
Method Article
Simplified Volumetric Models as an Effective Strategy for Segmenting Actin Networks in Cryo-Electron Tomograms
In This Article
Summary
Here, we present a protocol to place simplified volumetric models into noisy, complex, tomographic 3D volumes. This allows the fast segmentation of actin filament densities, detection of systematic filament bending and of gaps in hair bundle filaments, as well as convenient quantification of volumetric model properties, such as distances.
Abstract
Efficient methods for the extraction of features of interest remain one of the biggest challenges for the interpretation of cryo-electron tomograms. Various automated approaches have been proposed, many of which work well for high-contrast datasets where the features of interest can be easily detected and are clearly separated from one another. Our inner ear stereocilia cryo-electron tomographic datasets are characterized by a dense array of hexagonally packed actin filaments that are frequently cross-connected. These features make automated segmentation very challenging, further aggravated by the high-noise environment of cryo-electron tomograms and the high complexity of the densely packed features. Using prior knowledge about the actin bundle organization, we have placed layers of a highly simplified ball-and-stick actin model to first obtain a global fit to the density map, followed by regional and local adjustments of the model. We show that volumetric model building not only allows us to deal with the high complexity, but also provides precise measurements and statistics about the actin bundle. Volumetric models also serve as anchoring points for local segmentation, such as in the case of the actin-actin cross connectors. Volumetric model building, particularly when further augmented by computer-based automated fitting approaches, can be a powerful alternative when conventional automated segmentation approaches are not successful.
Introduction
Cryo-electron tomography allows entire organelles or parts of cells and tissues to be visualized at nanometer resolutions in their near-native state1,2,3 using either plunge-freezing4 or high-pressure freezing ultra-rapid vitrification5. Since only a limited electron dose can be tolerated by the cryo-preserved, unstained, frozen-hydrated sample, the tomographic 3D data is very noisy. This noise often can be significantly reduced by a variety of noise filtering algorithms6,7, including nonlinear anisotropic diffusion8, bilateral filtering9, and recursive median filtering10.
Furthermore, tilt limitations of the microscope stage, resulting in a missing wedge of information, and the fact that the specimen thickness increases at high tilt angles, lead to 3D reconstructions with anisotropic resolution. This means that the density is smeared out in the third dimension due to lower resolution in the Z-direction. As a result, the shape of macromolecules appears distorted (i.e., less well defined and elongated in the third dimension).
Among the biggest challenges in the interpretation of tomographic data is the automated extraction of the relevant features, also known as segmentation11. With sufficient unique shape features and low noise, macromolecular machines in complex 3D volumes can be identified by template matching12,13,14; however, the success of template matching depends on tomogram resolution, a suitable search model, as well size and shape characteristics of the feature volumes. If the features of interest are sufficiently spaced apart and repeating motifs (such as large macromolecular machines) can readily be identified, tomogram subvolumes can be combined to increase the signal-to-noise ratio and to average out individual particle shape distortions. Automated segmentation of an actin filament network in electron tomograms of the thin edge of frozen-hydrated Dictyostelium discoideum cells by template matching has been reported15.
However, if features of interest are closely spaced, the data resolution anisotropy can lead to a smearing out of the map densities in the Z-direction (along the direction of the electron beam), resulting in an apparent merging of the density envelope of closely spaced macromolecular machines or supramolecular complexes. In such cases, automated approaches for segmentation, such as watershed16, boundary segmentation17, or a variety of machine learning-based classification approaches18,19, may not be able to recognize the features of interest or establish a correct boundary around an object of interest. Often, one ends up with either a few very large pieces or with a heavily over-segmented volume, where much effort is needed to merge many small pieces until the feature of interest is perceived to be complete. Such manual curation of segmentation results can be very labor-intensive and may even fail altogether when the structure of interest is an array of closely spaced filaments that are interconnected via short linkers. In this gigantic network of filamentous structures, it can be difficult to orient oneself. This is because, due to resolution anisotropy, densities appear to blend into one another, presenting a formidable challenge for both automated as well as for interactive manual segmentation approaches. As a consequence, one can easily "jump" between filaments when only visually inspecting small regions.
Fortunately, in the case of the actin bundle in inner ear hair cell stereocilia, we have knowledge about the overall actin bundle organization and the directionality of the actin filaments20,21. The actin bundle consists of hundreds of hexagonally, densely-packed actin filaments 6-8 nm in diameter, which are spaced about 12-13 nm apart from one another22.
This allowed us to take a rather different approach to segmentation that is based on simplified ball-and-stick models to represent actin filaments. The strategy involved simultaneously placing an idealized regular array of filament models into slabs of the cryo-electron tomography density maps to build up a 3D model of the actin bundle layer by layer. We ensured that the model had a general overall fit to the density map before making local adjustments to individual filament models or groups of filament models to closely match the density map. By automatic color-coding of the map density value at the filament model location, we were able to easily detect apparent gaps in the actin bundle. Volumetric models allow a quantitative analysis of volumetric properties, such as distances between actin filaments, and also lead to a simplified display of the overall 3D filamentous network organization.
In addition, models can also serve as anchoring structures for the segmentation of additional features, such as actin-actin linkers, as (portions of) individual filament models can be selected, around which appropriate radius map density zones can be generated for inspection and further segmentation.
We believe that our volumetric model-based segmentation approach is particularly useful for large filamentous structure networks that may contain gaps and inter-filament cross-connections. Segmentation algorithms tend to operate locally, whereas the human brain takes larger areas into consideration, and thus is superior to computers when it comes to recognizing filament structures, even in a complex, high-noise environment.
Protocol
The protocol follows the guidelines of Southeast University's human research ethics committee.
1. Cryo-electron tomography data source for volumetric model building
NOTE: The stereocilia cryo-electron tomographic reconstructions used for a volumetric model building have been previously published22,23, and were obtained as described by Metlagel et al.22.
The UCSF Chimera python scripts for stereocilia modeling are provided in Supplementary File 1, Supplementary Coding File 1, Supplementary Coding File 2, Supplementary Coding File 3, Supplementary Coding File 4, and Supplementary Coding File 5.
- Briefly, blot stereocilia from the apical surface of mouse utricle sensory epithelia onto the lacey carbon support film of an electron microscope (EM) grid. Then, perform ultra-rapid plunge-freezing vitrification and single-axis cryo-electron tomographic data collection on a cryogenic transmission electron microscopy (cryo-TEM), operated at 300 kV with a nominal defocus of 3.5-4.5 µm, using a CMOS-type camera in integration mode, with a 0.47 to 0.59 nm pixel size (see Table of Materials).
- A typical dose for single-axis data collection is typically 80-100 electrons/Å2. Reconstruct the tomogram 3D volumes using the software package IMOD24, either by weighted back-projection or the SIRT method25,26.
- Denoise the tomograms using recursive median or bilateral filtering in Priism27 or the nonlinear anisotropic diffusion option in IMOD.
NOTE: The key software package used for interactive visualization, volumetric model building, and quantitative analysis was the UCSF Chimera software28,29. All software packages used are listed in the Table of Materials.
2. Cryo-electron tomography data preparation for volumetric model building
- Tomogram rotation
NOTE: The purpose of this step is to find the "optimal angles" in which the actin core is aligned in two of the three axes (x and z), so that placing the models in 3D can be done with only one axis to worry about. The following steps are carried out within the IMOD software package.- Open 3dmod (IMOD) and open the 3D image stack file in .mrc file format. Make sure the 3D stack is in 16-bit grayscale mode and displays the correct X, Y, Z dimensions in its image file header. If necessary, make corrections using the alterheader command: alterheader -d (x_pixelspacing),(y_pixelspacing),(z_pixelspacing) inputfilename.mrc in the command prompt. If the 3D stack is in TIFF file format, use the command tif2mrc inputfilename.tif outputfilename.mrc in the command prompt to create an .mrc file.
- Visually inspect the tomogram and open "slicer" (by pressing backslash "\") to find the best angles of rotation in the X, Y, and Z dimensions for alignment of the actin filament plane with the Z-plane. Manipulate the X rotation, Y rotation, Z rotation bar to find out the optimal angle at which the actin filaments are aligned on the X- and Z-planes. Note that changing the average thickness for the display of the tomogram (Thick : Img) can help in finding an ideal amount of averaging of the density in the Z-plane and thus increased contrast. Zoom in and out of the tomogram by utilizing the + and - keys, respectively, on the keyboard.
- Once the ideal angles for rotation have been identified, rotate the tomogram by typing the IMOD command rotatevol -a (Z rotation angles),(Y rotation angles),(X rotation angles) -s (x-width),(y-height),(z-depth) inputfilename.mrc outputfilename.mrc into the command prompt. Make sure to give the map plenty of space to accommodate for the rotation to avoid cutting off portions of the map upon rotation.
NOTE: For more information on how to use the rotatevol command, use the option from the help menu, or visit the following URL (https://bio3d.colorado.edu/imod/doc/man/rotatevol.html). This command (rotatevol) does not alter the currently shown map in the IMOD window, but rather creates a new map file with a density map being rotated. - Using the slicer window, identify the X, Y, and Z coordinates of two opposite corners of a map to identify the cropping region containing the stereocilium using the trimvol command.
- Use the IMOD command Trimvol to crop the map at the previously obtained coordinates, by typing trimvol -x (x-coordiante 1),( x-coordiante 2) -y (y-coordiante 1),( y-coordiante 2) -z (z-coordiante 1),( z-coordiante 2) inputfilename.mrc outputfilename.mrc into the command prompt. Now, the map is much smaller, and thus easier and faster to work with from this point onward.
NOTE: For more information on how to use the trimvol command, use the option from the help menu, or visit the following URL (https://bio3d.colorado.edu/imod/doc/man/trimvol.html). These steps create a .mrc file that has been rotated and trimmed.
- Tomogram filtering
NOTE: This step uses a nonlinear anisotropic diffusion (NAD) filter provided by IMOD for noise reduction. We used as initial value the ones recommended in the IMOD help page for non-linear anisotropic diffusion filtering, as listed in the following URL (https://bio3d.colorado.edu/imod/doc/NADexample.html), and was used as the initial values.- Type etomo in the command prompt to initiate IMOD's etomo graphic user interface.
- Select the Nonlinear Anisotropic Diffusion option from the etomo menu and select the map file (Pick a volume).
- In order to find a K value and the number of iterations that provide the best filtering results, as judged by visual inspection, apply the NAD filter to a smaller test volume to extract the specified NAD-filter test volume. Click the Rubberband Tool on top of the 3dmod interface, left-click drag to select test volume, and click the Hi and Lo buttons to indicate the Z-slice to be the boundary of the test volume (Get Test Volume Range from 3dmod). Extract the test volume by clicking Extract Test Volume.
- Filter the test volume using various K values (Find a K Value for Test Volume > List of K values), such as 0.1, 1,5, 10, 15, 25, 30, 50, and 75. Perform NAD filtering with each of the specified K values (Run with Different K Values > View Different K Values Test Results). Use the slicer window to evaluate the performance of the NAD filter and select a K value that will be used for filtering the entire volume.
- Use the K value identified in step 2.2.4 and test different numbers of iterations, such as 2, 5, 8, 11, 15, and 21 (Find an Iteration Number for Test Volume > List of iterations). Evaluate the NAD filter performance for different iterations by either clicking View Different Iteration Test Results or using a slicer tool. Choose a value for a different number of iterations.
- Use the identified K value and the number of iterations (Filter Full Volume > K value and Iterations) and filter the entire volume (Filter Full Volume). A newly filtered volume will appear in the same directory where the loaded full volume is located with a .nad file extension attached to the .mrc file.
NOTE: These steps create a .mrc file that has been filtered with a NAD filter and displays a lower noise and thus an improved signal-to-noise ratio.
3. Volumetric model building
- 3D model placement preparation
NOTE: The purpose of this step is to prepare and analyze the map for placing 3D volumetric models into the rotated and denoised density map using the UCSF Chimera program.- Using the slicer window, obtain a cross-sectional view (slab of 30 slices/28.4 nm) to identify the Z-coordinates of the center of the density slab, into which the actin filament model is to be placed. Click on the center of the filament density and note the Z-coordinates in the IMOD main window.
- Keep note of the coordinates identified for future model placement in UCSF Chimera.
NOTE: The Z-coordinates noted from this step is used during step 4 to easily find and place pre-made actin filament models at the correct Z-heights, making the model placement faster.
- 3D model placement
NOTE: The purpose of this step is to place a 3D volumetric model into the prepared density map using UCSF Chimera.- In UCSF Chimera, open the filtered, rotated map (File > Open)
- Check whether the parameters of the map are set correctly by selecting Features > Planes and Feature > Coordinates in the Volume Viewer to open the feature and coordinates interface. Check Voxel Size to see if the correct voxel spacing is set by using the header command in the command prompt (in IMOD). If not, correct the Voxel Size interface to the correct voxel spacing. Center the map to set the camera at the center of the viewing window (Origin Index > Center).
- Open the Camera Control window (Tools > Viewing Control > Camera). Then, set the camera viewing to the orthographic view (projection > orthographic)
- Load the pre-made python script model (File > Open > ActinFilamentPlane.py) and open as many actin filament plane scripts as there are actin filament planes in the map.
- Relocate the loaded models by turning on the Movement Mouse Mode (Tools > Movement > Movement Mouse Mode). Enable the movement mouse option (Set Movement Mouse Mode > Move Selection). Use Ctrl + Drag to multi-select any models present in the session and right-click (or any other key that is specified in Preference > Mouse) to move the models in the desired direction.
NOTE: The models have been created with balls and sticks with certain thickness (radius of ActinFilamentPlane.py script) and with certain spacings (Lattice_Spacing of ActinFilamentPlane.py script). - To visualize just the model that one is interested in, open Model Panel (Tools > General Controls > Model Panel). Click checkboxes under S (Show) to either enable or disable the visibility of a given model.
- Activate the command line panel (Tools > General Controls > Command Line).
- After placing each actin filament plane model to its proper Z-height, delete any excess actin filaments in each actin filament plane model by selecting the excess portion of the models, and by typing del sel (Delete Selected) in the command line at the bottom of the UCSF Chimera main view window.
- Only visualize one actin filament plane at a time, by going to the Model Panel window and selecting and hiding all other actin filament models (Ctrl + Click > Hide).
- To visualize portions of the map density that are in the immediate proximity of the single actin filament plane model of interest, select the model (Model Panel > select) and open the zone control panel in Volume Viewer (Volume Viewer > Features > Zone). Set the zone radius to 100 Å, then click Zone to zone the map in a radius of 100 Å (= 10 nm) from the selected model. Note that the dimension in Chimera is set in Angstrøm (1 Å = 0.1 nm).
- If needed, adjust the model to fit to the density map by turning on the Movement Mouse Mode. Multi-select any models that are not in the proper position to move the models to be properly placed inside the zoned density map.
- To overcome a programming bug in ActinFilamentPlane.py, run the FixingMarkerID.py script, which fixes the missing MarkerID of the "atoms" (balls in the ball-and-stick model). Select the entire model and type runscript FixingMarkerID.py in the command line (the script should be specified by correct directory, such as C: \ directory \ Script.py).
- If the density map indicates that the actin filament is curved, make sure to divide the straight actin filament into multiple parts to accommodate for the curvature by selecting the desired models and typing runscript dividelinks.py number. Additional markers (specified by the number in the command) are placed inside the actin model equidistant from one another, making "joints" where the model can be bent.
- Adjust the joints to reflect the curvature by moving additional marker "joints" along the zoned map by using Movement Mouse Mode. Add additional markers to the model as needed by using the dividelinks.py script.
- To verify that all the all the actin filaments are properly placed inside the actin densities, use clipping bars by clicking Tools > Viewing Control > Camera in the UCSF Chimera drop-down menu to open the Camera Control window, then select Side View to see the two clipping bars (near- and far-clipping planes) of the camera. Drag two yellow bars into a narrow slit by left-clicking and drag the clipping portion by using the Middle Mouse Button to inspect the models one small bit at a time.
NOTE: The stereocilium actin core model is now complete.
- Membrane segmentation
NOTE: The purpose of this step is to create a surface model of the stereocilia membrane.- The segmentation of surfaces utilizes the Volume Tracer tool (Tools > Volume Data > Volume Tracer).
- In the Volume Tracer window, click Mouse, and only check the options Place Markers on Data Planes and Link new marker to selected marker. This allows precisely placing the markers on the same Z-plane (when a single Z-plane is shown). Also check Place Markers Using the Middle Mouse Button in the Volume Tracer window.
- Create a new marker set (File > New Marker set). Marker set 1 should be created and selected on the Volume Tracer window.
- Use Volume Viewer to display single Z-planes in the main viewing window (Axis to Z > One.)
- Using the Middle Mouse Button, trace out the membrane from one end of the visible membrane to another. All markers should be connected by links from one end to another.
- De-select the last-placed marker by pressing Ctrl + Click on the background where not a single selectable item is located. This ensures that the next marker to be placed is connected with links in the new plane, and not with a previous membrane trace of another Z-plane.
- Repeat the membrane tracing for every 10 nm in the same direction of tracing from one end of the visible membrane to another side.
- Once many parallel bands of membranes are segmented, go to the Volume Tracer window and click Features > Surfaces. This enables the surface interface in the Volume Tracer window. Click Create next to the Surfaces interface to create surfaces in between bands of membrane segmentation. This creates a thin flap of the surface model that fills the spaced-out membrane traces.
NOTE: The stereocilium membrane surface model creation is now complete.
- Crosslinker modelling
NOTE: The purpose of this step is to model the crosslinkers between actin filaments.- Crosslinkers can also be placed by utilizing the Volume Tracer tool.
- In the Volume Tracer window, left-click to check only Place Markers on Surfaces and Link new marker to selected marker. Then, check Place Markers Using the Middle Mouse Button in the Volume Tracer window.
- Only visualize densities around the single actin filament plane model of interest by using the zone function. Set the zone radius to 100 Å to visualize the map within a radius of 100 Å from the selected model.
- Place markers across visible densities of crosslinkers by clicking the Middle Mouse Button. Once two markers are placed (and thus a single crosslinker is modeled) make sure to de-select the second marker to prevent links continuously being connected to the next crosslinker model.
- Repeat the steps above for every actin filament plane and repeat the entire above process to all three major directions in which crosslinkers may be located. When zoning actin filament planes in the diagonal direction, manually select actin filament models individually.
NOTE: For each of the actin crosslinker models, we can record the position on the actin filaments to which the putative crosslinkers connect. This information could, in principle, be extracted for a follow-up analysis using sub-tomogram averaging, which is beyond the scope of this study.
4. Quantitative analysis of the 3D model
- Gap analysis
NOTE: The purpose of this step is to detect gaps in the actin filaments.- Selected items can be analyzed. Specify their parameters, like their color, using the Inspect Selection tool (Action > Inspect).
- The color of the selected item can be specified by selecting any selectable item in UCSF Chimera, and by changing the color next to the Color section of the Inspect Selection window.
- Specify the size of the markers and links by selecting any markers and links in UCSF Chimera, and by changing the values next to the radius section of the Inspect Selection window, where the units are in Angstrøm.
- Perform gap analysis by auto-detection of density gaps, utilizing the Values at Atom Positions function (Tools > Volume Data > Values at Atom Positions).
- Select the map in which the values will be read by selecting the desired map next to the Volume Data section of the Values at Atom Positions window. Select the model in which all the markers will be assigned a parameter by selecting the desired model next to the Molecule section.
- With the Histogram button at the bottom of the Values at Atom Positions window, a histogram is created in a new window termed Render/Select by Attribute. Ctrl + Click on the histogram will add or remove cutoff points, for which different colors can be selected. Uncheck Keep Opaque to apply a transparent color to the scene. Markers with a density below a given threshold, which represents gaps, can not only be colored but can also be selected, and thus allow us to count the number markers in the gap region. To select, click on Select > Attribute > value_mapname.
- Determining inter-actin distances
- For measuring inter-actin distances, place markers on a single XZ-plane (referred to in UCSF Chimera as a Y-plane) by using the Volume Tracer window, clicking Mouse, and checking only Place Markers on Data Planes and unchecking Link new marker to selected marker. This allows precisely placing the markers on the same Y-plane (when a single Z-plane is shown). Also check Place Markers Using Middle Mouse Button in the Volume Tracer window and place the markers on a target Y-plane of interest for inter-actin distance measurement.
- Once single Y-plane's actin positions are successfully marked, select all the markers and type the command findclash #model-spec test self overlapCutoff -200 hbondAllowance 0 log true Linewidth 10 pbColor black in the command line at the bottom of the actin filament plane. This finds all inter-marker clashes within a distance of 200 Å (overlapCutoff -200), and creates black lines (pbColor black) with a thickness of 10 Å (Linewidth 10) between the markers.
- Since there are overlapping markers within the given range of 200 Å, remove redundant measurements that are not links between only the closest, adjacent markers. Do this by using the RemoveCross.py script provided on UCSF Chimera website, which removes all the overlapping links that are not the shortest link with its adjacent markers.
- To measure the distance of the bonds that are remaining, select all pseudobonds (links that are created by findclash) and use the python script pblengths.py by typing runscript pblengths.py. See the measured values from the Reply Log (Tools > Utilities > Reply Log). Then, copy the obtained values from the Reply Log and transfer to a spreadsheet or any other suitable tool to create a histogram.
Results
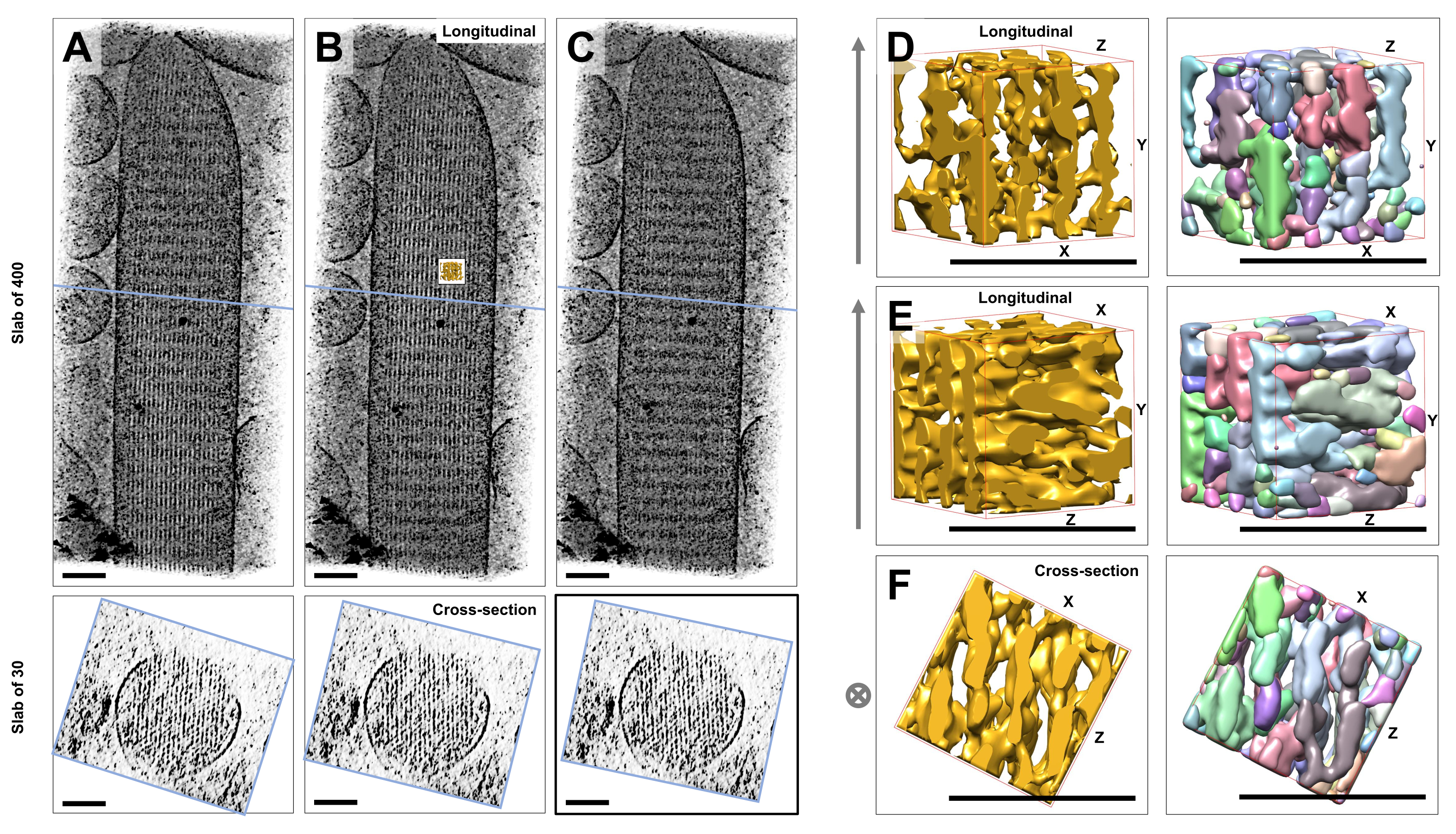
Using cryo-electron tomography of unstained, frozen-hydrated individual stereocilia embedded in vitreous ice, we obtained density maps of the actin bundle with its hexagonally arranged actin filaments, linked by cross-connector proteins23. The dimension of an individual voxel was 0.947 nm. Visual inspection in the IMOD slicer program of a volume rendering of the entire tomogram (400 slices/379 nm) indicated the presence of filamentous structures being aligned with the longitudinal axis of the stereocilia, as can be seen in longitudinal views (XY-plane; Figure 1A-C, top panels) as well as in cross-sectional views (XZ-plane; Figure 1A-C, bottom panels). We noticed that the projection view through the 400 slices/379 nm filament network became clearest when rotating the original reconstructed volume by -6° around the X-axis, -13.5° around the Y-axis, and 5° around the Z-axis. At this angle, all the filaments are aligned on top of one another, and hence the contrast is maximal, as can be appreciated from the cross-sectional views (Figure 1B). Since single cross-sectional slices do not have enough signal to unambiguously distinguish actin filaments, we chose to volume render a slab of 30 slices/28.4 nm, which clearly shows a hexagonal pattern in the cross-sectional view. The blue lines in Figure 1C (top panels) indicate the position of the middle of the corresponding 30 slices/28.4 nm cross-sectional slabs in the bottom panels.
Small deviations from this optimal viewing angle, by as little as ±2°, significantly reduced the perceived order of the actin filament network (Figure 1A,C), which is an indication how easily it is to get lost in the 3D volume of the tomogram.
To illustrate the challenge of using automated segmentation approaches, such as watershed segmentation, we chose a small subvolume (depicted as gold) for watershed segmentation, as implemented in the UCSF Chimera software package (Tools > Volume Data > Segger > Segment). The position of the subvolume relative to the entire stereocilia map is indicated by the small inset in Figure 1B.
Figure 1D-F shows the chosen subvolume in different orientations, with Figure 1D,E showing longitudinal viewing direction and Figure 1F showing a cross-sectional viewing direction. The arrows on the left-hand side of Figure 1D-F indicate the direction of the actin filaments.
Figure 1D-F (right panels) show the results of watershed segmentation. The subvolume is being color-coded by object identity, with colors being randomly assigned to the different objects. Different colors indicate a different object identity, hence it becomes apparent from Figure 1D-F that the map densities for filaments were both fragmented along the filament axis, whereas the same color and thus object identity were given to map densities that connected neighboring filaments. In other words, the watershed segmentation algorithm was not capable of following the density map of actin filaments for an extended period, and instead led to connecting densities from neighboring filaments. While it is possible to manually curate the selection (e.g., by deleting or merging of objects), this approach is rather labor-intensive and thus time-consuming.
Although it is not absolutely necessary for our volumetric model building strategy to work, it did help to reorient (rotate) the 3D map so that the actin filament network axis aligned with the Y-axis and the actin filament model planes aligned with the X-Y plane of the tomogram. We refer to this orientation as the standard orientation for stereocilia tomographic display.
We therefore decided to explore a different strategy for image segmentation, exploiting the fact that the actin filaments displayed an overall regular organization (hexagonal packing), with regular spacing and defined overall bundle orientation. Our strategy was to find an overall fit of models of an actin bundle, as an array of filaments, followed by regional and then local adjustments of the model position to fit the experimental density map. By placing an overall model first, we can overcome local map ambiguities, and detect regional trends of deviations of the model from its original organization, such as filament bending.
To place the model, we displayed slabs of the density (10 slices/9.47 nm) in the standard orientation that corresponded to a thickness of a single layer of the actin filaments, to which a layer of regularly-spaced, straight actin filament models was fitted. This is of course is an oversimplification of actin filaments, each of which consists of a linear array of actin monomers with helical symmetry. Figure 2A-C show three representative layers at different Z-heights, with the red-colored rods representing the actin filaments. The top panels, depicting ~30 slices/28.4 nm thick cross-sections, show at which Z-height an individual actin model layer of 19 rods was placed, whereas the lower panels show a longitudinal orientation (albeit shown in perspective view). Figure 2D shows the complete simplified model, both in the cross-sectional (top panel) and longitudinal perspective views (bottom panel). The cross-sectional orientation allowed us to position the filaments with good confidence. Here, our original move to reorient the entire volume to coincide with the tomograms major axes proved to be helpful, as it meant that the orientation of our model in our standard viewing direction was also parallel to the main axes. However, strictly speaking, our approach would also have worked without the reorientation of the tomogram, just the placement of the model to the density would have been more challenging.
Upon careful inspection of individual slabs of the density map, we noticed that a perfectly straight actin model did not fit the observed density map moving from the proximal end to the distal end (i.e., toward the tip) of the stereocilia (Figure 3A-C). Near the tip of the stereocilia, the map density for filaments was displaced by more than 13 nm (actin-actin spacing), which we could compensate for by adjusting the model while moving from the proximal to the distal portion of the stereocilia density map, thus introducing a small yet distinguishable gradual curvature into our actin model. Figure 3D shows a single slab of the map density of the actin filament, with a volumetric model fitted to the density map. A comparison between the straight (red) and the curved (yellow) model is shown in Figure 3E. This curvature is best appreciated by tilting a slab of the density map with the placed model by 80° around the X-axis, which allows one to have a perspective view along the direction of the actin filaments (Figure 3D,E).
The deviation of the two models, with the position of the actin model near the tip being shifted by about the same distance as the spacing of the actin filaments, could have caused much confusion if we had not proceeded the way we did. This "global" positioning of a layer of the actin filament model, followed by "regional" adjustment, allowed us to detect this curvature, which is barely noticeable in the longitudinal or cross-sectional view. However, overlaying the two models, as shown in Figure 3E, reveals the subtle difference.
Repeating this approach for multiple layers allows a complete 3D model to be obtained (Figure 3F), only limited by the data uncertainty at the very top and bottom of the stereocilia, when viewed in the cross-sectional orientation (Figure 3G). This lack of density is caused by the missing wedge in (single-axis) tomographic data collection and corresponding data resolution anisotropy, and its effect is indicated by the absence of a well-defined map density for stereocilia membranes.
Once we had a 3D model, we color-coded each location of the volumetric model according to the map density value at that position. Regions of the model with underlying weak map density were colored red, whereas regions of the model with strong map density signal were colored yellow (Figure 4A). We interpret such red-colored regions, which can extend to tens of nanometers, as gaps in the actin filament structures that, due to their extent, cannot be attributed to density variations frequently encountered in the high-noise environment of a cryo-EM map. Noise tends to affect individual voxels or small groups of voxels, but is unlikely to be the source for volumes consisting of hundreds of voxels, for which filament density is missing. Instead, such gaps are likely to be a real feature of the stereocilia actin meshwork and may constitute sites of actin turnover. Figure 4A has two different map density values, shown in light blue and dark blue. It should be explicitly noted that our volumetric model building approach, combined with an automated color-coding of our model in regions of weak density, is a fast and convenient way to detect and visualize the distribution of such gaps in the actin filament model, which would have been very difficult otherwise.
As shown in Figure 4B, portions of the volumetric model at locations with a comparably weak density can be easily hidden based on the results obtained in Figure 4A. This then results in a more fragmented model that may more realistically depict the actin model in stereocilia. The alternative of building small stretches of actin filaments would have been very labor-intensive and may have failed altogether, due to the problems discussed when describing Figure 1.
In addition, the volumetric model allows us to easily model the cross-connectors by simply placing a connection (shown in red) between model point positions of the actin filament model on either side of the cross-connection (Figure 4C). In our simplified approach, we do not need to make any assumption about the exact identity of each cross-connecting protein, which would require higher resolution and/or sophisticated labeling approaches. Instead, all we need to determine is whether a density exists that bridges adjacent actin filaments; if there is, we can place a short connection from one filament to its adjacent counterpart. In Figure 4D, a model of five actin filaments with their cross-connectors are shown, which gives an impression of the distribution of cross-connectors along the actin filament axis.
Another advantage building a volumetric model of the actin bundle is that one can quickly determine the spacing between adjacent actin filaments (Figure 4E-H). Figure 4E,F shows a cross-sectional view of the density map without and with a model fitted to the hexagonal lattice of the map density, respectively. Figure 4G shows the model with connections between the nearest neighboring balls. UCSF Chimera allows automatic computation of the distance of nearest neighbor centers, the result of which can then be plotted as a distance distribution (Figure 4H). Model building for two additional datasets are shown in Supplementary Figure 1 and Supplementary Figure 2.

Figure 1: The challenges encountered by watershed segmentation of hair cell stereocilia tomograms. (A-C) Longitudinal projections (400 slices/379 nm) through the tomographic 3D map in the XY-plane (top panels) and cross-sectional views (30 slices/28.4 nm) in the XZ-plane (bottom panels). (A) Tomographic map rotated -2° along the Y-axis from its optimal orientation. (B) Tomographic map in the optimal orientation, determined by adjustment of the rotation angles of the X-, Y- and Z-axes (X = -6˚, Y = -13.5˚, and Z= 5˚) and revealing a high degree of order in the density map, suggesting a highly-ordered actin filament network. (C) Tomographic map rotated +2° along the Y-axis from its optimal orientation; the rotation of as little as 2° around the Y-axis away from the optimal viewing direction severely compromises the perceived regularity of the density map. The bottom panels reveal the regularity of the actin filament array when viewed in the cross-sectional direction. The blue line in A-C indicates the position of the cross-sectional slab. (D-F) A cube of 50 nm x 50 nm x 50 nm viewed from three different directions before (left panels) and after (right panels) watershed segmentation. Note that watershed segmentation fails to detect continuous actin filament density, whereas adjacent actin filaments and their cross-connection share the same object identity, suggesting that watershed segmentation is not a suitable approach for tomogram segmentation. In panels D-F, the density map in Chimera is shown as map style "Surface". (A-C) Scale bars = 100 nm. (D-F) Scale bars = 50 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Building of a volumetric ball-and-stick actin filament model. (A-C) Top: cross-sectional 30 slice/28.4 nm views of the density map with a single-layer simplified actin filament model placed at a different Z-height. Bottom: a single 10 slice/9.47 nm slab of the density map with a single-layer simplified actin filament model. (D) Complete straight actin filament model in a 30 slice/28.4 nm cross-sectional view (top) and perspective view (bottom); Scale bars = 100 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Model adjustment to correct for the detected bending of actin filament density. (A-C) Careful visual inspection of the model, either in cross-sectional view (top panels) or longitudinal view (bottom panels), revealed a good fit of the model with the density map at the stereocilia's proximal end. However, as one moves toward the distal end of the stereocilia, the fit increasingly becomes worse for all actin filament models. This can be corrected for by moving the balls of the ball-and-stick model into the correct density map position, which results in a slightly curved actin filament model. The straight model is shown in red, and the corrected bent model is shown in yellow. (D) Single slab of the density map with the curved model fitted to it, revealing the curvature of the actin density toward the tip of the stereocilia. The actin model has been rotated 80° around the X-axis to better show this subtle yet significant bending of the actin filaments. (E) Comparison between the straight, uncorrected actin model shown in red and the curved, corrected actin model shown in yellow. For reasons of clarity, only one layer of the actin filament model is shown. (F-G) Actin bundle model with curved, corrected actin filaments shown in longitudinal (F) and cross-sectional (G) orientation. The segmented membrane is shown in blue. In panel D, the density map in Chimera is shown as map style "Mesh". Scale bars = 100 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Detection of gaps in the actin bundle using volumetric model building. Quantification of volumetric properties using ball-and-stick models. (A-C) A small region of a ~10 slice/9.47 nm thick longitudinal slab of the stereocilia density map is shown in blue, along with the curved, corrected actin filament model. (A) The density map is shown at a lower density threshold (light blue) and a higher density threshold (dark blue). There are regions of the actin filament model for which there is no corresponding density. At such locations, the model has been color-coded red to represent a lack of density. We interpret these locations as gaps in the actin filaments. (B) Model of the actin filaments that appears fragmented, reflecting the lack of actin filament density at such gap locations. (C) Model of the actin filaments with cross-connecting links (shown in red) added at locations where a strong density was found to bridge adjacent actin filaments. (D) Three selected model actin filaments are shown with cross-connectors to adjacent actin filaments (which are not shown for clarity). Note that many, but not all, possible inter-actin filament positions are being occupied by connector proteins; (E) A cross-sectional 30 slice/28.4 nm slab of the stereocilia density. (F) Model of actin filaments fitted to the cross-sectional 30 slice/28.4 nm stereocilia density slab. (G) Model of actin filaments without the underlying map density. Automatic detection of the nearest neighbor actin filament distances, as indicated by thin connections between actin filament model balls. (H) Histogram of actin filament model distances. In panels A-C, the density map in Chimera is shown as map style "Mesh". (A-C) Scale bars = 50 nm. (D) Scale bar = 25 nm. (E-G) Scale bars = 100 nm. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Figure 1: Model building for the first of two additional stereocilia datasets. (A-C) A small region of a ~10 slice/9.47 nm thick longitudinal slab of the stereocilia density map is shown in blue using mesh mode display. The initially placed model is shown in red and the corrected model shown in yellow. (A) Map density only. (B) Initial model placed into the density map. (C) Corrected model placed into the density map. (D-E) Larger stereocilia region without (D) and with (E) the corrected model fitted to a ~10 slice/9.47 nm thick longitudinal slab of the stereocilia density map. (F-G) The entire stereocilia tomogram region is shown. (F) Map only. (G) Map with the corrected model. (H) Superposition of the initial and corrected models. Scale bars = 100 nm Please click here to download this File.
Supplementary Figure 2: Model building for the second of two additional stereocilia datasets. (A-C) A small region of a ~10 slice/9.47 nm thick longitudinal slab of the stereocilia density map is shown in blue using mesh mode display. The initially placed model is shown in red and the corrected model shown in yellow; (A) Map density only. (B) Initial model placed into the density map. (C) Corrected model placed into the density map. (D-E) Larger stereocilia region without (D) and with (E) the corrected model fitted to a ~10 slice/9.47 nm thick longitudinal slab of the stereocilia density map. (F-G) The entire stereocilia tomogram region is shown. (F) Map only. (G) Map with the corrected model. (H) Superposition of the initial and corrected model. Scale bars = 100 nm. Please click here to download this File.
Supplementary File 1: UCSF Chimera python scripts for stereocilia modeling. Please click here to download this File.
Supplementary Coding File 1: pblengths.py. Please click here to download this File.
Supplementary Coding File 2: RemoveCross.py. Please click here to download this File.
Supplementary Coding File 3: ActinFilamentPlane.py. Please click here to download this File.
Supplementary Coding File 4: dividelinks.py. Please click here to download this File.
Supplementary Coding File 5: FixingMarkerID.py. Please click here to download this File.
Discussion
We have shown that automated approaches for segmentation, such as watershed segmentation, can fail in the high-noise and high-complexity environment of hair cell stereocilia cryo-electron tomograms. Distinguishing which part of this filamentous network represents actin filaments and what constitutes crosslinks on a local environment level seems challenging at best when just inspecting small tomographic subvolumes. The model building approach used in this study benefits from prior knowledge of the large-scale order of the actin bundle, which helps in developing an expectation about the orientation of actin filaments and the crosslinker densities. Perhaps even more significant is that a human brain can easily find patterns by considering the larger context beyond the local density distribution, whereas a computer algorithm only works for a relatively small region that is considered by the algorithm; hence, larger-scale trends cannot easily be taken into consideration. By fitting a model globally to a layer of density, we avoided the confusion that can occur when attempting to create a model for small portions of single actin filaments at a time. Of course, such global fitting assumes an order that extends over large distances. However, since we had an unexpected small but significant gradual bending of the actin filaments, the global fit was only an initial approximation, and required local adjustments of the model to fit the density map. Since the initial model was a good starting point, the adjustments could be made with high confidence. One big advantage of our approach was that we could choose to display only a defined zone of density, which helped to reduce the complexity of the scenery. Furthermore, viewing of the map density slab along the filament model axis helped to identify the unexpected curvature, which we would have most likely missed when simply displaying smaller subvolumes. Placement of the initial model also facilitated quick zooming in and out, to alternate between an overall view of the respective layer of actin filaments and the detailed views to make model adjustments.
The critical steps within the protocol included the rotation of the map after visual inspection, the creation and placement of the model into the density map, as well as the division of the filament model into smaller segments. The atom position of the segments could then be spatially adjusted to fit the density map, and/or color-coded to detect gaps.
This approach of actin model building can also be modified by placing a set of "atoms" (i.e., the balls of the ball-and-stick model) into filament densities by using a cross-sectional view of a 10-30 slice/9.47-28.4 nm averaged slab of density, which then can be connected by bonds (i.e., the sticks of the ball-and-stick model). We have used this approach, which is a modification from the protocol described here in detail, for the volumetric model building in the taper region of hair cell stereocilia23. Furthermore, as we have described here, our volumetric model building approach is also well suited for the segmentation and model building of membranes.
While volumetric model building can be applied to any density map that shows filamentous features, the technique we have described here is most efficient when we have an array of regularly spaced filaments, for which a global fit of a volumetric model can be obtained. It also depends on the filamentous features to alter their directionality in a gradual manner. If there are sudden kinks and sharp turns in the filamentous structures, our approach may not be particularly helpful for segmentation.
In the meantime, our collaborators have developed an automated approach for automated filament tracing that follows a similar concept used here for manual segmentation30,31. Going forward, the best approach may well be a hybrid of manual identification and placement of an initial sparse model (even only a few balls) into the density as a starting point, and then letting a search and fitting algorithm finish the tracing of the filaments.
Simplified volumetric models reduce the complexity of a system and allows certain patterns, such as actin filament bending near the tip, to be better appreciated. Also, the volumetric model can be used as an "anchor" to display a zone of density around the chosen anchoring ball-and-stick model, which allows the detection and visualization of crosslinker densities between adjacent actin filaments. The ability to select individual filaments and to set appropriate radii as a zone in which density is displayed again allows the overwhelming complexity of the scenery to be reduced to a manageable level.
One advantage of this volumetric model building approach of global fitting, followed by local adjustments, was that we were able to identify regions where actin filaments appeared to be interrupted, and significant gaps in actin filaments were indicated by the absence of map density. Since we had placed a volumetric ball-and-stick model, we could make use of a routine in the UCSF Chimera software package that color-codes each model ball position according to the map density value at that location. This approach allowed a fast detection and visualization of actin filament gaps in the actin bundle, which is a biologically significant feature we found in our cryo-electron tomogram, and which would have been very difficult to detect and visualize with traditional segmentation approaches. Yet another advantage of our volumetric model is that volumetric properties including lengths and distances can be easily obtained, which allows for actual numbers and thus a statistical analysis to be performed.
In summary, interactive manual model point placement, possibly further augmented by subsequent automated local fitting and filament tracing capabilities, is a rather promising approach for the visualization and quantitative analysis of electron tomographic subcellular volumes. This is because it uses the power of the human brain for pattern recognition and the power of computer science for model optimization.
Disclosures
The authors declare that they have no competing financial interests or other conflicts of interest.
Acknowledgements
We would like to thank Dr. Peter Barr-Gillespie and his team for their role in sample preparation and former members of the Auer lab and the Dr. Dorit Hanein lab for their role in tomographic data collection. We would also like to thank Tom Goddard of UCSF Resource for Biocomputing, Visualization, and Informatics (RBVI) for providing various UCSF Chimera scripts.
Materials
| Name | Company | Catalog Number | Comments |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/download.html | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=win64/chimera-1.16-win64.exe | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=mac64/chimera-1.16-mac64.dmg | |
| Chimera | RBVI | Version 1.16 https://www.cgl.ucsf.edu/chimera/cgi-bin/secure/chimera-get.py?file=linux_x86_64/chimera-1.16-linux_x86_64.bin | |
| Excel | Microsoft | Version 2211 https://www.office.com/?auth=1 | |
| Falcon II | Thermofisher | https://www.thermofisher.com/de/de/home/electron-microscopy/products/accessories-em/falcon-detector.html | |
| IMOD | University of Colorado | Version 4.11.1 https://bio3d.colorado.edu/imod/download.html | |
| PC Desktop | Intel | Windows 10, ver. 22H2 | |
| PC Laptop | Gigabyte | Windows 10, ver. 22H2 | |
| Powerpoint | Microsoft | Version 2211 https://www.office.com/?auth=1 | |
| Titan Krios Electron Microscope | Thermofisher | https://www.thermofisher.com/de/de/home/electron-microscopy/products/transmission-electron-microscopes/krios-g4-cryo-tem.html | |
| Word | Microsoft | Version 2211 https://www.office.com/?auth=1 |
References
- Downing, K. H., Sui, H., Auer, M. Electron tomography: A 3D view of the subcellular world. Analytical Chemistry. 79 (21), 7949-7957 (2007).
- Koning, R. I., Koster, A. J. Cellular nanoimaging by cryo electron tomography. Methods in Molecular Biology. 950, 227-251 (2013).
- Asano, S., Engel, B. D., Baumeister, W. In situ cryo-electron tomography: A post-reductionist approach to structural biology. Journal of Molecular Biology. 428 (2), 332-343 (2016).
- Serwas, D., Davies, K. M. Getting started with in situ cryo-electron tomography. Methods in Molecular Biology. 2215, 3-23 (2021).
- McDonald, K. L., Auer, M. High-pressure freezing, cellular tomography, and structural cell biology. Biotechniques. 41 (2), 137-143 (2006).
- Narasimha, R., et al. Evaluation of denoising algorithms for biological electron tomography. Journal of Structural Biology. 164 (1), 7-17 (2008).
- Frangakis, A. S. It's noisy out there! A review of denoising techniques in cryo-electron tomography. Journal of Structural Biology. 213 (4), 107804(2021).
- Frangakis, A. S., Hegerl, R. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. Journal of Structural Biology. 135 (3), 239-250 (2001).
- Jiang, W., Baker, M. L., Wu, Q., Bajaj, C., Chiu, W. Applications of a bilateral denoising filter in biological electron microscopy. Journal of Structural Biology. 144 (1-2), 114-122 (2003).
- vander Heide, P., Xu, X. -P., Marsh, B. J., Hanein, D., Volkmann, N. Efficient automatic noise reduction of electron tomographic reconstructions based on iterative median filtering. Journal of Structural Biology. 158 (2), 196-204 (2007).
- Volkmann, N. Methods for segmentation and interpretation of electron tomographic reconstructions. Methods in Enzymology. 483, 31-46 (2010).
- Böhm, J., et al. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proceedings of the National Academy of Sciences. 97 (26), 14245-14250 (2000).
- Frangakis, A. S., et al. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proceedings of the National Academy of Sciences. 99 (22), 14153-14158 (2002).
- Lebbink, M. N., et al. Template matching as a tool for annotation of tomograms of stained biological structures. Journal of Structural Biology. 158 (3), 327-335 (2007).
- Rigort, A., et al. Automated segmentation of electron tomograms for a quantitative description of actin filament networks. Journal of Structural Biology. 177 (1), 135-144 (2012).
- Volkmann, N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. Journal of Structural Biology. 138 (1-2), 123-129 (2002).
- Bajaj, C., Yu, Z., Auer, M. Volumetric feature extraction and visualization of tomographic molecular imaging. Journal of Structural Biology. 144 (1-2), 132-143 (2003).
- Yu, Z., Frangakis, A. S. Classification of electron sub-tomograms with neural networks and its application to template-matching. Journal of Structural Biology. 174 (3), 494-504 (2011).
- Moebel, E., et al. Deep learning improves macromolecule identification in 3D cellular cryo-electron tomograms. Nature Methods. 18 (11), 1386-1394 (2021).
- Tilney, L. G., Derosier, D. J., Mulroy, M. J. The organization of actin filaments in the stereocilia of cochlear hair cells. The Journal of Cell Biology. 86 (1), 244-259 (1980).
- Tilney, L. G., Tilney, M. S., DeRosier, D. J. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annual Review of Cell Biology. 8, 257-274 (1992).
- Metlagel, Z., et al. Electron cryo-tomography of vestibular hair-cell stereocilia. Journal of Structural Biology. 206 (2), 149-155 (2019).
- Song, J., et al. A cryo-tomography-based volumetric model of the actin core of mouse vestibular hair cell stereocilia lacking plastin 1. Journal of Structural Biology. 210 (1), 107461(2020).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Agulleiro, J. I., Fernandez, J. J. Fast tomographic reconstruction on multicore computers. Bioinformatics. 27 (4), 582-583 (2011).
- Fernandez, J. J. Computational methods for electron tomography. Micron. 43 (10), 1010-1030 (2012).
- Chen, H., Clyborne, W. K., Sedat, J. W., Agard, D. A. Priism: an integrated system for display and analysis of 3-D microscope images. Biomedical Image Processing and Three-Dimensional Microscopy. 1660, 784-790 (1992).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Goddard, T. D., Huang, C. C., Ferrin, T. E. Visualizing density maps with UCSF Chimera. Journal of Structural Biology. 157 (1), 281-287 (2007).
- Sazzed, S., et al. Tracing actin filament bundles in three-dimensional electron tomography density maps of hair cell stereocilia. Molecules. 23 (4), 882(2018).
- Sazzed, S., Scheible, P., He, P., Wriggers, J. Spaghetti tracer: A framework for tracing semiregular filamentous densities in 3D tomograms. Biomolecules. 12 (8), 1022(2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved