
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Caractérisation tridimensionnelle des sites de contact interorganelles dans les hépatocytes à l’aide de la microscopie électronique à section série
Dans cet article
Résumé
Un protocole simple et complet pour acquérir des détails tridimensionnels des sites de contact membranaire entre les organites dans les hépatocytes du foie ou les cellules d’autres tissus.
Résumé
La microscopie électronique à transmission a longtemps été considérée comme la référence en matière de visualisation de l’ultrastructure cellulaire. Cependant, l’analyse est souvent limitée à deux dimensions, ce qui entrave la capacité de décrire pleinement l’ultrastructure tridimensionnelle (3D) et la relation fonctionnelle entre les organites. La microscopie électronique volumique (vEM) décrit un ensemble de techniques qui permettent l’interrogation de l’ultrastructure cellulaire en 3D à des résolutions à méso-échelle, micro-échelle et nanométrique.
Ce protocole fournit une méthode accessible et robuste pour acquérir des données vEM à l’aide de la transmission de section série EM (TEM) et couvre les aspects techniques du traitement des échantillons jusqu’à la reconstruction 3D numérique dans un flux de travail unique et simple. Pour démontrer l’utilité de cette technique, la relation ultrastructurale 3D entre le réticulum endoplasmique et les mitochondries et leurs sites de contact dans les hépatocytes hépatiques est présentée. Les contacts interorganelles jouent un rôle essentiel dans le transfert d’ions, de lipides, de nutriments et d’autres petites molécules entre les organites. Cependant, malgré leur découverte initiale dans les hépatocytes, il reste encore beaucoup à apprendre sur leurs caractéristiques physiques, leur dynamique et leurs fonctions.
Les contacts interorganelles peuvent afficher une gamme de morphologies, variant dans la proximité des deux organites l’un à l’autre (généralement ~ 10-30 nm) et l’étendue du site de contact (des contacts ponctués aux contacts cisternels 3D plus grands). L’examen des contacts étroits nécessite une imagerie à haute résolution, et la section série TEM est bien adaptée pour visualiser l’ultrastructure 3D des contacts interorganelles lors de la différenciation des hépatocytes, ainsi que les altérations de l’architecture des hépatocytes associées aux maladies métaboliques.
Introduction
Depuis leur invention dans les années 1930, les microscopes électroniques ont permis aux chercheurs de visualiser les composants structurels des cellules et des tissus 1,2. La plupart des enquêtes ont fourni des informations 2D, car la construction de modèles 3D nécessite une collecte minutieuse de sections en série, une photographie manuelle, un traitement négatif, un traçage manuel et la création et l’assemblage de modèles 3D à partir de feuilles de verre, de plastique ou de polystyrène 3,4. Près de 70 ans plus tard, il y a eu des progrès considérables dans de nombreux aspects du processus, des performances des microscopes, de la collecte de sections en série, de l’imagerie numérique automatisée, des logiciels et du matériel sophistiqués pour la reconstruction, la visualisation et l’analyse 3D aux approches alternatives pour ce qui est maintenant collectivement appelé volume EM (vEM). Ces techniques de vEM sont généralement considérées comme fournissant des informations ultrastructurales 3D à des résolutions nanométriques à l’échelle du micron et englobent la microscopie électronique à transmission (TEM) et les nouvelles techniques de microscopie électronique à balayage (MEB); voir avis 5,6,7,8.
Par exemple, le SEM à faisceau d’ions focalisé (FIB-SEM) utilise un faisceau d’ions focalisés à l’intérieur d’un SEM pour fraiser la surface du bloc entre les balayages d’imagerie SEM séquentiels de la surface du bloc, ce qui permet le fraisage / imagerie automatisé répété d’un échantillon et la construction d’un ensemble de données 3D pour la reconstruction 9,10. En revanche, le SEM de face de bloc série (SBF-SEM) utilise un ultramicrotome à l’intérieur du SEM pour retirer du matériau de la face de bloc avant l’imagerie11,12, tandis que la tomographie en réseau est un processus non destructif qui nécessite la collecte de sections série, sur des couvercles, des plaquettes ou des bandes, avant de mettre en place un flux de travail automatisé d’imagerie de la région d’intérêt dans les sections séquentielles dans le SEM pour générer le jeu de données 3D13 . Semblable à la tomographie par matrice, la section série TEM (ssTEM) nécessite que les sections physiques soient collectées avant l’imagerie; cependant, ces sections sont collectées sur des grilles TEM et imagées dans un TEM 14,15,16. ssTEM peut être étendu en effectuant une tomographie d’inclinaison 17,18,19. La tomographie par inclinaison en série offre la meilleure résolution en x, y et z, et bien qu’elle ait été utilisée pour reconstruire des cellules entières20, elle est raisonnablement difficile. Ce protocole se concentre sur les aspects pratiques de ssTEM en tant que technique vEM la plus accessible disponible pour de nombreux laboratoires EM qui n’ont peut-être pas actuellement accès à des instruments de sectionnement ou de vEM spécialisés, mais qui bénéficieraient de la génération de données vEM 3D.
L’ultramicrotomie en série pour la reconstruction 3D a déjà été considérée comme difficile. Il était difficile de couper des rubans droits d’épaisseur de section uniforme, d’être capable d’organiser et de ramasser des rubans de la bonne taille, dans le bon ordre, sur des grilles avec un support suffisant, mais sans barres de grille obscurcissant les régions d’intérêt, et surtout, sans perdre de sections, car une série incomplète peut empêcher une reconstruction 3D complète21. Cependant, les améliorations apportées aux ultramicrotomes commerciaux, aux couteaux de coupe et de coupe au diamant 22,23, aux films de support électroniques sur les grilles21,24 et aux adhésifs pour faciliter l’adhérence des sections et la préservation du ruban13,21 ne sont que quelques-unes des avancées progressives au fil des ans qui ont rendu la technique plus courante dans de nombreux laboratoires. Une fois les sections série collectées, l’imagerie série dans TEM est simple et peut fournir des images EM avec des tailles px subnanométriques en x et y, permettant une interrogation à haute résolution des structures subcellulaires - une exigence potentielle pour de nombreuses questions de recherche. L’étude de cas présentée ici démontre l’utilisation de ssTEM et de la reconstruction 3D dans l’étude des contacts endoplasmiques réticulum (ER)-organites dans les hépatocytes hépatiques, où les contacts ER-organites ont été observés pour la première fois25,26.
Tout en étant contiguë à l’enveloppe nucléaire, l’ER établit également des contacts étroits avec de nombreux autres organites cellulaires, notamment les lysosomes, les mitochondries, les gouttelettes lipidiques et la membrane plasmique27. Les contacts ER-organites ont été impliqués dans le métabolisme des lipides28, la signalisation29 du phosphoinositide et du calcium, la régulation de l’autophagie et la réponse au stress30,31. Les contacts ER-organites et autres contacts interorganelles sont des structures très dynamiques qui répondent aux besoins métaboliques cellulaires et aux signaux extracellulaires. Il a été démontré qu’ils varient morphologiquement dans leur taille et leur forme et les distances entre les membranes des organites32,33. On pense que ces différences ultrastructurales sont susceptibles de refléter leurs différentes compositions protéiques / lipidiques et leur fonction34,35. Cependant, il reste difficile de définir les contacts interorganelles et de les analyser36. Par conséquent, un protocole fiable mais simple pour examiner et caractériser les contacts interorganelles est nécessaire pour des investigations plus approfondies.
Comme les contacts ER-organites peuvent aller de 10 à 30 nm dans la séparation membrane-membrane, l’étalon-or pour l’identification a toujours été TEM. Le TEM à section mince a révélé une localisation spécifique du sous-domaine pour les protéines ER résidentes à des contacts membranaires distincts37. Traditionnellement, cela a révélé des contacts ER-organites avec une résolution nm, mais n’a souvent présenté qu’une vue 2D de ces interactions. Cependant, les approches vEM révèlent la présentation ultrastructurale et le contexte de ces sites de contact en 3D, permettant une reconstruction complète des contacts et une classification plus précise des contacts (point vs tubulaire vs cisternal) et quantification38,39. En plus d’être le premier type de cellule où des contacts ER-organites ont été observés25,26, les hépatocytes ont un vaste système d’autres contacts interorganelles qui jouent un rôle vital dans leur architecture et leur physiologie28,40. Cependant, une caractérisation morphologique approfondie des contacts ER-organites et autres contacts interorganelles dans les hépatocytes fait encore défaut. En conséquence, la façon dont les contacts interorganelles se forment et se remodèlent pendant la régénération et la réparation est particulièrement pertinente pour la biologie des hépatocytes et la fonction hépatique.
Protocole
Tous les animaux ont été logés conformément aux directives du ministère de l’Intérieur du Royaume-Uni et la récolte des tissus a été effectuée conformément à la loi britannique de 1986 sur les animaux (procédures scientifiques).
1. Fixation et préparation de l’échantillon
- Disséquer le tissu hépatique en morceaux de taille appropriée, environ 8 mm x 8 mm x 3 mm, et placer les morceaux dans une solution saline chaude tamponnée au phosphate (PBS, 37 °C).
- Injecter un fixateur à température ambiante (20-25 °C) (1,5 % de glutaraldéhyde dans 1 % de saccharose, 0,1 M de cacodylate de sodium) dans les morceaux de foie et les transférer du PBS au fixateur jusqu’à 20 minutes à température ambiante. Gardez toujours le tissu immergé dans des solutions pour éviter le dessèchement.
REMARQUE: Les aldéhydes sont des irritants corrosifs et potentiellement cancérigènes. Le cacodylate de sodium est toxique s’il est avalé ou inhalé. Tous les fixateurs doivent être manipulés tout en portant un équipement de protection individuelle approprié, et l’expérience doit être effectuée dans une hotte aspirante. Une bonne fixation se traduira par un tissu plus ferme. - Configurez le microtome vibrant avec une lame, un bain de glace et un plateau tampon froid rempli de PBS. Montez le premier morceau de tissu hépatique fixe sur un porte-échantillon avec de la colle cyanoacrylate et transférez le bloc au microtome vibrant.
- En suivant les recommandations du fabricant, approchez le tissu et coupez le foie fixe en tranches de 100 μm d’épaisseur.
- Recueillir les tranches à l’aide d’une spatule ou d’un pinceau à cheveux naturels et les transférer dans un plat de 12 ou 24 puits contenant une solution glacée (1,5 % de glutaraldéhyde, 0,1 M de cacodylate de sodium) sur de la glace. Laissez les tranches sur de la glace jusqu’à ce que tous les échantillons aient été tranchés et soient prêts à être traités plus avant.
- Sélectionnez les tranches contenant les régions d’intérêt pour un traitement ultérieur et lavez-les avec une agitation douce. Effectuez trois lavages de 5 minutes avec un cacodylate de sodium de 0,1 M à température ambiante dans un plat de 12 ou 24 puits, en vous assurant que les tranches ont suffisamment de tampon pour se déplacer librement.
NOTE: En général, les régions d’intérêt sont sélectionnées en fonction des caractéristiques anatomiques liées à la question biologique de l’étude et guidées par des régions du tissu qui sont susceptibles d’être présentes dans toute la série, par exemple, pas sur le bord de la section, et qui sont bien conservées. - Dans une hotte, remplacer le cacodylate de sodium de 0,1 M par du tétroxyde d’osmium à 1 % et du ferricyanure de potassium fraîchement préparé. Placez le plat de 12 ou 24 puits dans un récipient scellé et transférez le récipient dans un réfrigérateur à produits chimiques dangereux pendant 1 h.
REMARQUE: L’osmium est extrêmement dangereux en cas d’ingestion, d’inhalation et de contact avec la peau. Le ferricyanure de potassium est un irritant et est nocif par inhalation et contact avec la peau. Manipulez toujours à l’aide d’un équipement de protection individuelle approprié et effectuez l’expérience dans une hotte aspirante. - Dans une hotte, retirer le tétroxyde d’osmium/ferricyanure de potassium dans une bouteille de déchets d’osmium dédiée et laver les échantillons pendant 5 min avec 0,1 M de cacodylate de sodium trois fois. Laisser les échantillons dans un récipient scellé pendant la nuit à 4 °C.
REMARQUE: Point de pause potentiel. Les échantillons peuvent être conservés dans du cacodylate de sodium de 0,1 M dans un récipient scellé à 4 °C pendant des semaines sans nuire à la conservation. Assurez-vous qu’il y a suffisamment de tampon pour empêcher le séchage. - Incuber les échantillons avec de l’acide tannique fraîchement préparé à 1% dans du cacodylate de sodium de 0,05 M pendant 45 min dans l’obscurité à température ambiante.
REMARQUE: L’acide tannique est un irritant et peut causer des dommages aux yeux. Portez un équipement de protection individuelle approprié et effectuez l’expérience dans une hotte aspirante. - Effectuez trois lavages de 5 minutes avec ddH2O avant la déshydratation et l’intégration.
2. Déshydratation de l’échantillon, incorporation de résine Epon et montage
- Préparer la résine Epon selon le rapport poids suivant (voir étape 2.2). Tare une balance avec un bécher en plastique jetable de 100 mL contenant une barre de remuage. Coupez les extrémités de 5 pipettes Pasteur en plastique jetables et utilisez-les pour transférer des composants en résine visqueuse dans le bécher.
- Ajouter séquentiellement 19,2 g de résine 812, 7,6 g de DDSA, 13,2 g de MNA et 0,8 g d’accélérateur DMP-30 dans le bécher. À l’aide de la cinquième pipette Pasteur en plastique propre, mélangez soigneusement les composants en résine à la main.
REMARQUE: Évitez d’introduire des bulles, mais assurez-vous d’un mélange suffisant de la résine inférieure avec le dessus pour obtenir un changement de couleur et un mélange brutal des couches de composants. Tous les composants de la résine sont irritants et nocifs par inhalation et contact avec la peau. Le DMP-30 est corrosif et peut provoquer une corrosion cutanée. Portez un équipement de protection individuelle approprié. - Placez le bécher sur un agitateur magnétique et laissez remuer doucement, en mélangeant périodiquement la résine manuellement.
- Laver les échantillons avec une agitation douce pendant 5 min avec 70% d’éthanol; répétez une fois.
- Laver les échantillons avec une agitation douce pendant 5 min avec de l’éthanol à 90%; répétez une fois.
- Laver les échantillons avec une agitation douce pendant 5 min avec 100% d’éthanol; répétez une fois.
- Pendant que les échantillons sont dans des lavages à 100% d’éthanol dans une hotte, préparez un mélange d’oxyde de propylène (PO) (PO) 50:50 (v/ v) dans un flacon en verre avec un couvercle en plastique résistant à l’oxyde de propylène (PO). Clipsez soigneusement mais solidement sur le couvercle du flacon en verre et, tout en tenant le couvercle et le flacon, secouez ou vortexez pour mélanger.
REMARQUE: L’oxyde de propylène est un irritant extrêmement toxique et inflammable qui dissout certains plastiques. Portez un équipement de protection individuelle approprié et effectuez l’expérience dans une hotte aspirante. - Après l’étape 2.6, incuber les échantillons avec PO:Epon pendant 1 h dans un récipient résistant aux PO (p. ex., plateaux en aluminium ou flacons en verre), avec un léger balancement/agitation dans la hotte.
- Dans la hotte, transférer les échantillons à 100% Epon. Incuber pendant 2 h à température ambiante dans la hotte avec basculement/rotation/agitation. Transférer le mélange PO:Epon dans une bouteille de déchets Epon en verre dédiée.
- Répétez l’étape 2.9 une fois.
- Montez les exemples pour l’incorporation. Selon la taille des tranches et la région d’intérêt, montez directement les tranches sur des tiges de résine prépolymérisée ou incorporez-les à plat pour les dissection et réincorporez-les à une date ultérieure.
REMARQUE: Pour l’intégration à plat, une « diapositive coulée » peut être utilisée pour incorporer plusieurs tranches à la fois. Les restes de résine peuvent être utilisés pour remplir des capsules de faisceau et cuits au four pour fabriquer des talons prépolymérisés ou congelés pour une utilisation ultérieure. - Une fois montés et recouverts de suffisamment de résine pour remplir la cavité « coulée à glissière », cuire les échantillons pendant la nuit dans un four à 60 °C.
REMARQUE: Point de pause potentiel. Les échantillons peuvent être conservés à température ambiante pendant des années. - Pour la réintégration, identifiez la région d’intérêt dans les tranches de tissu plat incorporé. À l’aide d’une scie à bijoutier, découper le morceau de tissu de taille appropriée (1 mm2 à 4 mm2) et réencastrer à l’aide de résine préparée, comme à l’étape 2.2, sur le dessus d’un bloc prépolymérisé et cuire toute la nuit dans un four à 60 °C.
REMARQUE: Alternativement, la pièce de tissu peut être collée à un talon ou à une épingle avec de la résine époxy en deux parties. Laisser reposer pendant la nuit. Point de pause potentiel.
3. Rognage et sectionnement en série des échantillons incorporés
REMARQUE: Le sectionnement est une compétence acquise; les utilisateurs doivent maîtriser la section ultramince avant de tenter la section série. Comme les contrôles exacts des microtomes varient d’un fabricant à l’autre, suivez les instructions et les directives du fabricant.
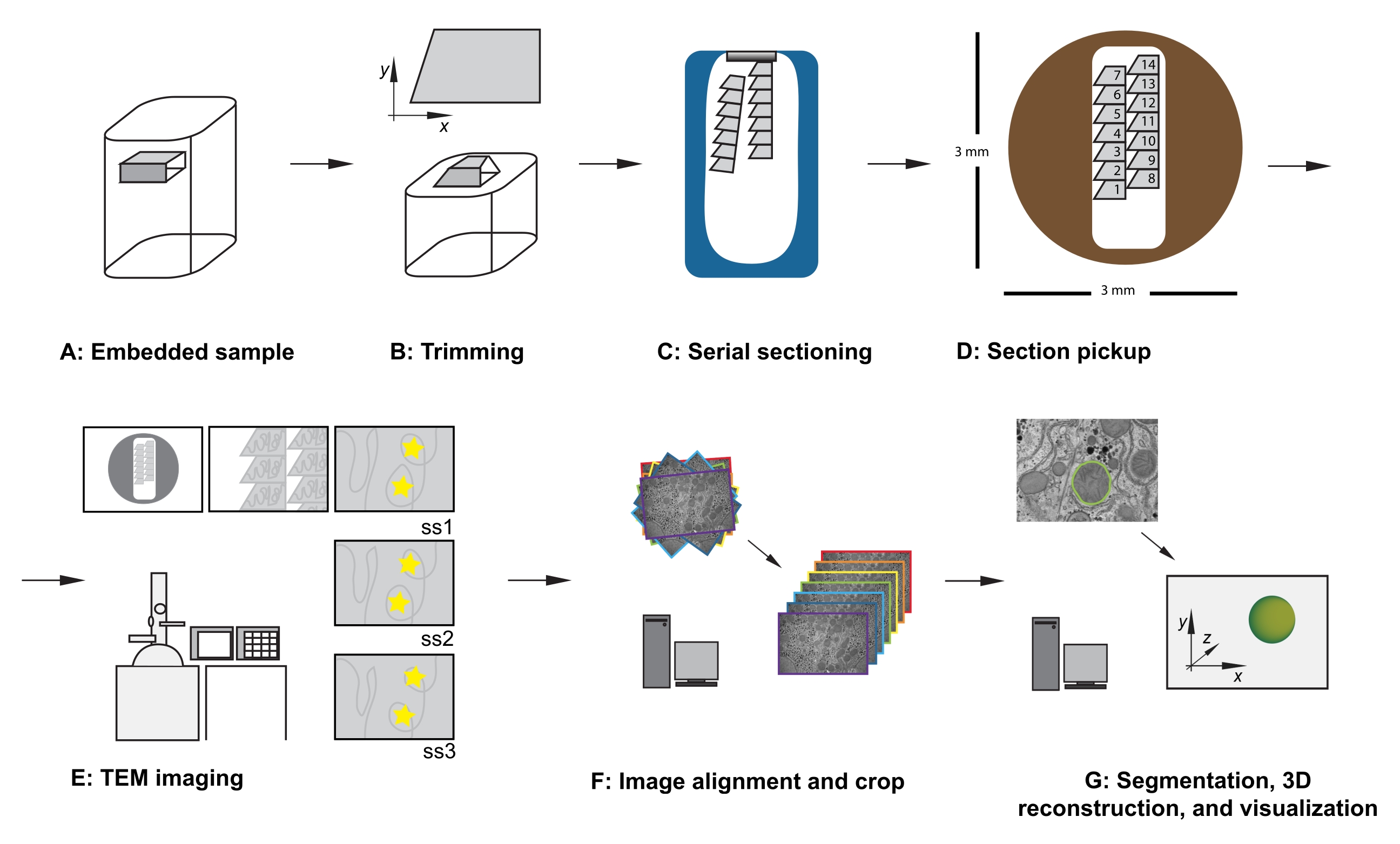
- Avec l’échantillon verrouillé dans l’adaptateur de coupe, utilisez une lame de rasoir pour couper soigneusement le tissu incorporé en résine afin de répondre aux critères suivants (voir Figure 1A, B) :
- Assurez-vous qu’il y a une surface plane et supérieure exposant le tissu autour de la région d’intérêt.
- Assurez-vous d’une forme trapézoïdale avec les bords supérieur et inférieur propres et parallèles.
- Assurer des dimensions hors tout de 200-500 μm en x, 100-500 μm en y.
- Assurez-vous d’une face de bloc asymétrique, par exemple, des coins droits d’environ 90 °, un coin supérieur gauche obtus et un coin inférieur gauche aigu.
REMARQUE: Un cryoknife de coupe peut être un outil alternatif pour une lame de rasoir. Les autres recommandations sont pour la commodité de l’utilisateur pour commander des sections lors de l’imagerie. Facultatif : Si les sections ne parviennent pas à former des rubans stables, un ciment de contact peut être appliqué sur le bord d’attaque de la face du bloc pour faciliter la formation du ruban. Les lames de rasoir sont tranchantes; prenez soin de tenir la lame de rasoir de manière à ce que des glissements accidentels ne soient pas susceptibles d’entraîner des dommages personnels.

Figure 1 : Flux de travail TEM de la section série. (A) Diagramme de l’échantillon dans le bloc de résine. (B) Bloc de coupe pour générer une forme trapézoïdale avec des bords adaptés à la section en série et une face de bloc asymétrique pour assurer une orientation connue. (C) Diagramme montrant des rubans de sections en série, flottant à la surface de l’eau dans le bateau à couteaux diamantés. (D) Schéma montrant l’organisation de la section et du ruban, dictant l’ordre des sections, sur une grille de fente TEM de 3 mm de diamètre. (E) Imagerie et navigation TEM. Afficher l’ordre du ruban et des sections et utiliser des « autocollants d’étoiles jaunes » sur le moniteur pour le référencement à l’écran afin d’assurer la réimagerie de la même région d’intérêt dans les sections suivantes. (F) Alignement et recadrage de l’image. (G) Segmentation, reconstruction 3D et visualisation. Abréviation : TEM = microscopie électronique à transmission. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Une fois coupé, transférer l’arc de l’échantillon, ainsi que le mandrin et l’échantillon, au bras de l’échantillon du microtome, en positionnant l’arc de l’échantillon de manière à ce que la plage de course de l’arc s’étende de haut en bas; fixez l’arc de l’échantillon en place.
- Placez et verrouillez le couteau à diamant dans le porte-couteau, en vous assurant que l’angle de coupe est réglé de manière appropriée au couteau. Verrouillez le porte-couteau dans la scène en toute sécurité.
- Avec l’éclairage de la scène allumé, utilisez l’avance du couteau tout en vérifiant constamment la relation entre la face du bloc et le bord du couteau. Avancez prudemment le couteau vers l’échantillon, en ajustant continuellement l’angle latéral du couteau, l’inclinaison de l’échantillon et la rotation de l’échantillon en ajustant les boutons pertinents jusqu’à ce que le bloc soit aligné sur le bord du couteau.
- Éteignez l’éclairage de la scène; allumer le downlighting de la scène; définir le haut et le bas de la fenêtre de coupe du bras de l’échantillon; et laissez le spécimen juste en dessous du bord du couteau.
- Remplissez le bateau à couteaux avec du ddH2O propre et assurez-vous que la surface de l’eau est au niveau du bord du couteau et légèrement concave.
- Facultatif : Trempez un cil dans le Triton X-100 à 0,1 %, puis dans l’eau du bateau à couteaux pour réduire la tension superficielle de l’eau afin de faciliter le chloroformage et le ramassage du ruban.
- Préparez le poste de travail avec des cils (cils collés à un bâton de cocktail), des grilles de fentes enduites de formvar, des pinces croisées étiquetées, du chloroforme, une solution Triton X-100 à 0,1%, de l’eau distillée, du papier filtre et une boîte à grille avec des notes de boîte de grille.
- Réglez la vitesse de coupe à 1 mm/s et l’épaisseur de coupe initiale à 100 nm et démarrez le cycle de coupe.
- Une fois la première section coupée, modifiez les paramètres de coupe à une vitesse de coupe de 0,8 mm/s et l’épaisseur de coupe à 70 nm, puis continuez à couper, ce qui permet aux sections de former un ruban se déplaçant le long de la surface du bateau à couteaux rempli d’eau (Figure 1C).
REMARQUE: Il est important d’être conscient de la couleur des sections produites car il s’agit souvent d’un guide plus précis de l’épaisseur des sections de résine. Les sections d’argent ont généralement une épaisseur d’environ 70 nm, tandis que les sections grises sont plus minces et les sections dorées sont plus épaisses. - Laissez le microtome continuer à couper des sections et le ruban s’allonger.
REMARQUE: Il est important d’éviter les fortes vibrations et les perturbations physiques dans la pièce. Les courants d’air peuvent provoquer le déplacement des sections à la surface de l’eau dans le bateau-couteau, et les vibrations physiques peuvent provoquer une coupe inégale du microtome. - Une fois que suffisamment de sections sont collectées et avant que le ruban n’arrive au bout du bateau, arrêtez la coupe (juste après que l’échantillon a passé le bord du couteau).
REMARQUE : Le nombre de sections nécessaires dépend de la taille de la face du bloc et de la taille du jeu de données à collecter. Ainsi, il est utile d’être conscient de la relation entre la taille du bloc et la grille de fentes lorsque les sections coupées se détachent. - À l’aide d’un cil dans chaque main, divisez doucement le ruban en rubans plus petits qui peuvent tenir dans la longueur de la grille de fente, en prenant soin de noter leurs positions relatives à partir de l’échantillon.
REMARQUE: Si leur largeur combinée convient, plusieurs rubans peuvent être délicatement placés les uns à côté des autres et ramassés ensemble dans une seule grille de fente. Si vous ramassez plusieurs rubans sur une seule grille d’emplacements, faites attention à l’ordre et à la position relative des rubans. Par exemple, placez toujours les rubans plus loin dans l’exemple à droite d’un ruban déjà présent dans l’exemple (Figure 1D). - Facultatif : À l’aide d’une tige applicatrice en verre, placez une goutte de chloroforme sur les sections pour les aplatir.
REMARQUE: Le chloroforme est toxique et irritant. Ne laissez pas le chloroforme toucher la surface ou les sections de l’eau. Si c’est le cas accidentellement, l’eau doit être retirée et le couteau lavé avant de retourner au sectionnement. Le chloroforme peut endommager les sections et dégrader la colle qui fixe le diamant dans le bateau à couteaux. - À l’aide de la première pince numérotée, ramassez la première grille de fente vide (sur le côté droit de la fente, côté formvar vers le bas), plongez doucement dans le Triton X-100, puis deux fois dans l’eau distillée avant d’éliminer l’excès d’eau du bord de la pince à l’aide d’un morceau de papier filtre.
- Avec un cil dans une main et les pinces dans l’autre main, abaissez doucement environ les 2/3 de la grille de fente enduite de formvar dans l’eau du bateau à couteaux (loin des sections), de sorte que le côté formvar soit tourné vers le bas et que le long bord droit de la fente soit à la surface de l’eau et parallèle au bord de l’eau.
- Agitez doucement la grille dans l’eau vers les rubans de sorte qu’au coup de retour, les sections dérivent vers la grille. Continuez à le faire dans des ondulations de plus en plus petites jusqu’à ce que le bord droit du ruban s’aligne avec le bord droit de la fente. Ensuite, avec la dernière vague, amenez doucement la grille vers le haut pour ramasser les sections dans la grille de fente.
- Laissez sécher la grille dans les pinces avant de la stocker dans la boîte de grille, annotée de manière appropriée sur la feuille de référence de la boîte de grille.
- Répétez l’étape 3.16 jusqu’à ce que tous les rubans soient collectés, en veillant à ce que l’ordre des rubans soit maintenu.
- Si d’autres sections sont nécessaires, rétractez le couteau à environ 150 nm, vérifiez le niveau d’eau dans le bateau et ajoutez-en d’autres si nécessaire. Recommencez le processus de coupe en suivant les étapes 3.11 à 3.18.
- Une fois toutes les sections rassemblées, assurez-vous que le bord du couteau est exempt de débris de section, retirez le couteau de la face du bloc et retirez et nettoyez le couteau.
4. Coloration de la grille
- Une fois secs, tachez les sections avec le citrate de plomb de Reynolds soit sur un parafilm sur le banc, soit dans une boîte de Pétri. Placez plusieurs pastilles d’hydroxyde de sodium sous le couvercle d’une boîte de Pétri pour fournir un environnement sans dioxyde de carbone. Puis, soigneusement, loin des granulés, pipettez 40 μL de gouttes de citrate de plomb de Reynolds sur le parafilm, une pour chaque grille.
REMARQUE: Ne pas tacher trop de grilles à la fois; Par exemple, le maximum devrait être de 6. Essayez de ne pas respirer directement sur le plat de coloration. Le dioxyde de carbone peut réagir avec le citrate de plomb et provoquer un précipité indésirable sur les grilles. - Inverser chaque grille (section côté vers le bas) sur la goutte de citrate de plomb et laisser protégé par le couvercle de la boîte de Petri pendant 7 à 10 min. Pendant que les grilles se tachent, préparez un deuxième morceau de parafilm plus grand sur le banc avec cinq gouttes d’eau distillée de 300 μL pour chaque grille.
- À la fin de l’incubation du citrate de plomb, transférer chaque grille dans une gouttelette d’eau distillée pour laver pendant 1 min sans respirer directement sur les grilles.
- Répétez l’étape 4.3 cinq fois au total.
- À l’aide de pinces croisées numérotées, ramassez la première grille, touchez le bord de la grille pour filtrer le papier afin d’évacuer la majeure partie de l’eau et laissez sécher dans la pince (pendant au moins 20 minutes). Répétez l’opération pour chaque grille.
5. Acquisition d’imagerie par TEM
REMARQUE: Comme les contrôles TEM exacts varient d’un fabricant à l’autre, suivez les instructions et les directives du fabricant. Les étapes suivantes doivent être effectuées par des utilisateurs qui maîtrisent déjà l’utilisation de la GDT.
- Avant l’imagerie, effectuez les vérifications habituelles, par exemple l’alignement du faisceau, les références de gain et l’eucentricité de l’échantillon.
- Chargez soigneusement la première grille de sections en série dans le porte-échantillon, en prenant soin d’aligner la fente (et donc les sections) sur l’axe vertical de l’étage du microscope.
REMARQUE: Cette précision n’est pas essentielle mais permet de gagner du temps lors de l’acquisition et des futures étapes de traitement des données. Lorsque vous insérez la grille (section vers le bas ou côté section vers le haut), veillez à imager toutes les grilles dans la même orientation. - À faible grossissement, observez l’ordre, l’emplacement et la position des sections série (Figure 1E). Accédez à la section centrale de la série sur la grille.
REMARQUE: Selon l’objectif exact de la recherche, les approches d’imagerie peuvent varier; cependant, ce qui suit est un point de départ utile. La forme des sections et la relation des rubans (comme indiqué à l’étape 3.14) dictent quelle section était la première et quelle section était la dernière sur la grille. - Parcourez l’échantillon et identifiez une région d’intérêt. Observez l’échantillon au grossissement souhaité et envisagez de collecter la série à un grossissement légèrement inférieur, car les sections ne sont souvent pas parfaitement alignées et les images peuvent devoir être recadrées plus tard.
- Prenez des images de référence à des grossissements plus faibles pour apprécier le contexte de la région d’intérêt, son emplacement approximatif à différents grossissements par rapport aux limites de section et les caractéristiques repères dans l’échantillon. Utilisez-les pour afficher la région d’intérêt dans d’autres sections.
- Facultatif : pour le référencement à l’écran, utilisez du mastic adhésif réutilisable, des autocollants ou un morceau de papier rétroprojecteur (OHP), scotché à l’écran, pour placer des marqueurs temporaires sur l’écran afin de permettre une réimagerie de routine des mêmes caractéristiques de la région d’intérêt au centre de l’image, dans tout le jeu de données (voir les étoiles jaunes de la figure 1E).
- À l’aide des images de référence, accédez à la région d’intérêt sur la première section de la grille et acquérez une image au grossissement souhaité.
Remarque : Lors de l’enregistrement d’images, notez le premier nom de fichier de la première image de la série et utilisez la nomenclature séquentielle des noms afin que tous les noms d’image suivent l’ordre séquentiel des sections série. - Accédez à la section suivante et répétez l’étape 5.7 jusqu’à ce que toutes les sections aient été imagées pour cette région d’intérêt.
6. Exportation d’images et enregistrement de l’alignement des sections série
- Exportez les fichiers image appartenant à la même pile dans un seul dossier. Assurez-vous que le dossier est trié par nom de fichier.
REMARQUE: Les images doivent idéalement avoir le même nom racine et suivre l’ordre dans lequel elles ont été acquises. - Ouvrez Fidji et cliquez sur Fichier | Importer | Séquence d’images.
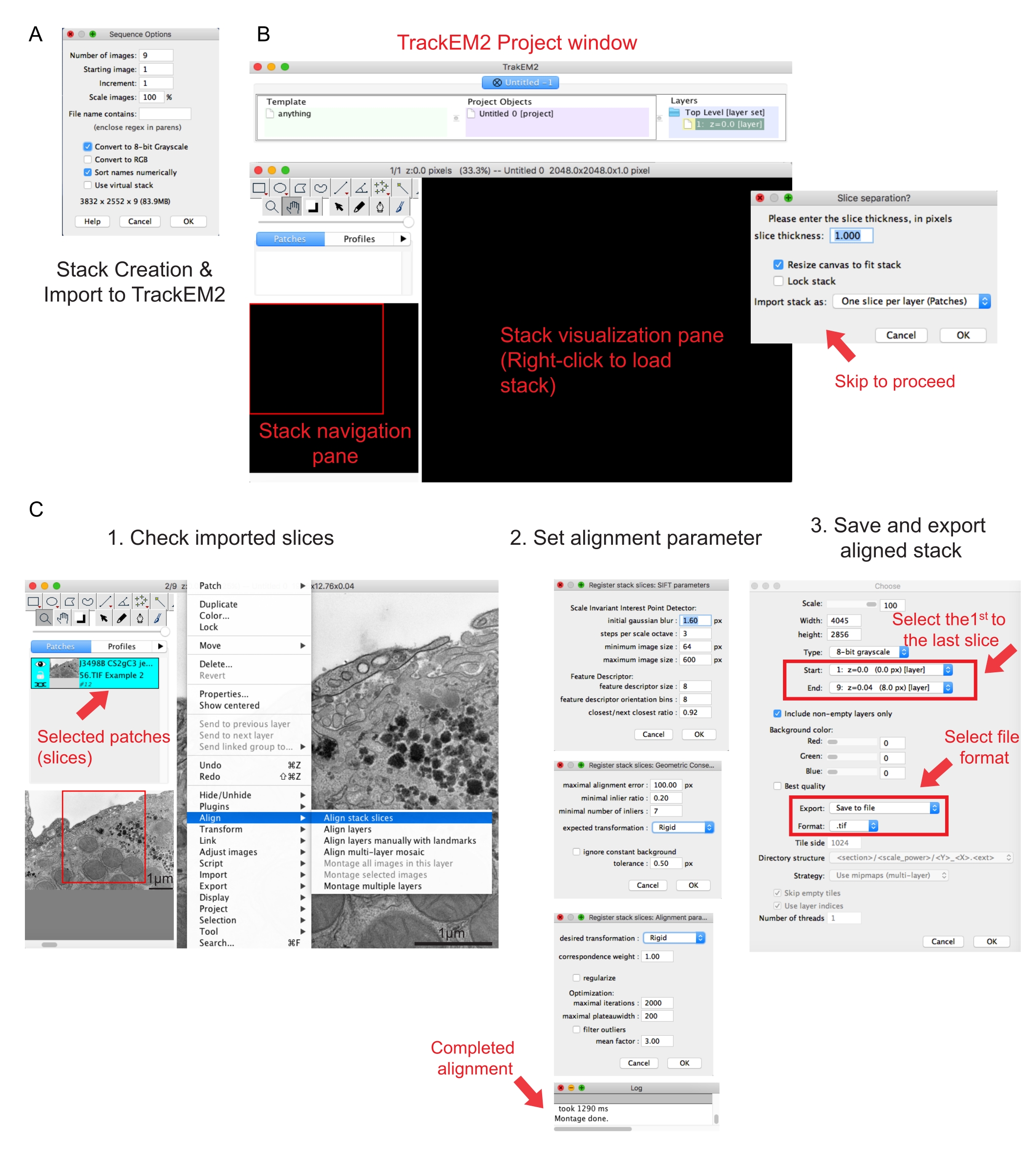
- Cliquez sur la première image du dossier et cliquez sur Ouvrir. Attendez qu’une fenêtre contextuelle options de séquence s’affiche (Figure 2A).

Figure 2 : Création d’une pile série et alignement de section série à l’aide de Fidji. (A) Capture d’écran montrant les options de séquence lors du chargement des images pour créer une pile série. (B) Capture d’écran du plugin TrackEM2 et des fenêtres clés du plugin. Appuyez sur OK dans la séparation des tranches pour poursuivre l’alignement. (C) Capture d’écran après avoir chargé avec succès la pile série dans le volet de visualisation. Trois fenêtres séquentielles de paramètres d’alignement apparaîtront une fois que les tranches de pile Aligner sont sélectionnées. Exportez la pile alignée une fois l’alignement terminé. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Cliquez sur Trier les noms numériquement | Convertir en option 8 bits . Appuyez sur OK.
REMARQUE: La conversion en 8 bits facilite l’importation des données dans Amira et réduit la taille du fichier, permettant des vitesses de traitement plus rapides dans les étapes ultérieures. - Vérifiez l’exhaustivité, la séquence et le grossissement de la pile créée. Enregistrez la pile créée en tant que fichier .tif.
REMARQUE: Les images doivent avoir été acquises au même grossissement. - Exécutez le plugin TrakEM241. Accédez à l'| de fichiers Nouvelle | TrackEM2 (vide).
REMARQUE: Le plugin demandera à l’utilisateur d’enregistrer les fichiers TrackEM2. Si nécessaire, enregistrez les fichiers TrackEM2 dans le dossier image. Trois fenêtres doivent apparaître : une fenêtre de projet, une fenêtre de navigation de pile (à gauche) et un volet de visualisation de pile (Figure 2B). - Cliquez avec le bouton droit de la souris sur le volet de visualisation noir. Cliquez sur Importer | Importez la pile et sélectionnez la pile créée précédemment.
- Cliquez sur OK pour charger la pile dans la fenêtre de navigation de la pile.
REMARQUE : Une fenêtre de séparation de tranche apparaîtra pour demander la relation entre les pixels et les dimensions. Pour l’alignement de la pile uniquement, cliquez sur OK pour ignorer cette étape. - Utilisez le curseur pour vérifier toutes les tranches de la pile. Recherchez la tranche chargée, qui apparaîtra sous la forme d’un patch dans le plan de navigation. Sélectionnez les correctifs qui seront inclus dans l’alignement suivant.
REMARQUE: Les patchs sélectionnés deviendront bleus. - Passez la souris sur le volet d’affichage. Cliquez avec le bouton droit sur l’image, sélectionnez Aligner | Aligner les tranches de pile (Figure 2C-1).
- Spécifiez les paramètres d’alignement via un ensemble de trois fenêtres séquentielles.
REMARQUE : Pour la plupart des données, commencez par un alignement rigide (permet la rotation et la translation, mais pas la transformation) et conservez les autres paramètres par défaut (Figure 2C-2). - Laissez l’alignement s’exécuter jusqu’à ce que le journal de lecture indique Montage terminé.
REMARQUE: L’exécution dépend du nombre de voxels et de la vitesse de l’ordinateur. - Vérifiez la pile alignée dans le volet d’affichage. Appuyez sur les touches Alt et - (sur PC) ou sur les touches Ctrl et - (sous Mac) pour obtenir un zoom arrière de la pile alignée.
- Si vous êtes satisfait de la pile alignée, cliquez avec le bouton droit sur Exporter | Créez une image plate pour enregistrer la pile alignée.
- Sélectionnez la première image comme début de la pile et la dernière image comme fin de la pile, cliquez sur OK (Figure 2C-3). Enregistrez la pile alignée sous forme de .tif.
REMARQUE : Pour réduire la taille du fichier, recadrez les données pour qu’elles contiennent uniquement la région d’intérêt nécessaire. - Si nécessaire, exécutez un alignement affine sur la pile alignée. Ouvrez la pile alignée dans Fidji, sélectionnez Plugin | | d’inscription StackReg.
- Choisissez l’option affine et appuyez sur OK. Attendez que le programme soit terminé.
- Enregistrez la pile alignée sur l’affine avec un nom de fichier différent.
7. Segmentation et reconstruction 3D
- Ouvrez Amira42. Cliquez sur | fichier Ouvrez Data pour charger la pile alignée.
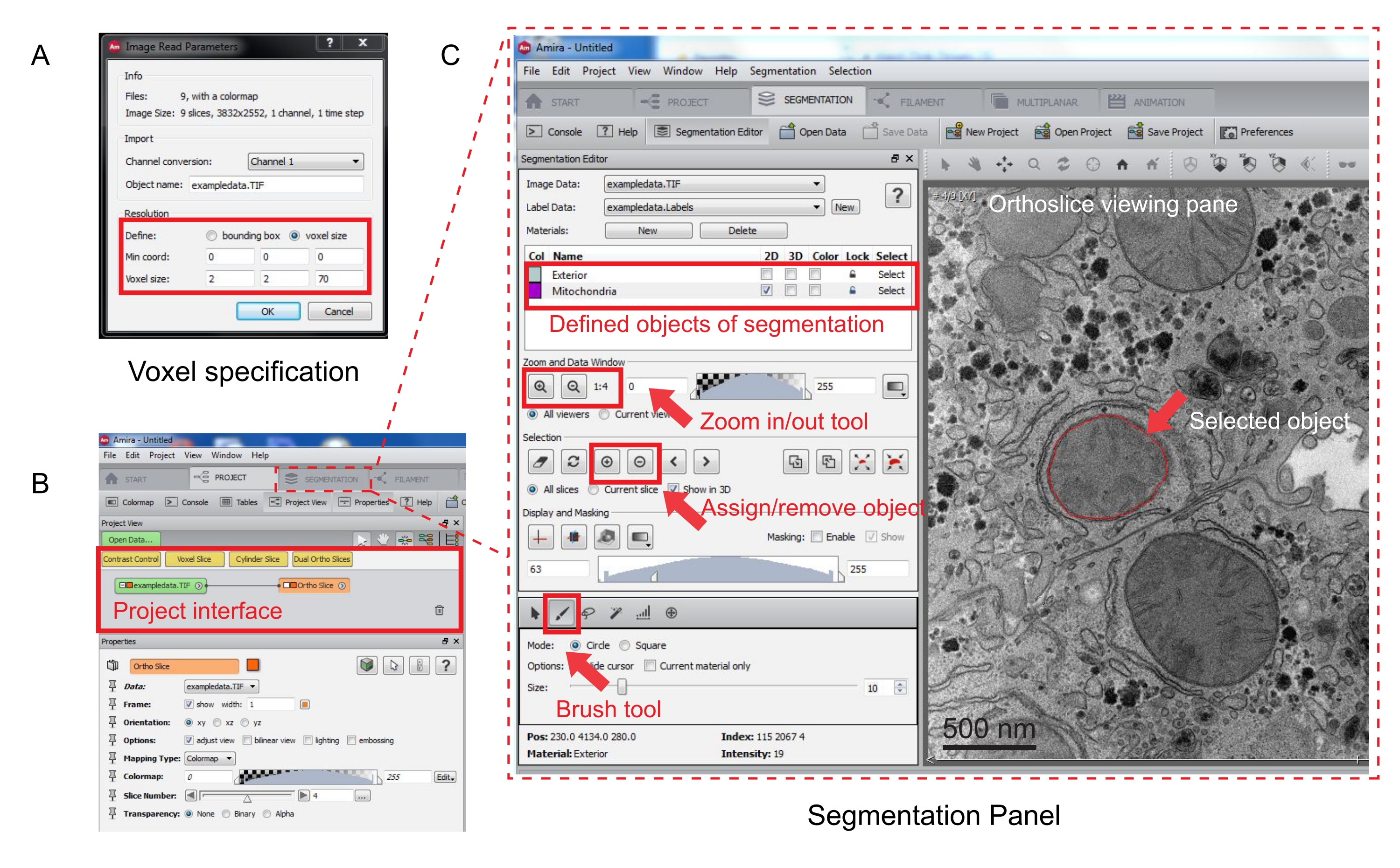
- Spécifiez les mesures voxel dans la nouvelle fenêtre contextuelle (Figure 3A).

Figure 3 : Segmentation de la pile série à l’aide d’Amira. (A) Fenêtre contextuelle de définition Voxel avant le chargement d’une pile alignée. (B) Capture d’écran de l’interface du projet après l’importation d’une pile. Sélectionnez l’onglet Segmentation pour lancer le suivi des objets dans le panneau Éditeur de segmentation. (C) Principales caractéristiques de l’onglet de segmentation. Définissez les objets à segmenter dans la section Éditeur de segmentation de l’onglet Segmentation. Utilisez la fonction de zoom pour faciliter l’identification des objets. Sélectionnez l’outil Pinceau et tracez la limite de l’objet. Cliquez sur le symbole + sous Sélection pour affecter la trace. Un objet affecté semble avoir une limite rouge dans le volet d’affichage de l’orthoslice. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Remarque : Un nœud de pile d’images apparaîtra dans l’interface du projet et une orthotranche apparaîtra dans le volet d’affichage à droite (Figure 3B).
- Pour lancer la segmentation, sélectionnez l’onglet Segmentation (Figure 3B).
REMARQUE : Il est recommandé d’enregistrer la progression de la segmentation avant et pendant la segmentation. Aller à Model | Enregistrer le modèle comme n’importe quel fichier .am qui convient. - Cliquez sur Nouveau dans le panneau de l’éditeur de segmentation pour définir de nouveaux objets dans la liste des matériaux. Cliquez avec le bouton droit de la souris pour modifier la couleur de l’objet et double-cliquez pour renommer l’objet.
- Pour la segmentation manuelle, choisissez l’outil de segmentation sous la liste des matériaux. Sélectionnez l’outil Pinceau par défaut pour mettre en surbrillance les pixels (Figure 3C).
Remarque : Vous pouvez également utiliser l’outil Pinceau pour tracer le contour de l’objet et appuyer sur Maj + F pour remplir l’objet. - Pour convertir l’outil Pinceau en gomme, appuyez continuellement sur Ctrl tout en sélectionnant les pixels à corriger. Annotez chaque tranche de la pile.
- Une fois confirmée, affectez la sélection à une étiquette en cliquant sur le signe + . Cliquez sur le signe - pour supprimer la sélection.
- Revenez à l’interface du projet une fois la segmentation terminée. Recherchez un nœud avec une extension « .label » connectée à la pile d’images.
- Cliquez avec le bouton droit sur l’extension « .label » et sélectionnez Générer surface | Appliquer pour créer un fichier .surf.
- Pour afficher le modèle 3D d’un objet segmenté, cliquez avec le bouton droit sur le fichier .surf et sélectionnez Vue Surface pour générer un modèle 3D dans le volet d’affichage.
- Enregistrez le modèle 3D pour une visualisation ou d’autres analyses quantitatives.
Résultats
Pour cette technique, les régions d’intérêt sont sélectionnées en fonction de l’objectif de la recherche biologique et identifiées avant la coupe et le découpage des tissus incorporés. De même, la taille de la face du bloc peut être dictée par la question de recherche; dans ce cas, l’échantillon a été coupé pour laisser une face de bloc d’environ 0,3 mm x 0,15 mm (figure 4A). Cela a permis deux grilles de 9 sections en série par grille, fournissant 18 sections en sér...
Discussion
Une technique vEM accessible pour visualiser la structure et les interactions des organites en 3D est décrite dans ce protocole. La morphologie des contacts interorganelles dans les hépatocytes est présentée ici comme une étude de cas. Cependant, cette approche a également été appliquée pour étudier une variété d’autres échantillons et domaines de recherche, y compris les interactions cellule-endothéliale de Schwann dans les nerfs périphériques45, la biogenèse du corps de Weibel...
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Nous remercions Joanna Hanley, Rebecca Fiadeiro et Ania Straatman-Iwanowska pour leur assistance technique experte. Nous remercions également les membres du laboratoire Stefan et Ian J. White pour leurs discussions utiles. J.J.B. est soutenu par le financement du MRC au Laboratoire de biologie cellulaire moléculaire du MRC à l’UCL, code de récompense MC_U12266B. C.J.S. est soutenu par le financement du MRC au MRC Laboratory of Molecular Cell Biology University Unit de l’UCL, code de récompense MC_UU_00012/6. P.G. est financé par le Conseil européen de la recherche, code de subvention ERC-2013-StG-337057.
matériels
| Name | Company | Catalog Number | Comments |
| 0.22 µm syringe filter | Sarstedt | 83.1826.001 | |
| Aluminum trays | Agar Scientific | AGG3912 | |
| Amira v6 | ThermoFisher | https://www.thermofisher.com | |
| Chloroform | Fisher | C/4960/PB08 | |
| DDSA/Dodecenyl Succinic Anhydride | TAAB | T027 | Epon ingredient |
| Diamond knife | DiaTOME | ultra 45° | |
| DMP-30/2,4,6-tri (Dimethylaminomethyl) phenol | TAAB | D032 | Epon ingredient |
| Dumont Tweezers N5 | Agar Scientific | AGT5293 | |
| Fiji | https://imagej.net/ | ||
| Fiji TrakEM2 plugin | https://imagej.net/ | ||
| Formaldehyde 36% solution | TAAB | F003 | |
| Formvar coated slot grid | Homemade | Alternative: EMS diasum (FF2010-Cu) | |
| Glass bottle with applicator rod | Medisca | 6258 | |
| Glass vials | Fisher Scientific | 15364769 | |
| Gluteraldehyde 25% solution | TAAB | G011 | |
| MNA/Methyl Nadic Anhydride | TAAB | M011 | Epon ingredient |
| Osmium Tetroxide 2% solution | TAAB | O005 | |
| Potassium Ferricyanide | Sigma-Aldrich | P-8131 | |
| Propylene oxide | Fisher Scientific | E/0050/PB08 | |
| Reuseable adhesive | Blue Tack | ||
| Reynolds Lead Citrate | TAAB | L037 | Section stain |
| Sodium Cacodylate | Sigma-Aldrich | C-0250 | to make 0.1 M Caco buffer |
| Super Glue | RS Components | 918-6872 | Cyanoacrylate glue, Step 1.3 |
| TAAB 812 Resin | TAAB | T023 | Epon ingredient |
| Tannic acid | TAAB | T046 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Two part Epoxy Resin | RS Components | 132-605 | Alternative: Step 2.13 |
| Ultramicrotome | Leica | UC7 | |
| Vibrating microtome | Leica | 100 µm thick slices, 0.16 mm/s cutting at 1 mm amplitude . | |
| Weldwood Original Contact cement | DAP | 107 | Contact adhesive: Step 3.1.4 |
Références
- Knoll, M., Ruska, E. Das elektronenmikroskop. Zeitschrift für Physik. 78 (5), 318-339 (1932).
- von Ardenne, M. Daselektronen-rastermikroskop. Zeitschrift für Physik. 109 (9), 553-572 (1938).
- Bang, B. H., Bang, F. B. Graphic reconstruction of the third dimension from serial electron microphotographs. Journal of Ultrastructure Research. 1 (2), 138-139 (1957).
- Birch-Andersen, A. Reconstruction of the nuclear sites of Salmonella typhimurium from electron micrographs of serial sections. Journal of General Microbiology. 13 (2), 327-329 (1955).
- Denk, W., Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Peddie, C. J., Collinson, L. M. Exploring the third dimension: volume electron microscopy comes of age. Micron. 61, 9-19 (2014).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure. Biology of the Cell. 108 (11), 307-323 (2016).
- Kornfeld, J., Denk, W. Progress and remaining challenges in high-throughput volume electron microscopy. Current Opinion in Neurobiology. 50, 261-267 (2018).
- Heymann, J. A., et al. Site-specific 3D imaging of cells and tissues with a dual beam microscope. Journal of Structural Biology. 155 (1), 63-73 (2006).
- Knott, G., Marchman, H., Wall, D., Lich, B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. Journal of Neuroscience. 28 (12), 2959-2964 (2008).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Martone, M. E., Deerinck, T. J., Yamada, N., Bushong, E., Ellisman, M. H. Correlated 3D light and electron microscopy: use of high voltage electron microscopy and electron tomography for imaging large biological structures. Journal of Histotechnology. 23 (3), 261-270 (2000).
- Micheva, K. D., Smith, S. J. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Sjostrand, F. S. Ultrastructure of retinal rod synapses of the guinea pig eye as revealed by three-dimensional reconstructions from serial sections. Journal of Ultrastructure Research. 2 (1), 122-170 (1958).
- Ware, R. W. Three-dimensional reconstruction from serial sections. International Review of Cytology. 40, 325 (1975).
- Stevens, J. K., Davis, T. L., Friedman, N., Sterling, P. A systematic approach to reconstructing microcircuitry by electron microscopy of serial sections. Cognitive Brain Research. 2 (3), 265-293 (1980).
- Hoppe, W. Three-dimensional electron microscopy. Annual Review of Biophysics. 10, 563-592 (1981).
- Frank, J. . Electron tomography: methods for three-dimensional visualization of structures in the cell. , (2008).
- Baumeister, W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Current Opinion in Structural Biology. 12 (5), 679-684 (2002).
- Hoog, J. L., Schwartz, C., Noon, A. T., O'Toole, E. T. Organization of interphase microtubules in fission yeast analyzed by electron tomography. Developmental Cell. 12 (3), 349-361 (2007).
- Harris, K. M., Perry, E., Bourne, J., Feinberg, M., Ostroff, L., Hurlburt, J. Uniform serial sectioning for transmission electron microscopy. Journal of Neuroscience. 26 (47), 12101-12103 (2006).
- Jesior, J. C. Use of low-angle diamond knives leads to improved ultrastructural preservation of ultrathin sections. Scanning Microscopy Supplement. 3, 147-152 (1989).
- Studer, D., Gnaegi, H. Minimal compression of ultrathin sections with use of an oscillating diamond knife. Journal of Microscopy. 197, 94-100 (2000).
- Gay, H., Anderson, T. F. Serial sections for electron microscopy. Science. 120 (3130), 1071-1073 (1954).
- Bernhard, W., Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. The Journal of Biophysical and Biochemical Cytology. 2, 73-78 (1956).
- Palade, G. E. An electron microscope study of the mitochondrial structure. The Journal of Histochemistry & Cytochemistry. 1 (4), 188-211 (1953).
- Wu, H., Carvalho, P., Voeltz, G. K. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 361 (6401), (2018).
- Vance, J. E. Inter-organelle membrane contact sites: implications for lipid metabolism. Biology Direct. 15 (1), 24 (2020).
- Stefan, C. J. Endoplasmic reticulum-plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Current Opinion in Cell Biology. 63, 125-134 (2020).
- Zaman, M. F., Nenadic, A., Radojicic, A., Rosado, A., Beh, C. T. Sticking with it: ER-PM membrane contact sites as a coordinating nexus for regulating lipids and proteins at the cell cortex. Frontiers in Cell and Developmental Biology. 8, 675 (2020).
- van Vliet, A. R., Sassano, M. L., Agostinis, P. The unfolded protein response and membrane contact sites: tethering as a matter of life and death. Contact. 1, 1-15 (2018).
- Cohen, S., Valm, A. M., Lippincott-Schwartz, J. Interacting organelles. Current Opinion in Cell Biology. 53, 84-91 (2018).
- Hariri, H., et al. Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress. EMBO Reports. 19 (1), 57-72 (2018).
- Stefan, C. J., Trimble, W. S., Grinstein, S., Drin, G. Membrane dynamics and organelle biogenesis-lipid pipelines and vesicular carriers. BMC Biology. 15 (1), 102 (2017).
- Eisenberg-Bord, M., Shai, N., Schuldiner, M., Bohnert, M. A tether is a tether is a tether: tethering at membrane contact sites. Developmental Cell. 39 (4), 395-409 (2016).
- Scorrano, L., De Matteis, M. A., Emr, S., Giordano, F. Coming together to define membrane contact sites. Nature Communications. 10 (1), 1287 (2019).
- Lak, B., Li, S., Belevich, I., Sree, S. Specific subdomain localization of ER resident proteins and membrane contact sites resolved by electron microscopy. European Journal of Cell Biology. 100 (7), 151180 (2021).
- Collado, J., Kalemanov, M., Campelo, F., Bourgoint, C. Tricalbin-mediated contact sites control ER curvature to maintain plasma membrane integrity. Developmental Cell. 51 (4), 476-487 (2019).
- West, M., Zurek, N., Hoenger, A., Voeltz, G. K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. Journal of Cell Biology. 193 (2), 333-346 (2011).
- Ilacqua, N., Anastasia, I., Raimondi, A., Lemieux, P. A three-organelle complex made by wrappER contacts with peroxisomes and mitochondria responds to liver lipid flux changes. Journal of Cell Science. 135 (5), 259091 (2022).
- Cardona, A., Saalfeld, S., Schindelin, J., Arganda-Carreras, I. TrakEM2 software for neural circuit reconstruction. PLoS One. 7 (6), 38011 (2012).
- Stalling, D., Westerhoff, M., Hege, H. -. C. Amira: A highly interactive system for visual data analysis. The Visualization Handbook. 38, 749-767 (2005).
- Hsieh, T. S., Chen, Y. J., Chang, C. L., Lee, W. R., Liou, J. Cortical actin contributes to spatial organization of ER-PM junctions. Molecular Biology of the Cell. 28 (23), 3171-3180 (2017).
- Anastasia, I., Ilacqua, N., Raimondi, A., Lemieux, P. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Reports. 34 (11), 108873 (2021).
- Cattin, A. L., Burden, J. J., Van Emmenis, L., Mackenzie, F. E. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell. 162 (5), 1127-1139 (2015).
- Lopes-da-Silva, M., et al. A GBF1-dependent mechanism for environmentally responsive regulation of ER-Golgi transport. Developmental Cell. 49 (5), 786-801 (2019).
- Banushi, B., Forneris, F., Straatman-Iwanowska, A., Strange, A. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. Nature Communications. 7, 12111 (2016).
- Rey, S. A., et al. Ultrastructural and functional fate of recycled vesicles in hippocampal synapses. Nature Communications. 6, 8043 (2015).
- Belicova, L., Repnik, U., Delpierre, J., Gralinska, E. Anisotropic expansion of hepatocyte lumina enforced by apical bulkheads. Journal of Cell Biology. 220 (10), 202303003 (2021).
- Kizilyaprak, C., Daraspe, J., Humbel, B. M. Focused ion beam scanning electron microscopy in biology. Journal of Microscopy. 254 (3), 109-114 (2014).
- Xu, C. S., Hayworth, K. J., Lu, Z., Grob, P. Enhanced FIB-SEM systems for large-volume 3D imaging. Elife. 6, 1-36 (2017).
- Parlakgül, G., Arruda, A. P., Cagampan, E., Pang, S. High resolution 3D imaging of liver reveals a central role for subcellular architectural organization in metabolism. bioRxiv. , (2020).
- Guerin, C. J., Kremer, A., Borghgraef, P., Lippens, S. Targeted studies using serial block face and focused ion beam scan electron microscopy. The Journal of Visualized Experiments: JoVE. (150), e59480 (2019).
- Kremer, A., et al. A workflow for 3D-CLEM investigating liver tissue. Journal of Microscopy. 281 (3), 231-242 (2021).
- Hayat, M. . Principles and techniques of electron microscopy: biological applications. , (2000).
- Wisse, E., Braet, F., Duimel, H., Vreuls, C. Fixation methods for electron microscopy of human and other liver. World Journal of Gastroenterology. 16 (23), 2851-2866 (2010).
- Hanley, J., Dhar, D. K., Mazzacuva, F., Fiadeiro, R. Vps33b is crucial for structural and functional hepatocyte polarity. Journal of Hepatology. 66 (5), 1001-1011 (2017).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. Microscopy. 1, 6-8 (2010).
- Miranda, K., Girard-Dias, W., Attias, M., de Souza, W., Ramos, I. Three dimensional reconstruction by electron microscopy in the life sciences: An introduction for cell and tissue biologists. Molecular Reproduction and Development. 82 (7-8), 530-547 (2015).
- Yamaguchi, M., Chibana, H. A method for obtaining serial ultrathin sections of microorganisms in transmission electron microscopy. The Journal of Visualized Experiments: JoVE. (131), e56235 (2018).
- Hall, D. H., Hartwieg, E., Nguyen, K. C. Modern electron microscopy methods for C. elegans. Methods in Cell Biology. 107, 93-149 (2012).
- Hagler, H. K. Ultramicrotomy for biological electron microscopy. Methods in Molecular Biology. 369, 67-96 (2007).
- Arganda-Carreras, I., Beichel, R. R., Sonka, M. Consistent and elastic registration of histological sections using vector-spline regularization. Computer vision approaches to medical image analysis, CVAMIA 2006, Lecture Notes in Computer Science. 4241, 85-95 (2006).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218, 52-61 (2005).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD). Journal of Structural Biology. 116 (1), 71-76 (1996).
- Iudin, A., Korir, P. K., Salavert-Torres, J., Kleywegt, G. J., Patwardhan, A. EMPIAR: a public archive for raw electron microscopy image data. Nature Methods. 13 (5), 387-388 (2016).
- Xu, C. S., Pang, S., Shtengel, G., Muller, A. An open-access volume electron microscopy atlas of whole cells and tissues. Nature. 599 (7883), 147-151 (2021).
- Karabag, C., et al. Semantic segmentation of HeLa cells: An objective comparison between one traditional algorithm and four deep-learning architectures. PLoS One. 15 (10), 0230605 (2020).
- Heinrich, L., Bennett, D., Ackerman, D., Park, W. Whole-cell organelle segmentation in volume electron microscopy. Nature. 599 (7883), 141-146 (2021).
- Kim, J. S., Greene, M. J., Zlateski, A., Lee, K. Space-time wiring specificity supports direction selectivity in the retina. Nature. 509 (7500), 331-336 (2014).
- Spiers, H., Songhurst, H., Nightingale, L., de Folter, J. Deep learning for automatic segmentation of the nuclear envelope in electron microscopy data, trained with volunteer segmentations. Traffic. 22 (7), 240-253 (2021).
- Hasan, N. M., Gupta, A., Polishchuk, E., Yu, C. H. Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. The Journal of Biological Chemistry. 287 (43), 36041-36050 (2012).
- Stoeck, I. K., Lee, J. Y., Tabata, K., Romero-Brey, I. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. The Journal of Virology. 92 (1), 01196 (2018).
- Ma, X., Qian, H., Chen, A., Ni, H. M., Ding, W. X. Perspectives on mitochondria-ER and mitochondria-lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells. 10 (9), 2273 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.