaches Protokoll zur Untersuchung und Charakterisierung von interorganellen Kontakten für weitere Untersuchungen erforderlich.
Da ER-Organellenkontakte bei der Membran-zu-Membran-Trennung zwischen 10 und 30 nm liegen können, war der Goldstandard für die Identifizierung in der Vergangenheit TEM. Dünnschnittiges TEM hat eine spezifische Subdomänenlokalisation für residente ER-Proteine an verschiedenen Membrankontakten37 aufgedeckt. Traditionell hat dies ER-Organellen-Kontakte mit nm-Auflösung aufgedeckt, aber oft nur eine 2D-Ansicht dieser Wechselwirkungen präsentiert. vEM-Ansätze zeigen jedoch die ultrastrukturelle Darstellung und den Kontext dieser Kontaktstellen in 3D, was eine vollständige Rekonstruktion von Kontakten und eine genauere Klassifizierung von Kontakten (Punkt vs. röhrenförmig vs. zisternalähnlich) und Quantifizierung38,39 ermöglicht. Hepatozyten sind nicht nur der erste Zelltyp, bei dem ER-Organellenkontakte beobachtet wurden25,26, sondern haben auch ein umfangreiches System anderer interorganeller Kontakte, die in ihrer Architektur und Physiologie eine wichtige Rolle spielen 28,40. Eine gründliche morphologische Charakterisierung von ER-Organellen und anderen interorganellen Kontakten in Hepatozyten fehlt jedoch noch. Dementsprechend ist die Art und Weise, wie sich interorganelle Kontakte während der Regeneration und Reparatur bilden und umgestalten, von besonderer Relevanz für die Biologie und Leberfunktion der Hepatozyten.
Access restricted. Please log in or start a trial to view this content.
Protokoll
Alle Tiere wurden in Übereinstimmung mit den Richtlinien des britischen Innenministeriums untergebracht, und die Gewebeentnahme wurde in Übereinstimmung mit dem UK Animal (Scientific Procedures) Act 1986 durchgeführt.
1. Fixierung und Vorbereitung der Proben
- Sezieren Sie das Lebergewebe in Stücke von geeigneter Größe, etwa 8 mm x 8 mm x 3 mm, und legen Sie die Stücke in warme phosphatgepufferte Kochsalzlösung (PBS, 37 ° C).
- Injizieren Sie Raumtemperatur (20-25 °C) fixierendes Mittel (1,5% Glutaraldehyd in 1% Saccharose, 0,1 M Natriumcacodylat) in die Leberstücke und übertragen Sie sie von PBS auf Fixiermittel für bis zu 20 min bei Raumtemperatur. Halten Sie das Gewebe immer in Lösungen getaucht, um ein Austrocknen zu vermeiden.
HINWEIS: Aldehyde sind Reizstoffe, die ätzend und potenziell krebserregend sind. Natriumcacodylat ist giftig, wenn es geschluckt oder eingeatmet wird. Alle Fixiermittel müssen unter Tragen geeigneter persönlicher Schutzausrüstung gehandhabt werden, und das Experiment sollte in einem Abzug durchgeführt werden. Eine gute Fixierung führt zu einem festeren Gewebe. - Stellen Sie das vibrierende Mikrotom mit einer Klinge, einem Eisbad und einer kalten PBS-gefüllten Pufferschale auf. Montieren Sie das erste Stück festsitzendes Lebergewebe mit Cyanacrylatkleber auf einen Probenhalter und übertragen Sie den Block auf das vibrierende Mikrotom.
- Nähern Sie sich den Empfehlungen des Herstellers dem Gewebe und schneiden Sie die fixierte Leber in 100 μm dicke Scheiben.
- Sammeln Sie die Scheiben mit einem Spatel oder einem natürlichen Haarpinsel und geben Sie sie in eine 12- oder 24-Well-Schale mit eiskaltem Fix (1,5% Glutaraldehyd, 0,1 M Natriumcacodylat) auf Eis. Lassen Sie die Scheiben auf Eis, bis alle Proben in Scheiben geschnitten sind und bereit sind, weiterverarbeitet zu werden.
- Wählen Sie die Scheiben mit den gewünschten Bereichen für die weitere Verarbeitung aus und waschen Sie sie mit sanftem Rühren. Führen Sie drei, 5 Minuten Wäschen mit einer Raumtemperatur von 0,1 m Natriumcacodylat in einer 12- oder 24-Well-Schale durch, um sicherzustellen, dass die Scheiben genügend Puffer haben, um sich frei zu bewegen.
HINWEIS: Im Allgemeinen werden Regionen von Interesse auf der Grundlage anatomischer Merkmale ausgewählt, die mit der biologischen Frage der Studie zusammenhängen und von Regionen des Gewebes geleitet werden, die wahrscheinlich in der gesamten Serie vorhanden sind, z. B. nicht am Rand des Abschnitts, und die gut erhalten sind. - In einem Abzug 0,1 M Natriumcacodylat durch frisch zubereitetes 1% Osmiumtetroxid/1,5% Kaliumferricyanid ersetzen. Stellen Sie die 12- oder 24-Well-Schüssel in einen versiegelten Behälter und geben Sie den Behälter für 1 Stunde in einen Kühlschrank mit gefährlichen Chemikalien.
HINWEIS: Osmium ist extrem gefährlich bei Einnahme, Inhalation und Hautkontakt. Kaliumferricyanid ist ein Reizstoff und ist schädlich durch Inhalation und Hautkontakt. Behandeln Sie immer die entsprechende persönliche Schutzausrüstung und führen Sie das Experiment in einem Abzug durch. - Entfernen Sie in einem Abzug das Osmiumtetroxid / Kaliumferricyanid in eine spezielle Osmium-Abfallflasche und waschen Sie die Proben dreimal für 5 Minuten mit 0,1 M Natriumcacodylat. Lassen Sie die Proben über Nacht in einem verschlossenen Behälter bei 4 °C.
HINWEIS: Möglicher Pausenpunkt. Die Proben können in 0,1 M Natriumcacodylat in einem verschlossenen Behälter bei 4 °C wochenlang mit geringer Schädigung der Konservierung gelagert werden. Stellen Sie sicher, dass genügend Puffer vorhanden ist, um ein Austrocknen zu verhindern. - Die Proben mit frisch zubereiteter 1% Gerbsäure in 0,05 M Natriumcacodylat für 45 min im Dunkeln bei Raumtemperatur inkubieren.
HINWEIS: Gerbsäure ist reizend und kann Augenschäden verursachen. Tragen Sie geeignete persönliche Schutzausrüstung und führen Sie das Experiment in einem Abzug durch. - Führen Sie vor dem Austrocknen und Einbetten drei 5-minütige Wäschen mit ddH2O durch.
2. Dehydratisierung der Probe, Einbettung und Montage von Epon-Harz
- Epon-Harz wird nach dem folgenden Gewichtsverhältnis hergestellt (siehe Schritt 2.2). Tare eine Waage mit einem 100 ml Einweg-Plastikbecher mit einem Rührstab. Schneiden Sie die Enden aus 5 Einweg-Pasteur-Pipetten aus Kunststoff und verwenden Sie diese, um viskose Harzkomponenten in das Becherglas zu übertragen.
- Nacheinander 19,2 g Harz-812, 7,6 g DDSA, 13,2 g MNA und 0,8 g DMP-30-Beschleuniger in das Becherglas geben. Mit der fünften sauberen Kunststoffpipette Pasteur die Harzkomponenten gründlich von Hand mischen.
HINWEIS: Vermeiden Sie das Einbringen von Blasen, aber stellen Sie sicher, dass das untere Harz mit der Oberseite ausreichend gemischt wird, um eine Farbänderung und eine grobe Vermischung der Komponentenschichten zu erreichen. Alle Harzbestandteile sind reizend und durch Inhalation und Hautkontakt schädlich. DMP-30 ist korrosiv und kann zu Hautkorrosion führen. Tragen Sie geeignete persönliche Schutzausrüstung. - Legen Sie das Becherglas auf einen Magnetrührer und lassen Sie es vorsichtig rühren, wobei Sie das Harz regelmäßig manuell mischen.
- Waschen Sie die Proben mit sanftem Rühren für 5 min mit 70% Ethanol; Wiederholen Sie den Vorgang.
- Waschen Sie die Proben mit sanftem Rühren für 5 min mit 90% Ethanol; Wiederholen Sie den Vorgang.
- Waschen Sie die Proben mit sanftem Rühren für 5 min mit 100% Ethanol; Wiederholen Sie den Vorgang.
- Während sich die Proben in 100% Ethanolwäschen in einem Abzug befinden, bereiten Sie eine 50:50 (v/v) Propylenoxid (PO):Epon-Mischung in einem Glasfläschchen mit einem propylenoxid (PO)-beständigen Kunststoffdeckel vor. Behutsam, aber sicher den Deckel der Glasdurchstechflasche befestigen und während Sie sowohl den Deckel als auch die Durchstechflasche festhalten, schütteln oder wirbeln, um sie zu mischen.
HINWEIS: Propylenoxid ist ein akut giftiger, brennbarer Reizstoff, der einige Kunststoffe auflöst. Tragen Sie geeignete persönliche Schutzausrüstung und führen Sie das Experiment in einem Abzug durch. - Nach Schritt 2.6 werden die Proben mit PO:Epon für 1 h in einem PO-resistenten Behälter (z. B. Aluminiumschalen oder Glasfläschchen) mit sanftem Schaukeln/Rühren im Abzug inkubiert.
- Übertragen Sie die Proben im Abzug auf 100% Epon. Inkubation für 2 h bei Raumtemperatur im Abzug mit Schaukeln/Drehen/Rühren. Übertragen Sie die PO:Epon-Mischung in eine spezielle Epon-Abfallflasche aus Glas.
- Wiederholen Sie Schritt 2.9 einmal.
- Montieren Sie die Muster zum Einbetten. Abhängig von der Größe der Scheiben und dem interessierenden Bereich montieren Sie die Scheiben direkt auf vorpolymerisierte Harzstummel oder betten Sie sie zur Dissektion flach ein und betten Sie sie zu einem späteren Zeitpunkt wieder ein.
HINWEIS: Für die flache Einbettung kann eine "Cast-a-Folie" verwendet werden, um viele Scheiben gleichzeitig einzubetten. Übrig gebliebenes Harz kann verwendet werden, um Balkenkapseln zu füllen und zu backen, um vorpolymerisierte Stümpfe herzustellen oder für den späteren Gebrauch einzufrieren. - Sobald sie montiert und mit ausreichend Harz bedeckt sind, um den "Cast-a-Slide" -Hohlraum zu füllen, backen Sie die Proben über Nacht in einem 60 ° C-Ofen.
HINWEIS: Möglicher Pausenpunkt. Proben können jahrelang bei Raumtemperatur gelagert werden. - Identifizieren Sie für die erneute Einbettung den interessierenden Bereich in den flachen, eingebetteten Gewebeschnitten. Mit einer Juweliersäge das Stück Gewebe der entsprechenden Größe (1 mm 2 bis 4 mm 2) ausschneiden und mit Harz, das wie in Schritt2.2 vorbereitet wurde, wieder auf die Oberseite eines vorpolymerisierten Blocks einbetten und über Nacht in einem 60 °C heißen Ofen backen.
HINWEIS: Alternativ kann das Gewebestück mit zweiteiligem Epoxidharz an einen Stumpf oder Stift geklebt werden. Über Nacht gehen lassen. Potenzieller Pausenpunkt.
3. Beschneiden und serielles Schneiden von eingebetteten Proben
HINWEIS: Das Schneiden ist eine erlernte Fähigkeit; Benutzer sollten mit ultradünnen Schnitten vertraut sein, bevor sie versuchen, serielle Abschnitte zu schneiden. Da die genauen Mikrotomkontrollen von Hersteller zu Hersteller variieren, befolgen Sie die Anweisungen und Richtlinien des Herstellers.
- Wenn die Probe im Trimmadapter eingeschlossen ist, verwenden Sie eine Rasierklinge, um das in Harz eingebettete Gewebe sorgfältig zu trimmen, um die folgenden Kriterien zu erfüllen (siehe Abbildung 1A,B):
- Stellen Sie sicher, dass eine flache, obere Oberfläche vorhanden ist, die das Gewebe um die interessierende Region freilegt.
- Stellen Sie eine trapezförmige Form sicher, wobei die Ober- und Unterkante sauber und parallel sind.
- Stellen Sie Gesamtabmessungen von 200-500 μm in x, 100-500 μm in y sicher.
- Stellen Sie eine asymmetrische Blockfläche sicher, z. B. rechte Seitenecken von ~ 90 °, die obere linke Ecke stumpf und die linke untere Ecke akut.
HINWEIS: Ein Trimm-Kryomesser kann ein alternatives Werkzeug für eine Rasierklinge sein. Die anderen Empfehlungen dienen der Benutzerfreundlichkeit für die Bestellung von Abschnitten bei der Bildgebung. Optional: Wenn Abschnitte keine stabilen Bänder bilden, kann ein Kontaktzement auf die Vorderkante der Blockfläche aufgetragen werden, um die Bandbildung zu unterstützen. Rasierklingen sind scharf; Achten Sie darauf, die Rasierklinge so zu halten, dass versehentliche Ausrutscher wahrscheinlich keinen persönlichen Schaden verursachen.

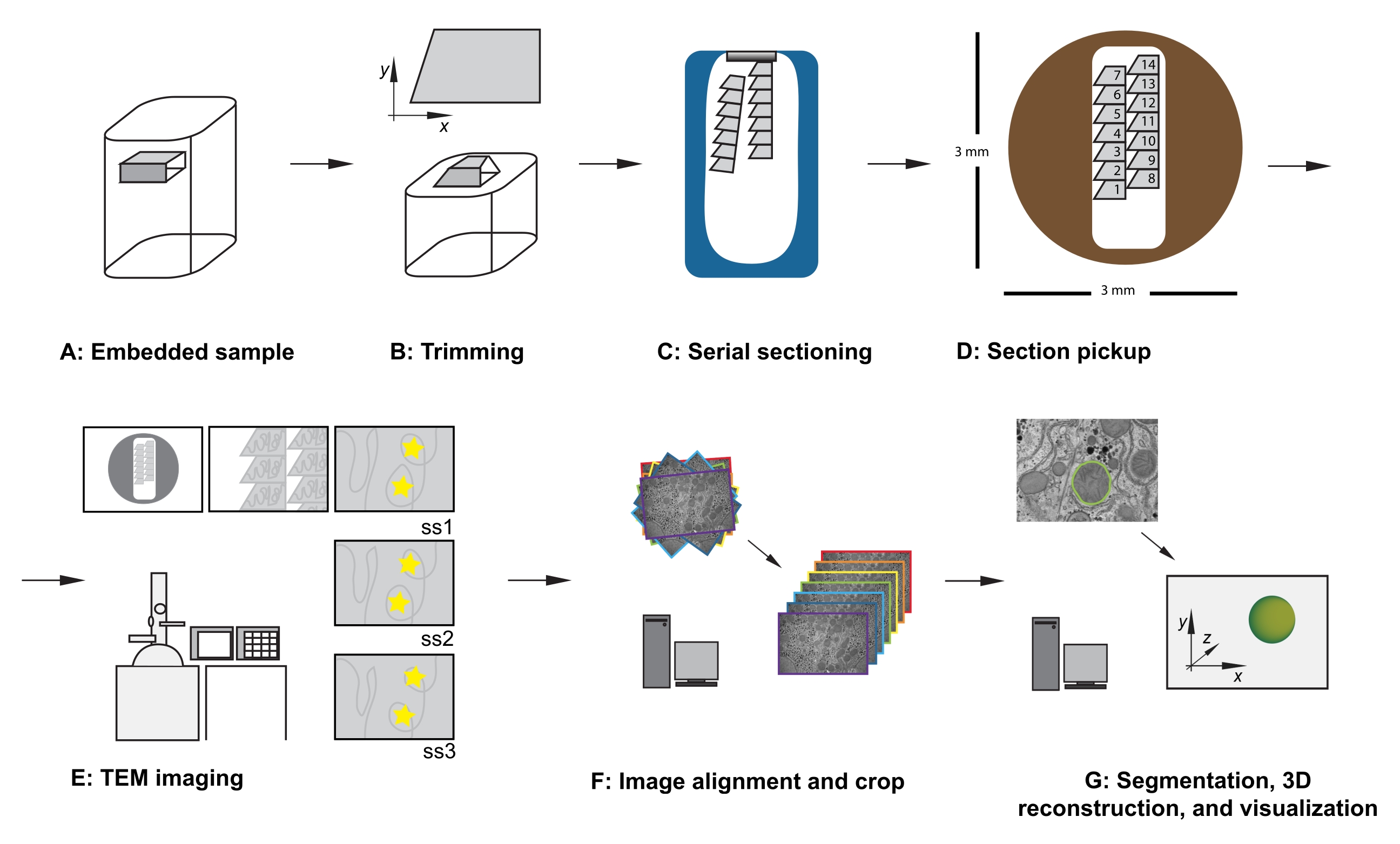
Abbildung 1: Serieller TEM-Workflow. (A) Diagramm der Probe im Harzblock. (B) Trimmen Sie den Block, um eine Trapezform mit Kanten zu erzeugen, die für die serielle Teilung geeignet sind, und einer asymmetrischen Blockfläche, um die bekannte Orientierung zu gewährleisten. (C) Diagramm mit Bändern aus seriellen Abschnitten, die im Diamantmesserboot auf der Wasseroberfläche schwimmen. (D) Diagramm, das die Organisation des Abschnitts und des Menübands zeigt, das die Reihenfolge der Abschnitte auf einem TEM-Schlitzraster mit einem Durchmesser von 3 mm vorgibt. (E) TEM-Bildgebung und -Navigation. Anzeigen der Menüband- und Abschnittsreihenfolge und Verwendung von "gelben Sternaufklebern" auf dem Monitor für die Bildschirmreferenzierung, um eine erneute Darstellung derselben Region von Interesse in nachfolgenden Abschnitten sicherzustellen. (F) Bildausrichtung und Zuschneiden. (G) Segmentierung, 3D-Rekonstruktion und Visualisierung. Abkürzung: TEM = transmission electron microscopy. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Nach dem Beschneiden übertragen Sie den Probenbogen zusammen mit dem Spannfutter und der Probe auf den Probenarm des Mikrotoms und positionieren den Probenbogen so, dass der Verfahrbereich des Lichtbogens von oben nach unten verläuft. Sichern Sie den Probenbogen an Ort und Stelle.
- Legen Sie das Diamantmesser in den Messerhalter und verriegeln Sie es, um sicherzustellen, dass der Schnittwinkel entsprechend dem Messer eingestellt ist. Verriegeln Sie den Messerhalter sicher in der Bühne.
- Verwenden Sie bei eingeschalteter Bühne den Messervorschub, während Sie ständig die Beziehung zwischen der Blockfläche und der Messerkante überprüfen. Bewegen Sie das Messer vorsichtig in Richtung der Probe und passen Sie kontinuierlich den seitlichen Winkel des Messers, die Neigung der Probe und die Rotation der Probe an, indem Sie die entsprechenden Knöpfe einstellen, bis der Block an der Messerkante ausgerichtet ist.
- Schalten Sie die Bühnenbeleuchtung aus; Schalten Sie das Downlighting der Bühne ein; Stellen Sie die Ober- und Unterseite des Schneidfensters des Probenarms ein; und lassen Sie das Exemplar knapp unter der Messerschneide.
- Füllen Sie das Messerboot mit sauberem ddH2O und stellen Sie sicher, dass die Wasseroberfläche auf Höhe der Messerkante und nur leicht konkav ist.
- Optional: Tauchen Sie eine Wimpern in den 0,1% Triton X-100 und dann in das Wasser des Messerboots, um die Oberflächenspannung des Wassers zu reduzieren, um die Chloroformung und die Bandaufnahme zu unterstützen.
- Bereiten Sie die Workstation mit Wimpern (Wimpern auf einen Cocktailstock geklebt), formvarbeschichteten Schlitzgittern, etikettierten Crossover-Pinzetten, Chloroform, 0,1% Triton X-100-Lösung, destilliertem Wasser, Filterpapier und Gitterbox mit Gitterbox-Notizen vor.
- Stellen Sie die Schnittgeschwindigkeit auf 1 mm/s und die anfängliche Schnittdicke auf 100 nm ein und starten Sie den Schneidzyklus.
- Nachdem der erste Abschnitt geschnitten wurde, ändern Sie die Schnittparameter auf eine Schnittgeschwindigkeit von 0,8 mm/s und die Schnittdicke auf 70 nm und setzen Sie das Schneiden fort, so dass die Abschnitte ein Band bilden können, das sich an der Oberfläche des mit Wasser gefüllten Messerboots bewegt (Abbildung 1C).
HINWEIS: Es ist wichtig, sich der Farbe der hergestellten Abschnitte bewusst zu sein, da dies oft ein genauerer Anhaltspunkt für die Dicke der Harzabschnitte ist. Silberschnitte sind in der Regel etwa 70 nm dick, während graue Abschnitte dünner und Goldabschnitte dicker sind. - Lassen Sie das Mikrotom weiterhin Abschnitte schneiden und das Band länger werden.
HINWEIS: Es ist wichtig, große Vibrationen und körperliche Störungen im Raum zu vermeiden. Zugluft kann dazu führen, dass sich die Abschnitte auf der Wasseroberfläche im Messerboot bewegen, und physische Vibrationen können dazu führen, dass das Mikrotom ungleichmäßig schneidet. - Sobald genügend Abschnitte gesammelt sind und bevor das Band das Ende des Bootes erreicht, stoppen Sie das Schneiden (kurz nachdem die Probe die Messerkante passiert hat).
HINWEIS: Die Anzahl der benötigten Abschnitte hängt von der Größe der Blockfläche und der Größe des zu sammelnden Datensatzes ab. Daher ist es nützlich, sich der Beziehung zwischen der Größe des Blocks und dem Schlitzgitter bewusst zu sein, wenn sich die geschnittenen Abschnitte lösen. - Verwenden Sie eine Wimpern in jeder Hand, brechen Sie das Band vorsichtig in kleinere Bänder, die in die Länge des Schlitzgitters passen, und achten Sie darauf, ihre relativen Positionen innerhalb der Probe zu notieren.
HINWEIS: Wenn ihre kombinierte Breite passt, können mehrere Bänder vorsichtig nebeneinander platziert und zusammen in einem einzigen Schlitzraster aufgenommen werden. Wenn Sie mehrere Farbbänder in einem einzigen Schlitzraster aufnehmen, achten Sie auf die Reihenfolge und die relative Position der Farbbänder. Platzieren Sie die Farbbänder beispielsweise immer weiter in der Stichprobe rechts neben einem bereits im Beispiel vorhandenen Menüband (Abbildung 1D). - Optional: Bewegen Sie mit einem Glasapplikatorstab einen Tropfen Chloroform über die Abschnitte, um sie abzuflachen.
HINWEIS: Chloroform ist giftig und reizend. Lassen Sie Chloroform nicht die Wasseroberfläche oder -abschnitte berühren. Wenn dies versehentlich der Fall ist, muss das Wasser entfernt und das Messer gewaschen werden, bevor zum Schneiden zurückkehrt. Das Chloroform kann die Abschnitte beschädigen und den Kleber, der den Diamanten sichert, in das Messerboot abbauen. - Nehmen Sie mit der ersten nummerierten Pinzette das erste leere Schlitzgitter auf (auf der rechten Seite des Schlitzes, Formvar-Seite nach unten), tauchen Sie vorsichtig in den Triton X-100 und dann zweimal in das destillierte Wasser, bevor Sie überschüssiges Wasser mit einem Stück Filterpapier von der Pinzettenkante entfernen.
- Mit einer Wimpern in der einen Hand und der Pinzette in der anderen Hand senken Sie vorsichtig etwa 2/3des formvarbeschichteten Schlitzgitters in das Wasser des Messerbootes (weg von den Abschnitten), so dass die Formvar-Seite nach unten zeigt und die rechte lange Kante des Schlitzes an der Wasseroberfläche und parallel zur Wasserkante liegt.
- Wehen Sie das Gitter sanft im Wasser in Richtung der Bänder, so dass beim Rückwärtsstrich die Abschnitte zum Gitter driften. Fahren Sie damit fort, dies in immer kleineren Wafts zu tun, bis der rechte Rand des Menübands mit dem rechten Rand des Steckplatzes übereinstimmt. Bringen Sie dann mit dem letzten Waft das Raster vorsichtig nach oben, um die Abschnitte in das Slot-Gitter aufzunehmen.
- Lassen Sie das Raster in der Pinzette trocknen, bevor Sie es in der Rasterbox aufbewahren, die auf dem Referenzblatt der Rasterbox entsprechend kommentiert ist.
- Wiederholen Sie Schritt 3.16, bis alle Menübänder gesammelt sind, und stellen Sie sicher, dass die Reihenfolge der Menübänder beibehalten wird.
- Wenn weitere Abschnitte erforderlich sind, ziehen Sie das Messer um etwa 150 nm ein, überprüfen Sie den Wasserstand im Boot und fügen Sie bei Bedarf weitere hinzu. Starten Sie den Schneidvorgang erneut, indem Sie die Schritte 3.11-3.18 ausführen.
- Sobald alle Abschnitte gesammelt sind, stellen Sie sicher, dass die Messerkante frei von Abschnittsablagerungen ist, ziehen Sie das Messer von der Blockfläche weg und entfernen und reinigen Sie das Messer.
4. Rasterfärbung
- Nach dem Trocknen färben Sie die Abschnitte mit Reynolds' Bleicitrat entweder auf Parafilm auf der Bank oder in einer Petrischale. Legen Sie mehrere Pellets Natriumhydroxid unter einen Petrischalendeckel, um eine kohlendioxidfreie Umgebung zu schaffen. Dann, vorsichtig, weg von den Pellets, pipettieren Sie 40 μL Tropfen von Reynolds' Bleicitrat auf den Parafilm, einen für jedes Gitter.
HINWEIS: Färben Sie nicht zu viele Gitter auf einmal; z.B. sollte das Maximum 6 sein. Versuchen Sie, nicht direkt auf der Färbeschale zu atmen. Kohlendioxid kann mit dem Bleicitrat reagieren und unerwünschte Ausfällungen auf den Gittern verursachen. - Drehen Sie jedes Gitter (Abschnittsseite nach unten) auf den Bleicitratabwurf um und lassen Sie es 7 bis 10 Minuten durch den Petrischalendeckel geschützt. Während die Gitter färben, bereiten Sie ein größeres zweites Stück Parafilm auf der Bank mit fünf 300 μL Tropfen destilliertem Wasser für jedes Gitter vor.
- Am Ende der Bleicitrat-Inkubation übertragen Sie jedes Gitter in einen Tröpfchen destilliertes Wasser, um es 1 Minute lang zu waschen, ohne direkt auf den Gittern zu atmen.
- Wiederholen Sie Schritt 4.3 insgesamt fünfmal.
- Nehmen Sie mit nummerierten Crossover-Pinzetten das erste Gitter auf, berühren Sie den Rand des Gitters, um Papier zu filtern, um den größten Teil des Wassers abzuleiten, und lassen Sie es in der Pinzette trocknen (für mindestens 20 Minuten). Wiederholen Sie den Vorgang für jedes Raster.
5. Bilderfassung durch TEM
HINWEIS: Da die genauen TEM-Kontrollen von Hersteller zu Hersteller variieren, befolgen Sie die Anweisungen und Richtlinien des Herstellers. Die folgenden Schritte sollten von Benutzern ausgeführt werden, die bereits über TEM-Kenntnisse verfügen.
- Führen Sie vor der Bildgebung die üblichen Überprüfungen durch, z. B. Strahlausrichtung, Verstärkungsreferenzen und Probeneuzentrizität.
- Laden Sie vorsichtig das erste Gitter serieller Abschnitte in den Probenhalter und achten Sie darauf, den Schlitz (und damit die Abschnitte) an der vertikalen Achse des Mikroskoptisches auszurichten.
HINWEIS: Diese Genauigkeit ist nicht unbedingt erforderlich, spart jedoch Zeit bei der Erfassung und zukünftigen Datenverarbeitungsphasen. Achten Sie beim Einfügen des Rasters (Schnittseite nach unten oder Schnittseite nach oben) darauf, alle Raster in der gleichen Ausrichtung abzubilden. - Beobachten Sie bei geringer Vergrößerung die Reihenfolge, Position und Position der seriellen Abschnitte (Abbildung 1E). Navigieren Sie zum mittleren Abschnitt der Serie im Raster.
HINWEIS: Abhängig vom genauen Forschungsziel können die Ansätze für die Bildgebung variieren. Das Folgende ist jedoch ein nützlicher Ausgangspunkt. Die Form der Abschnitte und die Beziehung der Farbbänder (wie in Schritt 3.14 aufgegriffen) bestimmen, welcher Abschnitt der erste und welcher Abschnitt der letzte im Raster war. - Durchsuchen Sie das Beispiel, und identifizieren Sie eine Region von Interesse. Beobachten Sie die Probe bei der gewünschten Vergrößerung und erwägen Sie, die Reihe mit einer etwas niedrigeren Vergrößerung zu sammeln, da die Abschnitte oft nicht perfekt ausgerichtet sind und die Bilder möglicherweise später zugeschnitten werden müssen.
- Nehmen Sie Referenzbilder bei geringerer Vergrößerung auf, um den Kontext der Region von Interesse, ihre ungefähre Position bei verschiedenen Vergrößerungen in Bezug auf Abschnittsgrenzen und die Markierungsmerkmale innerhalb der Stichprobe zu schätzen. Verwenden Sie diese, um die Region von Interesse in anderen Abschnitten zu kennzeichnen.
- Optional: Verwenden Sie für die Bildschirmreferenzierung wiederverwendbaren Klebekitt, Aufkleber oder ein Stück Overheadprojektorpapier (OHP), das auf den Bildschirm geklebt wird, um temporäre Markierungen auf dem Bildschirm zu platzieren, die eine routinemäßige Neudarstellung derselben Merkmale des interessierenden Bereichs in der Mitte des Bildes im gesamten Datensatz ermöglichen (siehe gelbe Sterne in Abbildung 1E).
- Navigieren Sie mithilfe der Referenzbilder zu dem interessierenden Bereich im ersten Abschnitt des Rasters und erfassen Sie ein Bild mit der gewünschten Vergrößerung.
HINWEIS: Notieren Sie sich beim Speichern von Bildern den ersten Dateinamen des ersten Bildes der Serie und verwenden Sie die sequentielle Namensnomenklatur, damit alle Bildnamen der sequenziellen Reihenfolge der seriellen Abschnitte folgen. - Navigieren Sie zum nächsten Abschnitt, und wiederholen Sie Schritt 5.7, bis alle Abschnitte für diese Region von Interesse abgebildet sind.
6. Bildexport und serielle Ausrichtungsregistrierung
- Exportieren Sie die Bilddateien, die zum selben Stapel gehören, in einen einzigen Ordner. Stellen Sie sicher, dass der Ordner nach Dateinamen sortiert ist.
HINWEIS: Die Bilder sollten idealerweise den gleichen Stammnamen haben und der Reihenfolge folgen, in der sie erworben wurden. - Öffnen Sie Fidschi und klicken Sie auf Datei | | importieren Bildreihenfolge.
- Klicken Sie auf das erste Bild des Ordners und klicken Sie auf Öffnen. Warten Sie, bis ein Popup-Fenster mit Sequenzoptionen angezeigt wird (Abbildung 2A).

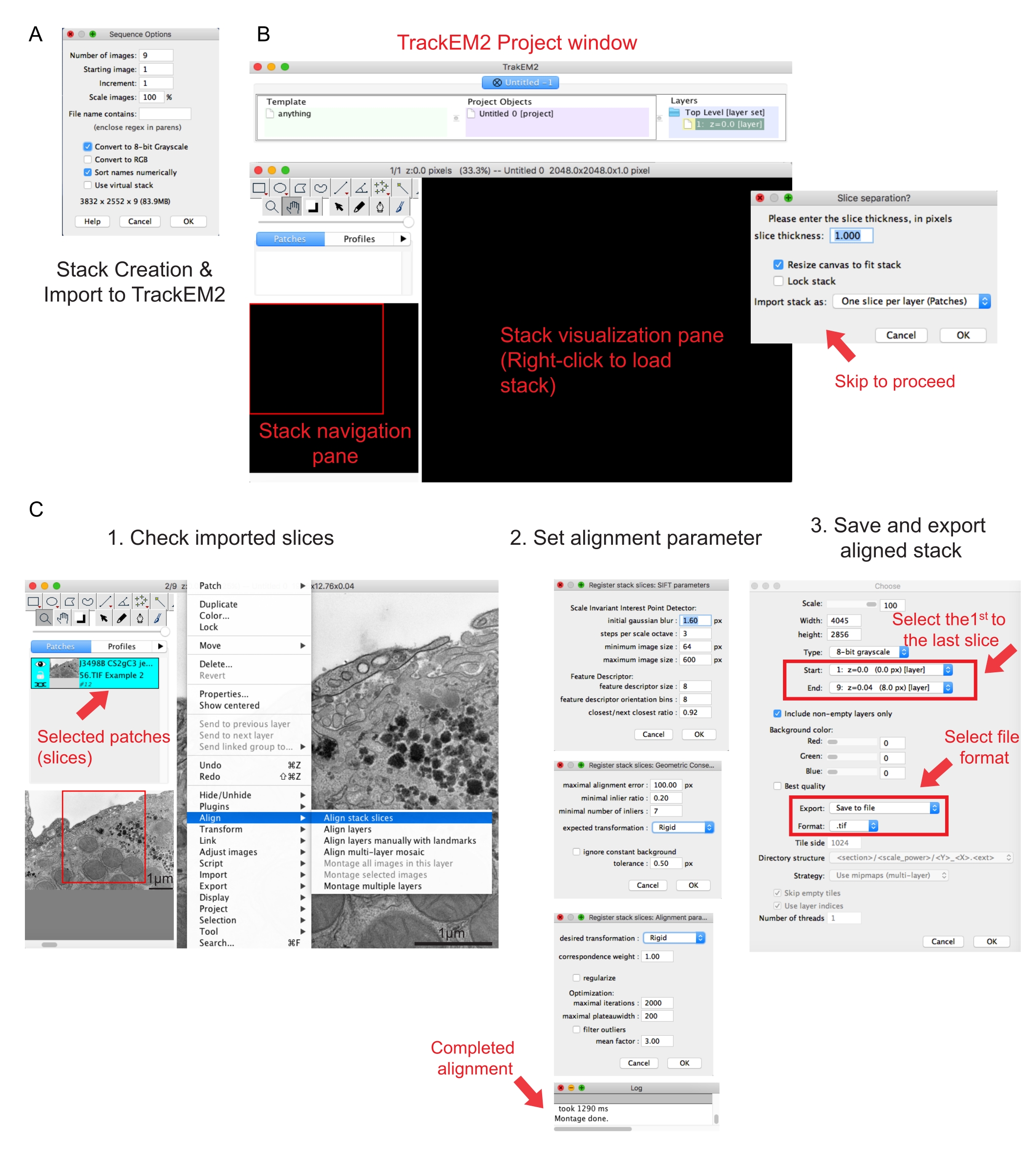
Abbildung 2: Erstellung eines seriellen Stacks und serieller Abschnittsausrichtung mit Fidschi. (A) Screenshot mit den Sequenzoptionen beim Laden der Bilder zum Erstellen eines seriellen Stacks. (B) Screenshot des TrackEM2-Plugins und der Schlüsselfenster des Plugins. Drücken Sie OK in der Slice-Trennung , um mit der Ausrichtung fortzufahren. (C) Screenshot nach erfolgreichem Laden des seriellen Stacks in den Visualisierungsbereich. Drei aufeinanderfolgende Fenster mit Ausrichtungsparametern werden angezeigt, sobald die Stapelsegmente ausrichten ausgewählt sind. Exportieren Sie den ausgerichteten Stapel, sobald die Ausrichtung abgeschlossen ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Klicken Sie auf Namen numerisch sortieren | Konvertieren Sie in 8-Bit-Option. Drücken Sie OK.
HINWEIS: Die Konvertierung in 8-Bit erleichtert den Import der Daten in Amira und reduziert die Dateigröße, was eine schnellere Verarbeitungsgeschwindigkeit in späteren Schritten ermöglicht. - Überprüfen Sie die Vollständigkeit, Reihenfolge und Vergrößerung des erstellten Stacks. Speichern Sie den erstellten Stapel als .tif Datei.
HINWEIS: Bilder sollten mit der gleichen Vergrößerung aufgenommen worden sein. - Führen Sie das TrakEM2-Plugin41 aus. Gehe zu Datei | Neue | TrackEM2 (leer).
HINWEIS: Das Plugin fordert den Benutzer auf, die TrackEM2-Dateien zu speichern. Speichern Sie bei Bedarf die TrackEM2-Dateien im Bildordner. Es sollten drei Fenster angezeigt werden: ein Projektfenster, ein Stapelnavigationsfenster (links) und ein Stapelvisualisierungsbereich (Abbildung 2B). - Klicken Sie mit der rechten Maustaste auf den schwarzen Visualisierungsbereich. Klicken Sie auf | importieren Importieren Sie den Stack und wählen Sie den zuvor erstellten Stack aus.
- Klicken Sie auf OK, um den Stapel in das Stapelnavigationsfenster zu laden.
HINWEIS: Ein Fenster zur Trennung von Slice wird angezeigt, in dem Sie nach der Beziehung zwischen Pixel und Dimension gefragt werden. Klicken Sie für nur die Stapelausrichtung auf OK, um diesen Schritt zu überspringen. - Verwenden Sie den Schieberegler, um alle Segmente des Stapels zu überprüfen. Suchen Sie nach dem geladenen Segment, das als Patch im Navigationsplan angezeigt wird. Wählen Sie die Patches aus, die in der folgenden Ausrichtung enthalten sein sollen.
HINWEIS: Ausgewählte Patches werden blau. - Bewegen Sie den Mauszeiger über den Anzeigebereich. Klicken Sie mit der rechten Maustaste auf das Bild, wählen Sie | ausrichten Ausrichten von Stapelsegmenten (Abbildung 2C-1).
- Geben Sie die Ausrichtungsparameter über einen Satz von drei aufeinanderfolgenden Fenstern an.
HINWEIS: Beginnen Sie bei den meisten Daten mit einer starren Ausrichtung (ermöglicht Rotation und Verschiebung, aber keine Transformation) und behalten Sie andere Parameter als Standard bei (Abbildung 2C-2). - Lassen Sie die Ausrichtung laufen, bis das Ausleseprotokoll Montage beendet anzeigt.
HINWEIS: Die Laufzeit hängt von der Anzahl der Voxel und der Geschwindigkeit des Computers ab. - Überprüfen Sie den ausgerichteten Stapel im Anzeigebereich. Drücken Sie die Tasten Alt und - (im PC) oder die Tasten Strg und - (im Mac), um eine Verkleinerungsansicht des ausgerichteten Stapels zu erhalten.
- Wenn der ausgerichtete Stapel zufrieden ist, klicken Sie mit der rechten Maustaste auf | exportieren Erstellen Sie ein flaches Bild , um den ausgerichteten Stapel zu speichern.
- Wählen Sie das erste Bild als Anfang des Stapels und das letzte Bild als Ende des Stapels aus, und klicken Sie auf OK (Abbildung 2C-3). Speichern Sie den ausgerichteten Stapel als .tif.
HINWEIS: Um die Dateigröße zu reduzieren, schneiden Sie die Daten so zu, dass sie nur die erforderliche Region von Interesse enthalten. - Führen Sie bei Bedarf eine affine Ausrichtung auf dem ausgerichteten Stapel aus. Öffnen Sie den ausgerichteten Stapel in Fidschi, wählen Sie Plugin | Registrierung | StackReg.
- Wählen Sie die affine Option und drücken Sie OK. Warten Sie, bis das Programm abgeschlossen ist.
- Speichern Sie den affine-ausgerichteten Stapel mit einem anderen Dateinamen.
7. Segmentierung und 3D-Rekonstruktion
- Öffnen Sie Amira42. Klicken Sie auf Datei | Öffnen Sie Daten , um den ausgerichteten Stack zu laden.
- Geben Sie die Voxelmessungen im neuen Popup-Fenster an (Abbildung 3A).

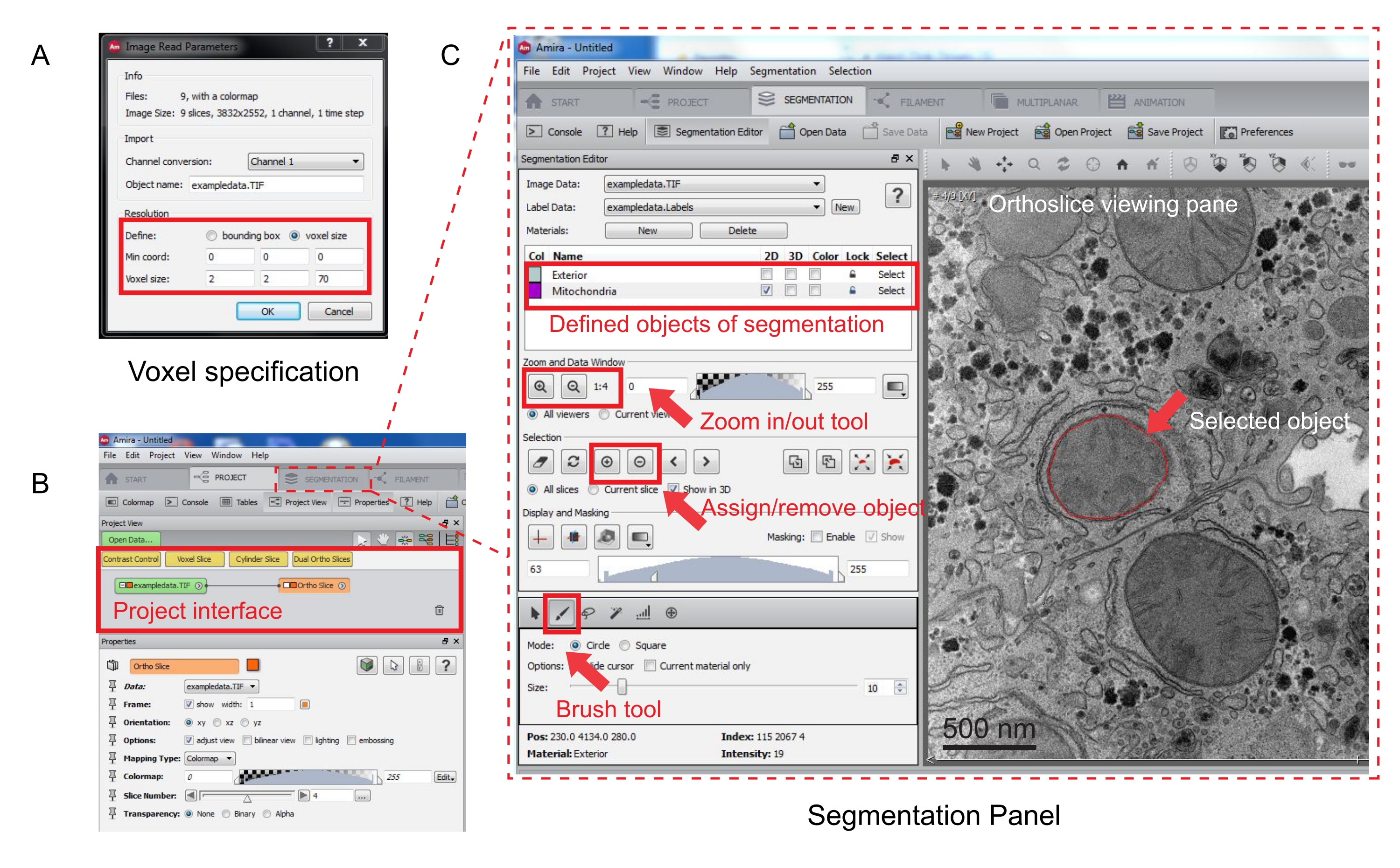
Abbildung 3: Segmentierung des seriellen Stacks mit Amira. (A) Das Voxel-Definitions-Popup-Fenster vor dem Laden eines ausgerichteten Stacks. (B) Screenshot der Projektoberfläche nach dem Import eines Stacks. Wählen Sie die Registerkarte Segmentierung aus, um die Objektverfolgung im Bedienfeld "Segmentierungs-Editor" zu starten. (C) Hauptmerkmale der Registerkarte Segmentierung. Definieren Sie die Objekte für die Segmentierung im Abschnitt Segmentierungs-Editor der Registerkarte Segmentierung. Verwenden Sie die Zoom-Funktion, um die Identifizierung von Objekten zu unterstützen. Wählen Sie das Pinselwerkzeug aus, und zeichnen Sie die Begrenzung des Objekts nach. Klicken Sie auf das +-Symbol unter Auswahl, um die Ablaufverfolgung zuzuweisen. Ein zugewiesenes Objekt scheint im Orthoslice-Anzeigebereich eine rote Berandung zu haben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
HINWEIS: Ein Bildstapelknoten wird in der Projektoberfläche und ein Orthoslice im Anzeigebereich auf der rechten Seite angezeigt (Abbildung 3B).
- Um die Segmentierung zu starten, wählen Sie die Registerkarte Segmentierung (Abbildung 3B).
HINWEIS: Es wird empfohlen, den Segmentierungsfortschritt vor und während der Segmentierung zu speichern. Zum Modell| Speichern Sie das Modell Als eine beliebige AM-Datei, die geeignet ist. - Klicken Sie im Bedienfeld des Segmentierungseditors auf Neu , um neue Objekte in der Materialliste zu definieren. Klicken Sie mit der rechten Maustaste, um die Farbe des Objekts zu ändern, und doppelklicken Sie, um das Objekt umzubenennen.
- Für die manuelle Segmentierung wählen Sie das Segmentierungswerkzeug unterhalb der Materialliste. Wählen Sie das Standardpinsel-Werkzeug aus, um die Pixel hervorzuheben (Abbildung 3C).
HINWEIS: Alternativ können Sie das Pinselwerkzeug verwenden, um den Umriss des Objekts zu verfolgen, und Umschalt + F drücken, um das Objekt zu füllen. - Um das Pinselwerkzeug in einen Radiergummi umzuwandeln, drücken Sie ständig die Strg-Taste , während Sie Pixel für die Korrektur auswählen. Kommentieren Sie jedes Segment im Stapel.
- Weisen Sie nach der Bestätigung die Auswahl einer Beschriftung zu, indem Sie auf das +- Zeichen klicken. Klicken Sie auf das Zeichen - , um die Auswahl zu entfernen.
- Kehren Sie zur Projektoberfläche zurück, sobald die Segmentierung abgeschlossen ist. Suchen Sie nach einem Knoten mit der Erweiterung ".label", die mit dem Image-Stack verbunden ist.
- Klicken Sie mit der rechten Maustaste auf die Erweiterung ".label", und wählen Sie Surface | generieren aus. Anwenden , um eine .surf-Datei zu erstellen.
- Um das 3D-Modell eines segmentierten Objekts zu rendern, klicken Sie mit der rechten Maustaste auf die SURF-Datei, und wählen Sie Oberflächenansicht aus, um im Anzeigebereich ein 3D-Modell zu generieren.
- Speichern Sie das 3D-Modell zur Visualisierung oder weiteren quantitativen Analysen.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Für diese Technik werden interessante Regionen auf der Grundlage des biologischen Forschungsziels ausgewählt und vor dem Trimmen und Schneiden von eingebettetem Gewebe identifiziert. Ebenso kann die Größe der Blockfläche durch die Forschungsfrage diktiert werden; In diesem Fall wurde die Probe so beschnitten, dass eine Blockfläche von etwa 0,3 mm x 0,15 mm übrig blieb (Abbildung 4A). Dies ermöglichte zwei Gitter von 9 seriellen Abschnitten pro Gitter, die 18 serielle Abschnitte liefe...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Eine zugängliche vEM-Technik zur Visualisierung der Organellenstruktur und -interaktionen in 3D wird in diesem Protokoll beschrieben. Die Morphologie der interorganellen Kontakte in Hepatozyten wird hier als Fallstudie vorgestellt. Dieser Ansatz wurde jedoch auch zur Untersuchung einer Vielzahl anderer Proben und Forschungsbereiche angewendet, darunter Schwann-Zell-Endothel-Interaktionen in peripheren Nerven 45, Weibel-Palade-Körper-Biogenese in Endothelzellen46, Frachtsekretion in Nierenzellen ...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Wir danken Joanna Hanley, Rebecca Fiadeiro und Ania Straatman-Iwanowska für die fachkundige technische Unterstützung. Wir danken auch den Stefan-Labormitgliedern und Ian J. White für die hilfreichen Gespräche. J.J.B. wird durch MRC-Mittel für das MRC Laboratory of Molecular Cell Biology am UCL, Award Code MC_U12266B, unterstützt. C.J.S. wird durch MRC-Mittel für das MRC Laboratory of Molecular Cell Biology University Unit am UCL, Vergabecode MC_UU_00012/6, unterstützt. P.G. wird gefördert durch den Europäischen Forschungsrat, Förderkennzeichen ERC-2013-StG-337057.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.22 µm syringe filter | Sarstedt | 83.1826.001 | |
| Aluminum trays | Agar Scientific | AGG3912 | |
| Amira v6 | ThermoFisher | https://www.thermofisher.com | |
| Chloroform | Fisher | C/4960/PB08 | |
| DDSA/Dodecenyl Succinic Anhydride | TAAB | T027 | Epon ingredient |
| Diamond knife | DiaTOME | ultra 45° | |
| DMP-30/2,4,6-tri (Dimethylaminomethyl) phenol | TAAB | D032 | Epon ingredient |
| Dumont Tweezers N5 | Agar Scientific | AGT5293 | |
| Fiji | https://imagej.net/ | ||
| Fiji TrakEM2 plugin | https://imagej.net/ | ||
| Formaldehyde 36% solution | TAAB | F003 | |
| Formvar coated slot grid | Homemade | Alternative: EMS diasum (FF2010-Cu) | |
| Glass bottle with applicator rod | Medisca | 6258 | |
| Glass vials | Fisher Scientific | 15364769 | |
| Gluteraldehyde 25% solution | TAAB | G011 | |
| MNA/Methyl Nadic Anhydride | TAAB | M011 | Epon ingredient |
| Osmium Tetroxide 2% solution | TAAB | O005 | |
| Potassium Ferricyanide | Sigma-Aldrich | P-8131 | |
| Propylene oxide | Fisher Scientific | E/0050/PB08 | |
| Reuseable adhesive | Blue Tack | ||
| Reynolds Lead Citrate | TAAB | L037 | Section stain |
| Sodium Cacodylate | Sigma-Aldrich | C-0250 | to make 0.1 M Caco buffer |
| Super Glue | RS Components | 918-6872 | Cyanoacrylate glue, Step 1.3 |
| TAAB 812 Resin | TAAB | T023 | Epon ingredient |
| Tannic acid | TAAB | T046 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Two part Epoxy Resin | RS Components | 132-605 | Alternative: Step 2.13 |
| Ultramicrotome | Leica | UC7 | |
| Vibrating microtome | Leica | 100 µm thick slices, 0.16 mm/s cutting at 1 mm amplitude . | |
| Weldwood Original Contact cement | DAP | 107 | Contact adhesive: Step 3.1.4 |
Referenzen
- Knoll, M., Ruska, E. Das elektronenmikroskop. Zeitschrift für Physik. 78 (5), 318-339 (1932).
- von Ardenne, M. Daselektronen-rastermikroskop. Zeitschrift für Physik. 109 (9), 553-572 (1938).
- Bang, B. H., Bang, F. B. Graphic reconstruction of the third dimension from serial electron microphotographs. Journal of Ultrastructure Research. 1 (2), 138-139 (1957).
- Birch-Andersen, A. Reconstruction of the nuclear sites of Salmonella typhimurium from electron micrographs of serial sections. Journal of General Microbiology. 13 (2), 327-329 (1955).
- Denk, W., Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biology. 2 (11), 329(2004).
- Peddie, C. J., Collinson, L. M. Exploring the third dimension: volume electron microscopy comes of age. Micron. 61, 9-19 (2014).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure. Biology of the Cell. 108 (11), 307-323 (2016).
- Kornfeld, J., Denk, W. Progress and remaining challenges in high-throughput volume electron microscopy. Current Opinion in Neurobiology. 50, 261-267 (2018).
- Heymann, J. A., et al. Site-specific 3D imaging of cells and tissues with a dual beam microscope. Journal of Structural Biology. 155 (1), 63-73 (2006).
- Knott, G., Marchman, H., Wall, D., Lich, B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. Journal of Neuroscience. 28 (12), 2959-2964 (2008).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Martone, M. E., Deerinck, T. J., Yamada, N., Bushong, E., Ellisman, M. H. Correlated 3D light and electron microscopy: use of high voltage electron microscopy and electron tomography for imaging large biological structures. Journal of Histotechnology. 23 (3), 261-270 (2000).
- Micheva, K. D., Smith, S. J. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Sjostrand, F. S. Ultrastructure of retinal rod synapses of the guinea pig eye as revealed by three-dimensional reconstructions from serial sections. Journal of Ultrastructure Research. 2 (1), 122-170 (1958).
- Ware, R. W. Three-dimensional reconstruction from serial sections. International Review of Cytology. 40, 325(1975).
- Stevens, J. K., Davis, T. L., Friedman, N., Sterling, P. A systematic approach to reconstructing microcircuitry by electron microscopy of serial sections. Cognitive Brain Research. 2 (3), 265-293 (1980).
- Hoppe, W. Three-dimensional electron microscopy. Annual Review of Biophysics. 10, 563-592 (1981).
- Frank, J. Electron tomography: methods for three-dimensional visualization of structures in the cell. , Springer. New York, NY. (2008).
- Baumeister, W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Current Opinion in Structural Biology. 12 (5), 679-684 (2002).
- Hoog, J. L., Schwartz, C., Noon, A. T., O'Toole, E. T. Organization of interphase microtubules in fission yeast analyzed by electron tomography. Developmental Cell. 12 (3), 349-361 (2007).
- Harris, K. M., Perry, E., Bourne, J., Feinberg, M., Ostroff, L., Hurlburt, J. Uniform serial sectioning for transmission electron microscopy. Journal of Neuroscience. 26 (47), 12101-12103 (2006).
- Jesior, J. C. Use of low-angle diamond knives leads to improved ultrastructural preservation of ultrathin sections. Scanning Microscopy Supplement. 3, 147-152 (1989).
- Studer, D., Gnaegi, H. Minimal compression of ultrathin sections with use of an oscillating diamond knife. Journal of Microscopy. 197, 94-100 (2000).
- Gay, H., Anderson, T. F. Serial sections for electron microscopy. Science. 120 (3130), 1071-1073 (1954).
- Bernhard, W., Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. The Journal of Biophysical and Biochemical Cytology. 2, 73-78 (1956).
- Palade, G. E. An electron microscope study of the mitochondrial structure. The Journal of Histochemistry & Cytochemistry. 1 (4), 188-211 (1953).
- Wu, H., Carvalho, P., Voeltz, G. K. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 361 (6401), (2018).
- Vance, J. E. Inter-organelle membrane contact sites: implications for lipid metabolism. Biology Direct. 15 (1), 24(2020).
- Stefan, C. J. Endoplasmic reticulum-plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Current Opinion in Cell Biology. 63, 125-134 (2020).
- Zaman, M. F., Nenadic, A., Radojicic, A., Rosado, A., Beh, C. T. Sticking with it: ER-PM membrane contact sites as a coordinating nexus for regulating lipids and proteins at the cell cortex. Frontiers in Cell and Developmental Biology. 8, 675(2020).
- van Vliet, A. R., Sassano, M. L., Agostinis, P. The unfolded protein response and membrane contact sites: tethering as a matter of life and death. Contact. 1, 1-15 (2018).
- Cohen, S., Valm, A. M., Lippincott-Schwartz, J. Interacting organelles. Current Opinion in Cell Biology. 53, 84-91 (2018).
- Hariri, H., et al. Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress. EMBO Reports. 19 (1), 57-72 (2018).
- Stefan, C. J., Trimble, W. S., Grinstein, S., Drin, G. Membrane dynamics and organelle biogenesis-lipid pipelines and vesicular carriers. BMC Biology. 15 (1), 102(2017).
- Eisenberg-Bord, M., Shai, N., Schuldiner, M., Bohnert, M. A tether is a tether is a tether: tethering at membrane contact sites. Developmental Cell. 39 (4), 395-409 (2016).
- Scorrano, L., De Matteis, M. A., Emr, S., Giordano, F. Coming together to define membrane contact sites. Nature Communications. 10 (1), 1287(2019).
- Lak, B., Li, S., Belevich, I., Sree, S. Specific subdomain localization of ER resident proteins and membrane contact sites resolved by electron microscopy. European Journal of Cell Biology. 100 (7), 151180(2021).
- Collado, J., Kalemanov, M., Campelo, F., Bourgoint, C. Tricalbin-mediated contact sites control ER curvature to maintain plasma membrane integrity. Developmental Cell. 51 (4), 476-487 (2019).
- West, M., Zurek, N., Hoenger, A., Voeltz, G. K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. Journal of Cell Biology. 193 (2), 333-346 (2011).
- Ilacqua, N., Anastasia, I., Raimondi, A., Lemieux, P. A three-organelle complex made by wrappER contacts with peroxisomes and mitochondria responds to liver lipid flux changes. Journal of Cell Science. 135 (5), 259091(2022).
- Cardona, A., Saalfeld, S., Schindelin, J., Arganda-Carreras, I. TrakEM2 software for neural circuit reconstruction. PLoS One. 7 (6), 38011(2012).
- Stalling, D., Westerhoff, M., Hege, H. -C. Amira: A highly interactive system for visual data analysis. The Visualization Handbook. 38, 749-767 (2005).
- Hsieh, T. S., Chen, Y. J., Chang, C. L., Lee, W. R., Liou, J. Cortical actin contributes to spatial organization of ER-PM junctions. Molecular Biology of the Cell. 28 (23), 3171-3180 (2017).
- Anastasia, I., Ilacqua, N., Raimondi, A., Lemieux, P. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Reports. 34 (11), 108873(2021).
- Cattin, A. L., Burden, J. J., Van Emmenis, L., Mackenzie, F. E. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell. 162 (5), 1127-1139 (2015).
- Lopes-da-Silva, M., et al. A GBF1-dependent mechanism for environmentally responsive regulation of ER-Golgi transport. Developmental Cell. 49 (5), 786-801 (2019).
- Banushi, B., Forneris, F., Straatman-Iwanowska, A., Strange, A. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. Nature Communications. 7, 12111(2016).
- Rey, S. A., et al. Ultrastructural and functional fate of recycled vesicles in hippocampal synapses. Nature Communications. 6, 8043(2015).
- Belicova, L., Repnik, U., Delpierre, J., Gralinska, E. Anisotropic expansion of hepatocyte lumina enforced by apical bulkheads. Journal of Cell Biology. 220 (10), 202303003(2021).
- Kizilyaprak, C., Daraspe, J., Humbel, B. M. Focused ion beam scanning electron microscopy in biology. Journal of Microscopy. 254 (3), 109-114 (2014).
- Xu, C. S., Hayworth, K. J., Lu, Z., Grob, P. Enhanced FIB-SEM systems for large-volume 3D imaging. Elife. 6, 1-36 (2017).
- Parlakgül, G., Arruda, A. P., Cagampan, E., Pang, S. High resolution 3D imaging of liver reveals a central role for subcellular architectural organization in metabolism. bioRxiv. , (2020).
- Guerin, C. J., Kremer, A., Borghgraef, P., Lippens, S. Targeted studies using serial block face and focused ion beam scan electron microscopy. The Journal of Visualized Experiments: JoVE. (150), e59480(2019).
- Kremer, A., et al. A workflow for 3D-CLEM investigating liver tissue. Journal of Microscopy. 281 (3), 231-242 (2021).
- Hayat, M. Principles and techniques of electron microscopy: biological applications. , Cambridge University Press. (2000).
- Wisse, E., Braet, F., Duimel, H., Vreuls, C. Fixation methods for electron microscopy of human and other liver. World Journal of Gastroenterology. 16 (23), 2851-2866 (2010).
- Hanley, J., Dhar, D. K., Mazzacuva, F., Fiadeiro, R. Vps33b is crucial for structural and functional hepatocyte polarity. Journal of Hepatology. 66 (5), 1001-1011 (2017).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. Microscopy. 1, 6-8 (2010).
- Miranda, K., Girard-Dias, W., Attias, M., de Souza, W., Ramos, I. Three dimensional reconstruction by electron microscopy in the life sciences: An introduction for cell and tissue biologists. Molecular Reproduction and Development. 82 (7-8), 530-547 (2015).
- Yamaguchi, M., Chibana, H. A method for obtaining serial ultrathin sections of microorganisms in transmission electron microscopy. The Journal of Visualized Experiments: JoVE. (131), e56235(2018).
- Hall, D. H., Hartwieg, E., Nguyen, K. C. Modern electron microscopy methods for C. elegans. Methods in Cell Biology. 107, 93-149 (2012).
- Hagler, H. K. Ultramicrotomy for biological electron microscopy. Methods in Molecular Biology. 369, 67-96 (2007).
- Arganda-Carreras, I., et al. Consistent and elastic registration of histological sections using vector-spline regularization. Computer vision approaches to medical image analysis, CVAMIA 2006, Lecture Notes in Computer Science. Beichel, R. R., Sonka, M., et al. 4241, Springer Berlin Heidelberg. Berlin, Heidelberg. 85-95 (2006).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biology. 14 (1), 1002340(2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218, 52-61 (2005).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD). Journal of Structural Biology. 116 (1), 71-76 (1996).
- Iudin, A., Korir, P. K., Salavert-Torres, J., Kleywegt, G. J., Patwardhan, A. EMPIAR: a public archive for raw electron microscopy image data. Nature Methods. 13 (5), 387-388 (2016).
- Xu, C. S., Pang, S., Shtengel, G., Muller, A. An open-access volume electron microscopy atlas of whole cells and tissues. Nature. 599 (7883), 147-151 (2021).
- Karabag, C., et al. Semantic segmentation of HeLa cells: An objective comparison between one traditional algorithm and four deep-learning architectures. PLoS One. 15 (10), 0230605(2020).
- Heinrich, L., Bennett, D., Ackerman, D., Park, W. Whole-cell organelle segmentation in volume electron microscopy. Nature. 599 (7883), 141-146 (2021).
- Kim, J. S., Greene, M. J., Zlateski, A., Lee, K. Space-time wiring specificity supports direction selectivity in the retina. Nature. 509 (7500), 331-336 (2014).
- Spiers, H., Songhurst, H., Nightingale, L., de Folter, J. Deep learning for automatic segmentation of the nuclear envelope in electron microscopy data, trained with volunteer segmentations. Traffic. 22 (7), 240-253 (2021).
- Hasan, N. M., Gupta, A., Polishchuk, E., Yu, C. H. Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. The Journal of Biological Chemistry. 287 (43), 36041-36050 (2012).
- Stoeck, I. K., Lee, J. Y., Tabata, K., Romero-Brey, I. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. The Journal of Virology. 92 (1), 01196(2018).
- Ma, X., Qian, H., Chen, A., Ni, H. M., Ding, W. X. Perspectives on mitochondria-ER and mitochondria-lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells. 10 (9), 2273(2021).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten