
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Citometría de Flujo Protocolos de superficie y intracelular de antígenos Los análisis de los tipos de células neuronales
En este artículo
Resumen
We provide a detailed description of a protocol for flow cytometric analysis of surface antigens and/or intracellular antigens in neural cell types. Critical aspects of experimental planning, step-by-step methodological procedures, and fundamental principles of flow cytometry are explained in order to enable neurobiologists to exploit this powerful technology.
Resumen
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Introducción
La citometría de flujo se ha explotado ampliamente en la inmunología, hematología y oncología para definir poblaciones de células a través de las propiedades intrínsecas de dispersión, antígeno de superficie de la célula de expresión, y otros parámetros de fluorescencia 1-3. Nuestros conocimientos sobre el desarrollo linaje de sangre y la enfermedad son el resultado de un grado significativo de el refinamiento continuo de esta metodología después de su aplicación inicial de 4,5. Una mayor conciencia del potencial analítico cuantitativo y global de citometría de flujo ha animado recientemente su uso más generalizado en la investigación con células madre y puede permitir igualmente profunda progreso en un marco de tiempo más corto 6. Sin embargo, la aplicación de citometría de flujo para analizar específicamente y aislar poblaciones neuronales durante mucho tiempo ha sido percibido como un reto. En contraste con las células hematopoyéticas que existen naturalmente en suspensión, tipos de células neuronales se cosechan típicamente de fuentes excesivamente complejos que pueden incluir células gliales y varios other células circundantes, así como una intrincada red de neuronas de procesos que soportan. En consecuencia, la neurobiología todavía tiene que aplicar la versatilidad de citometría de flujo a su potencial completo en las rutinas diarias de investigación. Sin embargo, siempre y cuando una suspensión de una sola célula viable puede ser generado (y protocolos se han ideado y optimizado para este fin 7), citometría de flujo y células activadas por fluorescencia (FACS) puede ser considerado como un valioso elemento del repertorio analítico en neurobiología 8-11.

. Figura 1. Principio de análisis de citometría de flujo y componentes de un citómetro de flujo flujo citómetros comprenden tres sistemas principales: hidráulica, óptica y electrónica. Un flujo aerodinámico de células en suspensión (preparado a partir de tejido primario o en cultivo in vitro) se lleva a cabo por el FLUI vainad través enfoque hidrodinámico, restringiendo la muestra a su núcleo central. Las partes ópticas se componen de láseres que iluminan la corriente de células y filtros ópticos que dirigen la señal a los detectores apropiados. Las señales de luz detectadas se convierten en señales electrónicas, posteriormente procesadas por una computadora y visualizados en un monitor para el análisis de datos y conmutación. Haga clic aquí para ver una versión más grande de esta figura.
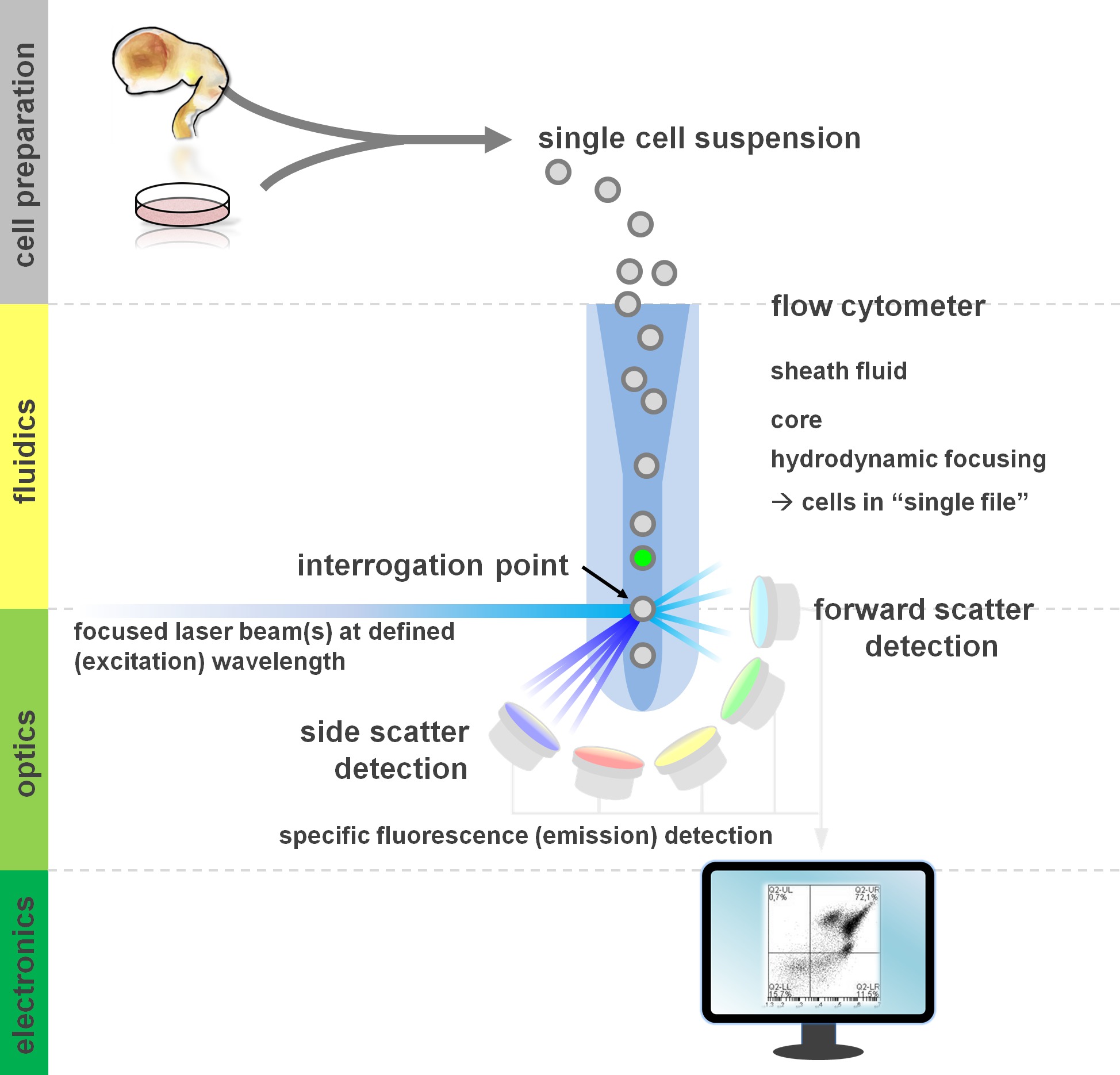{kind=link}
Los usuarios de los métodos de citometría de flujo de ganancias de al menos un conocimiento básico de los fundamentos subyacentes, como bloques de construcción de un citómetro (para revisión ver 12,13; también ver Figura 1). Un rayo láser se cruza con una corriente de fluido hidrodinámicamente enfocada que contiene las células en suspensión, que a su vez pasan a través del haz de láser en 'solo archivo' uno tras otro. El interceptien de una célula (o cualquier otra partícula, para el caso) con los resultados de láser en la dispersión de la luz desde este punto de interrogación. La luz dispersada se puede detectar en la continuación de la dirección láser (dispersión hacia adelante, asociado con el tamaño de la partícula), así como perpendicular a su dirección (dispersión lateral; reflejando la granulosidad de la partícula / célula). Estas propiedades de dispersión antes mencionados no requieren etiquetado específico, por lo que una muestra sin etiqueta (o también restos celulares, burbujas de aire, etc.) generará una señal (evento) en la dispersión hacia adelante frente bivariado lado gráfico de dispersión comúnmente utilizado para gating inicial. Mediante el uso de los láseres y filtros específicos para el correspondiente espectros de excitación y emisión adecuadas, una célula puede ser analizada por su positividad, el nivel de intensidad, o ausencia de marcadores fluorescentes. La mayoría de las aplicaciones de citometría de flujo se han centrado en la caracterización a través de antígenos de superficie celular. A diferencia de la lineag hematopoyéticoe, el linaje neural ha permanecido menos ampliamente definida de acuerdo con los patrones de expresión de superficie epítopo 5. Una ventaja de la explotación de los antígenos de superficie es que las células vivas pueden ser sometidos a la celda paradigmas de clasificación, tales como FACS. En contraste, la tinción de antígeno intracelular requiere etapas de fijación y permeabilización de mediar en la interacción-epítopo de anticuerpo, lo que impide aplicaciones posteriores que requieren células viables. De nota, estos enfoques aún permiten para numerosos ensayos cuantitativos 14, así como los análisis aguas abajo para ARN y expresión de la proteína 15. Hematología, inmunología y oncología han utilizado a menudo más de una docena de marcadores en conjunto para definir determinadas subpoblaciones 16. Además, la citometría de masa o CyTOF ahora se pueden utilizar para analizar hasta 30 parámetros simultáneamente 17,18.
Para aplicaciones de células madre neurales, así como cultivos primarios 14,19,20 la heterogeneidad de las células envitro es un fenómeno común 21-23. Las células no representan a la población diana de interés encarnan un factor potencialmente de confusión para la lectura experimental 24,25. Convenientemente, los diferentes subconjuntos celulares presentes dentro de una suspensión de células heterogénea tienen perfiles de expresión de antígeno diferentes (conocidas o aún para ser descifrados), que pueden ser utilizados para definir estos diferentes poblaciones. La citometría de flujo por lo tanto puede desempeñar un papel crucial en la resolución de la heterogeneidad celular y, de ese modo, facilitar aplicaciones biomédicas (ensayos in vitro, la terapia celular) y optimizar lectura cuantitativa, centrándose en el subconjunto más relevantes 24,26. Varias combinaciones del antígeno de superficie han sido identificados en los últimos años para permitir la cuantificación y el aislamiento de tipos específicos de células neuronales. Esto incluye CD133 para el enriquecimiento de células madre neurales 27, una combinación de los CD15 / CD24 / CD29 antígenos de superficie para el aislamiento de NSC, Differentiated neuronas y las células de la cresta neural 28 o CD15 / CD24 / CD44 / CD184 / CD271 para aislar los nervios y subconjuntos gliales 25, entre otras firmas 29,30. Más allá de las neuronas, marcadores gliales incluyen A2B5 31, CD44 25, NG2 32 y GLAST 33. Una publicación reciente ha explotado el cerebro medio marcador precursor floorplate CORIN 34,35 para enriquecer precursores dopaminérgicos en el trasplante de células Parkinson paradigmas 36. Moléculas de CD no son únicos marcadores, pero funcionalmente relevantes mediadores de las interacciones célula-célula y de la capacidad de una célula para responder a las señales de las moléculas de la matriz extracelular y factores de crecimiento 37. Una estrategia de mejorar aún más el arsenal de antígenos CD combinatorias para caracterizar el desarrollo del linaje neural es el uso de marcadores intracelulares conocidos para detectar y definir combinaciones de antígenos CD para un tipo de célula particular de interés. Hemos explotado recientemente un enfoque de este tipo e identificado CD49f - / CD200 altos patrones de expresión combinatoria como un enfoque novedoso para enriquecer subconjuntos neuronales a partir de sistemas de cultivo de células madre pluripotentes inducidas neuralmente diferenciados 38. Aquí, incluimos y discutimos este último protocolo (y variaciones opcionales de los mismos) en los que tinción de la superficie y la tinción intracelular se pueden utilizar simultáneamente para definir subpoblaciones de células neuronales por citometría de flujo.

Figura 2. Diagrama de flujo de opciones de protocolo experimental. La figura representa una representación esquemática de las principales etapas implicadas en el protocolo. Pasos opcionales (tinte CFSE o etiquetado antígeno intracelular) se indican con cajas de color gris claro. Después de la cosecha, es esencial para evaluar la viabilidad celular y número de suspensiones de células neurales antes de la tinción de la superficie celular. Positivo comoasí como controles negativos se deben incluir además de las muestras de interés. Las muestras pueden ser analizadas mediante análisis de citometría de flujo y / o se utilizan en los paradigmas de clasificación de células. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Si bien hemos utilizado anteriormente anticuerpo primario en combinación con el anticuerpo secundario para la tinción intracelular 38, introducimos ahora etiquetado no covalente del anticuerpo primario a través de fragmentos Fab fluorescentes (etiquetado Zenon) como una ligera variación, reduciendo así las etapas de la manipulación de células 39. Por otra parte, como un ejemplo más de la versatilidad del protocolo, empleamos un etiquetado facultativo de un subconjunto experimental por carboxifluoresceına succinimidiléster (CFSE) antes de salir a la superficie tinción antígeno. Tal pre-etiquetado CFSE permite la comparación inmediata directa de dos líneas celulares o condiciones experimentales (CFSE marcado vs. sin etiqueta) dentro de un único tubo de muestra, la reducción de la varianza o sutiles diferencias en el tiempo de incubación y el ahorro de anticuerpo. CFSE es un colorante fluorescente establecido que se utiliza comúnmente para el seguimiento de la célula 40, en la proliferación de 41,42 y 43,44 códigos de barras experimentos. Por último, mientras que los pasos reales (FACS, separación celular inmunomagnética o immunopanning) no forman parte de este protocolo, en principio, la recolección y el etiquetado que se describen aquí hacen muestras de rendimiento que se pueden someter a la superficie de antígeno o aplicaciones intracelulares basado etiquetado clasificación 15 25,28.
Con este artículo, nos proponemos: resumir una superficie viable protocolo de tinción antígeno 25,28, resumir un protocolo para la detección de dianas intracelulares, así como la superficie combinada y el análisis del antígeno intracelular de 38 años, presentar un paso CFSE etiquetado tinte intracelular 41,45 como opción experimental para comcomparativa análisis de poblaciones de células neuronales, y resumir los enfoques para el análisis de citometría de flujo (controles apropiados 13,46, apertura de puerta estrategia y datos de presentación 47).
Protocolo
1. Neural cosecha celular
- Evaluación por microscopio:
- Antes de iniciar un experimento, comprobar el estado de la cultura con la de campo claro o microscopía de contraste de fase.
NOTA: Mientras que el tejido neural primaria obtenida de disecciones es, en principio, igualmente susceptibles de análisis de citometría de flujo 14,28, por favor, tenga en cuenta que el enfoque del protocolo está en las células obtenidas de los sistemas de células neurales in vitro.
- Antes de iniciar un experimento, comprobar el estado de la cultura con la de campo claro o microscopía de contraste de fase.
- Recolección de las células 7:
- Lave suavemente el plato / recipiente con células adherentes con Mg 2 + / Ca 2+ de buffer fosfato salino (PBS) libre a temperatura ambiente (por ejemplo, 5 ml por frasco T75, 3 ml por pocillo de placa de 6 pocillos, o 10 ml de placa de 10 cm).
NOTA: El PBS utiliza en todo el protocolo es Mg 2 + / Ca 2+ libre.- Para el ejemplo de vídeo, utilice un matraz T75 de células de neuroblastoma SH-SY5Y en 80% de confluencia. Aplicar etapas de lavado adicionales en los casos enescombros considerable está presente en el plato.
- Considere lavados con PBS 48 que contiene albúmina, Percoll o Ficoll 49 gradientes de centrifugación de suero y / o cuentas disponibles en el mercado 50,51. La eliminación de la mielina y otro lípido u otros contaminantes es crítica, especialmente cuando se utilizan fuentes de tejidos primarios adultos.
- Añadir precalentado (37 ° C) la sustitución de tripsina en un volumen apropiado que cubre toda la superficie del recipiente de cultivo de tejidos.
NOTA: Como alternativa, considere Accutase u otras opciones de digestión enzimática. Este paso crítico puede afectar negativamente a la superficie epítopo expresión (véase 7). - Incubar la placa / matraz a 37 ° C durante 2 - 5 min (dependiendo del tipo de célula) para permitir que las células se despeguen. Golpee suavemente el recipiente de cultivo de tejidos o al ras con una pipeta serológica para desalojar las células. Evitar el exceso de digestión (ya que esto puede causar la pérdida de células y la coagulación en los pasos posteriores).
- Quench la sustitución de la tripsina mediante la adición de dos veces el volumen de tampón de flujo (2% de FBS en PBS) y recoger las células en un tubo cónico de 15 ml.
- Tritura suavemente la suspensión celular usando una pipeta de microlitro (100 - 1000 l) o una pipeta de 5 ml serológica para preparar una suspensión de células individuales.
- Centrifugar las células a 220 xg durante 5 min a 25 ° C. Aspirar cuidadosamente el sobrenadante dejando el sedimento atrás.
- Vuelva a suspender el sedimento en un volumen apropiado de tampón de flujo, dependiendo del tamaño de la pastilla (por ejemplo, para una confluentes T75 matraz de células SH-SY5Y el rendimiento típico es de al menos 10 x 10 6 células, en cuyo caso las células se resuspenden en 5 ml de tampón de flujo).
NOTA: Si se observan trozos más grandes o de coagulación, se filtra a través de un 30 - 100 micras de malla.
- Lave suavemente el plato / recipiente con células adherentes con Mg 2 + / Ca 2+ de buffer fosfato salino (PBS) libre a temperatura ambiente (por ejemplo, 5 ml por frasco T75, 3 ml por pocillo de placa de 6 pocillos, o 10 ml de placa de 10 cm).
- El recuento de células 52:
- Transferir una pequeña alícuota de la suspensión celular a un tubo de microcentrífuga y se diluye en una proporción definida en un volumen de trypan azul o un colorante de viabilidad alternativa antes de la transferencia a un hemocitómetro o sistema de conteo de células automatizado.
- Diluir la suspensión celular a una concentración de 1 x 10 6 células viables / ml mediante la adición del volumen apropiado de tampón de flujo o PBS con 0,1% de BSA (si proceder con el etiquetado CFSE).
NOTA: yoduro de propidio, 7-aminoactinomicina D, anexina V y kits de ensayo de viabilidad pueden solucionar disponibles comercialmente representan opciones alternativas con el fin de evaluar la viabilidad de las células. Además, ensayos de apoptosis usando la caspasa-3 de fluorescencia como se describió anteriormente 53 puede ser utilizado. Canales fluorescentes serán "ocupadas" por estos reactivos que pueden limitar las opciones para los pasos posteriores si se incluye en todas las muestras.
2. intracelular Tinte Etiquetado Usando CFSE (Figura 3)
- Diluir la CFSE a una concentración de stock deseada que puede ser utilizado fácilmente.
NOTA: Para estos experimentos una concentración madre deSe utilizó 0,01 mM. Determinar la concentración de trabajo óptimo de CFSE empíricamente. - Añadir 10 l de solución 0,01 mM CFSE por ml de células (Sección 1.3.2, la concentración de 1 x 10 6 células / ml en PBS + BSA al 0,1%) para una concentración final de 0,1 mM. Vórtice brevemente para mezclar bien.
NOTA: Las concentraciones CFSE utilizados aquí son aproximadamente diez veces menor que las que comúnmente aplicados en ensayos de proliferación, por lo tanto, la toxicidad celular del colorante es mínima. No observamos efectos negativos sobre la viabilidad celular. - Incubar durante 5 min a temperatura ambiente con agitación constante (200 rpm). Proteger de la luz.
- Se inactiva el colorante mediante la adición de 5 volúmenes de tampón de flujo a los tubos. Centrifugar a 94 xg durante 5 min a TA.
- Descartar el sobrenadante dejando el sedimento atrás. Volver a suspender las células con 5 volúmenes de tampón de flujo.
- Centrifugar a 94 xg durante 5 min a TA. Descartar el sobrenadante y volver a suspender las células en tampón de flujo a una concentración de 1 x 10 6 células / ml.
- Añadir un número igual de células no teñidas de interés para la suspensión de células manchadas. Proceder a la superficie protocolo de tinción de antígeno (Sección 3).

Figura 3. Detección de la expresión diferencial de antígeno de superficie de CD entre dos líneas de células a través de células de neuroblastoma SH-SY5Y CFSE tinte etiquetado. Son pre-marcado con CFSE para su posterior identificación en comparación con fibroblastos BJ no marcados. Co-tinción de la muestra mixta (paneles de la derecha) con marcadores de superficie CD24 o CD54 (tanto conjugado con APC) demuestra que las líneas celulares son fácilmente distinguibles debido a la tinción CFSE (flechas = SH-SY5Y; puntas de flecha = BJ fibroblastos). La mayoría de las células SH-SY5Y expresan CD24 pero no CD54 (ICAM-1). En contraste, los fibroblastos BJ (CFSE-negativo) son positivos para CD54 pero en gran medida negativa para CD24.Objetivo "https://www.jove.com/files/ftp_upload/52241/52241fig3highres.jpg" = "_ blank"> Haga clic aquí para ver una versión más grande de esta figura.
3. celular tinción de la superficie
- Tubos de etiquetas incluidas las muestras y los controles críticos (ver Tabla 1).
| Tubo no. | Nombre de la muestra | Antígeno-fluoróforo | Dilución |
| 1 | Células sin teñir | - | |
| 2 | Manchada Individual | CD24-APC | 1:50 |
| 3 | Manchada Individual | Tuj1-Alexa Fluor 488 nm | 1: 2000 |
| 4 | Doble manchada | CD24-APC | 1:50 |
| Tuj1-Alexa Fluor 488 nm | 1: 2000 | ||
| 5 | Manchada Individual | Secundaria solamente: Alexa fluor 488 nm | 1: 2000 |
Tabla 1. L ist de tubos para ser incluido en un flujo típico de citometría de experimento. La tabla muestra un conjunto mínimo de tubos de muestra requerido para un experimento de co-tinción descrito en este artículo de vídeo. Un experimento ideales debe incluir todos los controles necesarios (controles de compensación negativos y positivos, así como) para la correcta interpretación de los resultados obtenidos.
- Añadir 100 l de suspensión celular (de la Sección 2.7 o 1.3.2) a cada tubo de microcentrífuga de 1,5 ml.
NOTA: Asegúrese de que un mínimo de 0,1 x 10 6 células presentes por cada 100 l de suspensión celular. - Añadir fluoróforo anticuerpo conjugado a la muestra a una dilución apropiada.
NOTA: Determinar la dilución de trabajo para cada anticuerpo antes del experimento. Ver Tabla 2 para una lista of antígenos de superficie neural.
| Antígeno | Tipo de la célula | Referencia |
| CD15 | Las células madre neurales | [28, 67] |
| CD24 | Las células neuronales | [28, 68] |
| CD29 | Las células madre neurales | [28, 69, 70] |
| CD44 | Las células gliales | [25] |
| CD49f | Las células madre neurales | [38] |
| CD56 (NCAM) | Las células neuronales | [71] |
| CD133 | Las células madre neurales | [27] |
| CD184 | Las células madre neurales y células gliales | [25] |
| CD200 | Las células neuronales | [38] |
| CD271 | Las células madre de la cresta neural | [25] |
| A2B5 | Las células gliales | [31] |
| CORIN | Precursores dopaminérgicos | [35, 36] |
| FORSE1 | Las células madre neurales (NSC) | [72] |
| GLAST | Las células gliales | [33] |
| NG2 | Las células gliales | [32] |
Tabla 2. Selección de los antígenos de superficie neuronales. Esta tabla proporciona una lista de los epítopos de superficie encontró que se expresa por varios tipos de células neuronales para ejemplificar el panel cada vez mayor de los antígenos de superficie utilizados para caracterizar el linaje neural. Tenga en cuenta que esta selección está lejos de ser completa y que la mayoría de estos marcadores también se expresan por una serie de otras células neuronales y no neuronales. En consecuencia, se requerirá una combinación de varios marcadores para definir y aislar los subconjuntos neuronales indicadas mejor.
- Incubar durante 30 min en un agitador orbital (200 rpm) en la oscuridad.
- Tampón de lavado de flujo
- Añadir 1 ml de tampón de flujo a los tubos. Se centrifuga a 380 g durante 4 min a 4 ° C.
- Descartar el sobrenadante dejando el sedimento atrás.
- Repita el paso de lavado de nuevo.
- Después del segundo lavado, decantar el sobrenadante y volver a suspender las células en tampón de flujo a un volumen final de 100 microlitros.
- Utilice la muestra para el análisis de citometría de flujo. Como alternativa, proceda con la Sección 4 y 5.
NOTA: Si las células se deben ordenar y poner de nuevo en la cultura post-FACS (es decir, la suspensión de células viables sin fijación o permeabilización), aplicar técnicas asépticas durante la cosecha, las manchas y las medidas analíticas.
4. Fijación y permeabilización 38
- Fijación utilizando paraformaldehído (PFA):
- Prepare el buffer de fijación que contienen 2% PFA en PBS.
NOTA: PFA es perjudicial para los seres humanos y el medio ambiente. Use el equipo de protección personal adecuado y desechar los residuos de acuerdo con las regulaciones locales. - Añadir 500 l de tampón de fijación a 100 l de suspensión celular.
- Incubar los tubos a temperatura ambiente durante 15 min en un agitador orbital (100 rpm) en la oscuridad.
- Prepare el buffer de fijación que contienen 2% PFA en PBS.
- PBS de lavado:
- Añadir 1 ml de PBS al tubo. Se centrifuga a 380 g durante 3 min a 4 ° C.
- Desechar / decantar el sobrenadante dejando aproximadamente 100 l en el tubo.
- La permeabilización con Tween-20:
- Preparar tampón de permeabilización que contiene 0,7% de Tween-20 en PBS.
- Añadir 500 l de tampón de permeabilización a 100 l de suspensión celular.
- Incubar los tubos a temperatura ambiente durante 15 min en un agitador orbital (100 rpm) en la oscuridad.
- Se lavan las células una vez con PBS (como se describe en la Sección 4.2) y eliminar el sobrenadante de los tubos completely, dejando sólo el pellet atrás.
5. intracelular de antígeno de tinción 38 (Figura 4)
- Preparación de las soluciones de anticuerpos primarios:
- Diluir los anticuerpos primarios en tampón de dilución que contiene 1% de albúmina de suero bovino, 10% de suero (por ejemplo, burro o suero de cabra normal) y 0,5% de Tween-20 en PBS.
NOTA: Elija el suero para ser utilizados en función de la especie de los anticuerpos secundarios se plantearon. - Alternativamente, utilizar Zenon etiquetado fluoresceína del anticuerpo primario de acuerdo con las instrucciones del fabricante.
- Preparar 1 mg de anticuerpo primario en PBS a una dilución apropiada (volumen total ≤ 20 l).
- Añadir 5 l de reactivo de Zenon fluoresceína IgG etiquetado (Componente A) a la solución de anticuerpo.
- Incubar la mezcla durante 5 min a TA.
- Añadir 5 l de Zenon reactivo de bloqueo (Componente B) a la mezcla de reacción.
- Incubarla mezcla durante 5 min a TA. Aplicar el anticuerpo a la muestra dentro de 30 min.
- Diluir los anticuerpos primarios en tampón de dilución que contiene 1% de albúmina de suero bovino, 10% de suero (por ejemplo, burro o suero de cabra normal) y 0,5% de Tween-20 en PBS.
- Tinción de anticuerpos primario:
- Añadir 100 l de la solución de anticuerpo primario al sedimento celular y se tritura suavemente para mezclar.
- Alternativamente, añadir Zenon fluoresceína anticuerpo marcado a la suspensión celular a la dilución apropiada.
- Incubar los tubos a temperatura ambiente durante 30 min en un agitador orbital (200 rpm), protegido de la luz. Se lavan las células una vez con PBS (como se describe en la Sección 4.2) y eliminar el sobrenadante de los tubos completamente, dejando sólo el pellet atrás.
- Tinción de anticuerpos secundaria (no es necesario para los anticuerpos Zenon fluoresceína etiquetados):
- Diluir la secundaria de anticuerpos en PBS a una concentración apropiada.
- Añadir 100 l de la solución de anticuerpo secundario al sedimento celular y se tritura suavemente para mezclar. Incubar los tubos a temperatura ambiente durante 30 min en un agitador (200 rpm) en la oscuridad.
- Lávese las muestras dos veces con PBS (como se describe en la Sección 4.2).
- Lavar una vez con tampón de flujo (ver sección 3.5).
- Vuelva a suspender las células en aproximadamente 150 l de tampón de flujo y analizar en el citómetro de flujo.

Figura 4. Co-tinción de la superficie y las proteínas intracelulares. Citometría de flujo de datos es un ejemplo de una comparación entre primaria + secundaria basada en anticuerpos frente a la tinción intracelular basado fluoresceína Zenon en combinación con tinción de la superficie. Positividad exclusiva en el eje y (cuadrante superior izquierdo) y el eje x (cuadrante inferior derecho) muestra células teñidas para MAP2 y CD24, respectivamente. Después de co-tinción, MAP2 compartida y la expresión CD24 pueden verse en el cuadrante superior derecho (paneles de la derecha). Comparación de utilizarAlexa fluor 488. (Arriba; AF488) versus Zenon fluoresceína (abajo) para rendimientos MAP2-etiquetado resultados similares Haga clic aquí para ver una versión más grande de esta figura.
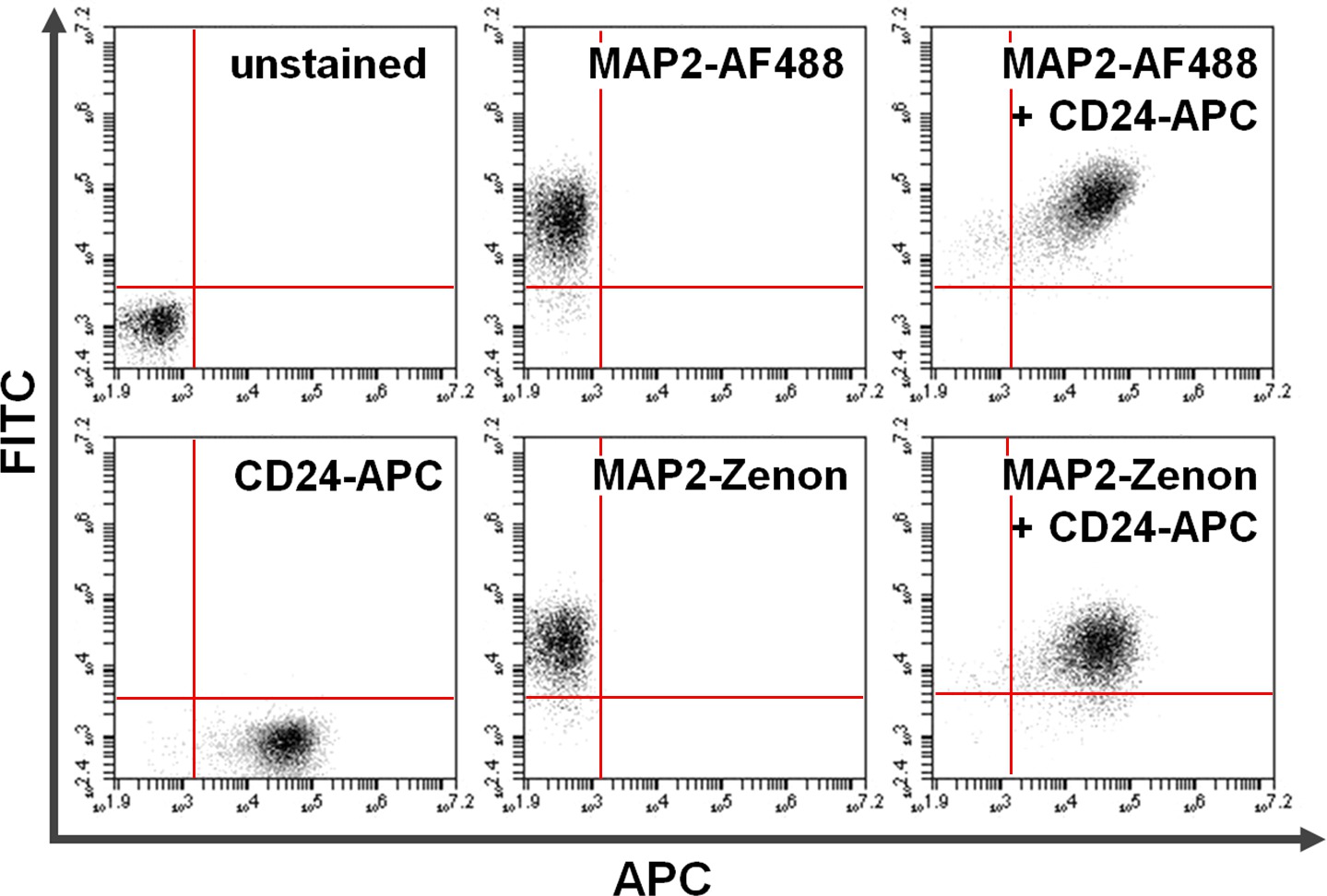{kind=link}
6. Análisis de citometría de flujo
- Realizar un análisis de citometría de flujo inmediatamente después de la finalización del protocolo de tinción usando un citómetro de flujo con filtros adecuados para la detección de la señal. Utilice 488 nm láser rojo azul nm y 640 con FL-1 (533/30), FL-2 (585/40), y filtros FL-4 (675/25) de paso de banda.
- Configure puertas principales basados en la dispersión frontal y lateral con exclusión de los desechos y células muertas.
- Establecer puertas de fluorescencia para la superficie y el antígeno intracelular a ≤0.5% sobre la base de las muestras y la compensación no teñidas de solapamiento espectral utilizando controles manchadas individuales.
Resultados
El protocolo que aquí se presenta permite enfoques versátil experimentales (Figura 2). En su versión más corta (pasos 1, 3 y 6), que puede considerarse una guía para la tinción simple de los antígenos de superficie. En su forma más compleja, una serie de paradigmas co-etiquetado con una gama de antígenos intracelulares puede ser perseguido (pasos opcionales 2 y / o 4 a 5). Por otra parte, el pa...
Discusión
El protocolo presentado aquí está bien establecido para los cultivos de células neurales derivadas de células madre humanas, pero se puede aplicar igualmente a otras fuentes de células neuronales incluyendo tejido primario o líneas celulares neuronales. Además de las fuentes embrionarias, las células madre o progenitoras neurales pueden ser extraídos de las regiones neurogénicas de cerebro adulto de 27. Además, la citometría de flujo FACS y se pueden explotar para cuantificar, analizar y aislar di...
Divulgaciones
The authors declare no potential conflicts of interest.
Agradecimientos
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
Materiales
| Name | Company | Catalog Number | Comments |
| DMEM/F12 (1:1) (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) |  Life Technologies Life Technologies | 11330057 | |
| DPBS without Ca2+ Mg2+ | Life Technologies | 14190169 | |
| Fetal bovine serum, qualified, E.U.-approved, South America origin (FBS) | Life Technologies | 10270-106 | |
| MEM Non-essential amino acids (100x) | Life Technologies | 11140035 | |
| TrypLE Express | Life Technologies | 12604013 | |
| Trypan blue solution, 0.4% | Life Technologies | 15250061 | |
| Paraformaldehyde | Carl Roth | 335.3 | |
| Bovine serum albumin (BSA) Fraction V | PAA Laboratories, Coelbe | K41-001 | |
| Tween-20 Detergent | Calbiochem | 655205 | |
| Carboxyfluorescein succinimidyl ester (CFSE) | eBioscience | 65-0850-84 | |
| DMSO | AppliChem | A1584 | |
| Bottle top filters express plus 0.22 µm, 250 ml | Millipore | SCGPU02RE | |
| Cell culture treated flasks (T 25) | NUNC | 156367 | |
| Cell culture treated flasks (T 75) | NUNC | 156499 | |
| Conical tubes (15 ml) | Greiner Bio-One | 188271 | |
| Conical tubes (50 ml) | Greiner Bio-One | 227261 | |
| Pasteur pipet, glass (150 mm) | STEIN Labortechnik, Remchingen | S03710150 | |
| Pipet tips (0.1-10 µl) | Corning | 4125 | |
| Pipet tips (1-200 µl) | Corning | 4126 | |
| Pipet tips (100-1000 µl) | Corning | 4129 | |
| Serological pipets, 5 ml | Corning | 4051 | |
| Serological pipets, 10 ml | Corning | 4101 | |
| Serological pipets, 25 ml | Corning | 4251 | |
| Microcentrifuge tubes (0.5 ml) | Sarstedt | 72,699 | |
| Microcentrifuge tubes (1.5 ml) | Greiner Bio-One | 616201 | |
| Microcentrifuge tubes (2.0 ml) | Sarstedt | 72,695,500 | |
| Anti-Human CD24 APC monoclonal antibody | eBioscience | 17-0247-42 | Working dilution 1:50 |
| Anti-Human CD54 PE monoclonal antibody | eBioscience | 12-0549-42 | Working dilution 1:50 |
| Neuronal Class III β-Tubulin (Tuj1) polyclonal antibody | Covance | PRB-435P | Working dilution 1:2,000 |
| Alexa Fluor 488 Donkey anti Rabbit | Life Technologies | A21206 | Working dilution 1:2,000 |
| Zenon® Fluorescein Rabbit IgG Labeling Kit | Life Technologies | Z-25342 | |
| Neubauer-Improved counting chamber | Marienfeld | 0640010 | |
| Vortex | Scientific Industries | G560E | |
| Thermomixer comfort | Eppendorf | 5355 000.001 | |
| Accuri C6 flow cytometer | Becton Dickinson (BD) | 653118 | |
| Microcentrifuge refrigerated, PerfectSpin 24 R | Peqlab | 91-PSPIN-24R | |
| Orbital shaker, Unimax 1010 | Heidolph | 543-12310-00 | |
| Centrifuge refrigerated, Rotanta 96 RC | Hettich | 4480-50 | |
| Class II Biological safety cabinet Safe 2020 | Thermo Scientific | 51026640 | |
| CO2 Incubator, Heracell 240i | Thermo Scientific | 51026331 | |
| Vacuum system, Vacusafe comfort | Integra Biosciences | 158320 | |
| Microscope, Axiovert 40 CFL | Zeiss | 451212 | |
| Pipet controller, accu-jet pro | Brand | 26303 | |
| Micropipet, Pipetman neo P20N (2-20 µl) | Gilson | F144563 | |
| Micropipet, Pipetman neo P200N (20-200 µl) | Gilson | F144565 | |
| Micropipet, Pipetman neo P1000N (100-1000 µl) | Gilson | F144566 |
Referencias
- Herzenberg, L. A., et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 48 (10), 1819-1827 (2002).
- Chattopadhyay, P. K., Roederer, M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif). 57 (3), 251-258 (2012).
- Jaye, D. L., Bray, R. A., Gebel, H. M., Harris, W. A. C., Waller, E. K. Translational applications of flow cytometry in clinical practice). J. Immunol. 188 (10), 4715-4719 (2012).
- Henel, G., Schmitz, J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 38 (7), 428-436 (2007).
- Seita, J., Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2 (6), 640-653 (2010).
- Ulrich, H., Bocsi, J. Phenotypes of stem cells from diverse origin. Cytometry. A. 77 (1), 6-10 (2010).
- Panchision, D. M., et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 25 (6), 1560-1570 (2007).
- Meyer, R. A., Zaruba, M. E., McKhann, G. M. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 2 (1), 66-74 (1980).
- Junger, H., Junger, W. G. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 9 (16), 3749-3754 (1998).
- McLaren, F. H., Svendsen, C. N., Vander Meide, P., Joly, E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 112 (1-2), 35-46 (2001).
- Wang, S., Roy, N. S., Benraiss, A., Goldman, S. A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 22 (1-2), 167-176 (2000).
- Tanke, H. J., vander Keur, M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 11 (2), 55-62 (1993).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243 (1-2), 77-97 (2000).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 129 (1), 73-79 (2003).
- Ernst, A., et al. Neurogenesis in the striatum of the adult human brain. Cell. 156 (5), 1072-1083 (2014).
- Perfetto, S. P., Chattopadhyay, P. K., Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 4 (8), 648-655 (2004).
- Bandura, D. R., et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81 (16), 6813-6822 (2009).
- Bendall, S. C., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332 (6030), 687-696 (2011).
- Neveu, I., Rémy, S., Naveilhan, P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 113 (4), 767-777 (2002).
- Pruszak, J., Just, L., Isacson, O., Nikkhah, G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2 (Unit 2D.5), (2009).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99 (22), 14506-14511 (2002).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1-2), 18-29 (2003).
- Pruszak, J., Isacson, O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 651, 112-123 (2009).
- Carson, C. T., Aigner, S., Gage, F. H. Stem cells: the good, bad and barely in control. Nat. Med. 12 (11), 1237-1238 (2006).
- Yuan, S. H., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 6 (3), e17540 (2011).
- Roy, N. S., Cleren, C., Singh, S. K., Yang, L., Beal, M. F., Goldman, S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 12 (11), 1259-1268 (2006).
- Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 97 (26), 14720-14725 (2000).
- Pruszak, J., Ludwig, W., Blak, A., Alavian, K., Isacson, O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 27 (12), 2928-2940 (2009).
- Peh, G. S. -. L., Lang, R. J., Pera, M. F., Hawes, S. M. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 18 (2), 269-282 (2009).
- Golebiewska, A., Atkinson, S. P., Lako, M., Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 27 (6), 1298-1308 (2009).
- Dietrich, J., Noble, M., Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 40 (1), 65-77 (2002).
- Nishiyama, A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 14 (1), 77-82 (2001).
- Jungblut, M., et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 60 (6), 894-907 (2012).
- Ono, Y., et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 134 (17), 3213-3225 (2007).
- Chung, S., et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 108 (23), 9703-9708 (2011).
- Doi, D., et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2 (3), 337-350 (2014).
- Solozobova, V., Wyvekens, N., Pruszak, J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 8 (3), (2012).
- Turaç, G., et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 8 (6), e68519 (2013).
- Buchwalow, I. B., Böcker, W. Chapter 2. Immunohistochemistry: Basics and Methods. , 9-17 (2010).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. (70), e4287 (2012).
- Lyons, A. B., Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171 (1), 131-137 (1994).
- Hawkins, E. D., et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2 (9), 2057-2067 (2007).
- Quah, B. J. C., Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 44, (2010).
- Sukhdeo, K., et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 8 (1), e53015 (2013).
- Jiang, L., et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 140, 90-99 (2014).
- Hulspas, R., et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 76 (6), 355-364 (2009).
- Moloney, M., Shreffler, W. G. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 101 (5), 544-549 (2008).
- Siebzehnrubl, F. A., et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 750, 61-77 (2011).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain). J. Neurosci. Methods. 203 (1), 10-18 (2012).
- Tham, C. -. S., Lin, F. -. F., Rao, T. S., Yu, N., Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 21 (8), 431-443 (2003).
- Nguyen, H. X., Beck, K. D., Anderson, A. J. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. (50), e2698 (2011).
- Marchenko, S., Flanagan, L. Counting human neural stem cells. J. Vis. Exp. (7), 262 (2007).
- Brunlid, G., Pruszak, J., Holmes, B., Isacson, O., Sonntag, K. C. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 25 (10), 2551-2558 (2007).
- Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 71 (2), 143-155 (1997).
- Cardona, A. E., Huang, D., Sasse, M. E., Ransohoff, R. M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1 (4), 1947-1951 (2006).
- Nielsen, J. A., Maric, D., Lau, P., Barker, J. L., Hudson, L. D. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 26 (39), 9881-9891 (2006).
- Daneman, R., et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 5 (10), e13741 (2010).
- Gräbner, R., Till, U., Heller, R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 40 (3), 238-244 (2000).
- Quah, B. J. C., Parish, C. R. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 379 (1-2), 1-14 (2012).
- Lathia, J. D., et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6 (1), 117-129 (2014).
- Ganat, Y. M., et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 122 (8), 2928-2939 (2012).
- Hedlund, E., et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 26 (6), 1526-1536 (2008).
- Maroof, A. M., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12 (5), 559-572 (2013).
- Chivet, M., Hemming, F., Pernet-Gallay, K., Fraboulet, S., Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145 (2012).
- Graner, M. W., et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23 (5), 1541-1557 (2009).
- Eldh, M., Lötvall, J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. (59), e3037 (2012).
- Capela, A., Temple, S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291 (2), 300-313 (2006).
- Nieoullon, V., Belvindrah, R., Rougon, G., Chazal, G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 28 (3), 462-474 (2005).
- Nagato, M., et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 80 (4), 456-466 (2005).
- Hall, P. E., Lathia, J. D., Miller, N. G. A., Caldwell, M. A., French-Constant, C. Integrins are markers of human neural stem cells. Stem Cells. 24 (9), 2078-2084 (2006).
- Hargus, G., et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107 (36), 15921-15926 (2010).
- Elkabetz, Y., et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152-165 (2008).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados