
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Citometria de Fluxo protocolos para a superfície e intracelular Antigen análises de tipos de células neurais
Neste Artigo
Resumo
We provide a detailed description of a protocol for flow cytometric analysis of surface antigens and/or intracellular antigens in neural cell types. Critical aspects of experimental planning, step-by-step methodological procedures, and fundamental principles of flow cytometry are explained in order to enable neurobiologists to exploit this powerful technology.
Resumo
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Introdução
A citometria de fluxo foi explorado extensivamente em imunologia, hematologia e oncologia para definir populações de células através de dispersão propriedades intrínsecas, a expressão do antigénio da superfície celular, e outros parâmetros de fluorescência 1-3. Os nossos insights sobre desenvolvimento linhagem de sangue e doença são o resultado de um significativo grau de refinamento contínuo desta metodologia após a sua implementação inicial de 4,5. O aumento da consciência do potencial analítico quantitativo e global de citometria de fluxo tem incentivado recentemente o seu uso mais difundido na pesquisa com células-tronco e pode permitir semelhante profunda progresso em um tempo mais curto 6. No entanto, a aplicação da citometria de fluxo para analisar especificamente e isolar populações neuronais tem sido entendida como um desafio. Em contraste com as células hematopoiéticas que existem naturalmente em suspensão, os tipos de células neuronais são tipicamente colhidas de fontes excessivamente complexas que podem incluir células gliais e vários outras células circundantes, bem como uma intrincada rede de neurônios portadores de processo. Consequentemente, neurobiologia ainda tem que implementar a versatilidade de citometria de fluxo para o seu potencial completo em rotinas do cotidiano da pesquisa. No entanto, desde que uma única suspensão de células viáveis podem ser gerados (e protocolos têm sido desenvolvidos e optimizados para esse fim 7), citometria de fluxo e de células activadas por fluorescência (FACS) pode ser considerado um elemento valioso do repertório analítico em neurobiologia 8-11.

. Figura 1. Princípio da análise de citometria de fluxo e componentes de um citômetro de fluxo Fluxo cytometers compreendem três sistemas principais: fluídicos, óptica e eletrônica. Um fluxo simplificado de células em suspensão (preparada a partir de tecido primário ou em cultura in vitro) é realizada pela bainha Fluid através de focagem hidrodinâmica, a limitação da amostra para o seu núcleo central. As peças ópticas são compostos de lasers que iluminam o fluxo de células e os filtros ópticos que encaminham o sinal para os detectores apropriados. Os sinais de luz detectados são convertidos em sinais eletrônicos, posteriormente processadas por um computador e visualizadas em um monitor para análise de dados e gating. Por favor, clique aqui para ver uma versão maior desta figura.
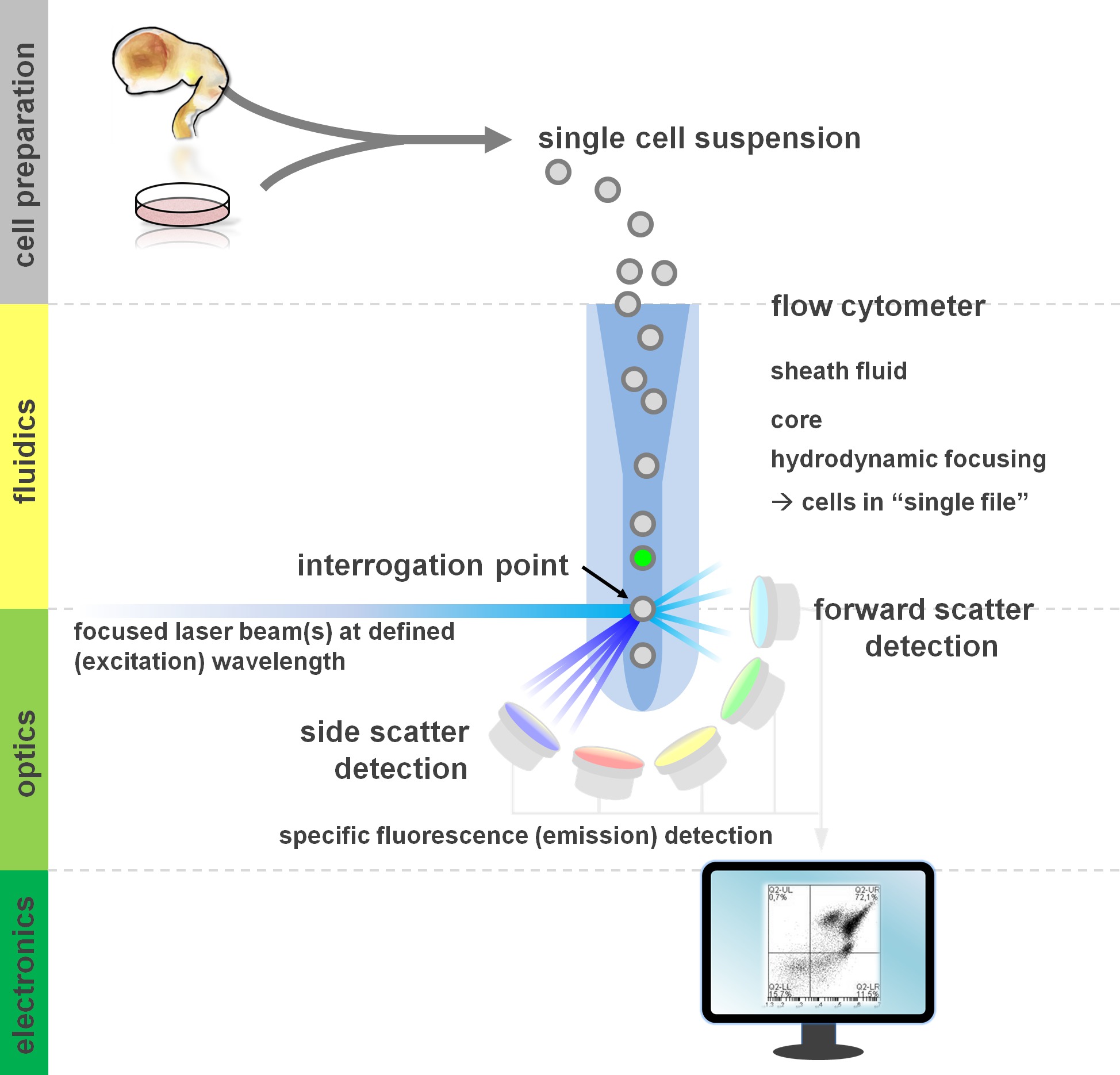{kind=link}
Usuários de métodos de citometria de fluxo de lucro de pelo menos um conhecimento básico dos fundamentos subjacentes, incluindo blocos de construção de um citómetro (para revisão ver 12,13; também veja a Figura 1). Um feixe de raios laser intersecta com um fluxo de fluidos hidrodinamicamente focado que contém as células em suspensão, que por sua vez, passam através do feixe de laser em um ficheiro único "um após o outro. O Interceptino de uma célula (ou qualquer outra partícula, para esse efeito) com os resultados de laser na dispersão de luz a partir deste ponto de interrogação. Luz dispersada pode ser detectada na continuação da direcção de laser (dispersão frontal, associada ao tamanho da partícula), bem como perpendiculares à sua direcção (dispersão lateral; reflectindo a granulosidade da partícula / célula). Estas propriedades de dispersão acima mencionados não exigem rotulagem específica, razão pela qual uma amostra não marcado (ou também os restos celulares, bolhas de ar, etc.) gerará um sinal (evento) na dispersão para a frente bivariada e lado dispersão comumente usado para gating inicial. Ao utilizar os lasers e filtros específicos para os espectros de excitação e de emissão correspondente adequadas, uma célula pode ser analisada quanto a sua positividade, o nível de intensidade, ou na ausência de marcadores fluorescentes. A maioria das aplicações de citometria de fluxo têm-se centrado na caracterização através de antigénios da superfície celular. Ao contrário do lineag hematopoiéticase, a linhagem neural permaneceu menos extensivamente definido de acordo com a superfície de expressão epitopo padrões 5. Uma vantagem de explorar os antigénios de superfície é que as células vivas podem ser submetidas a triagem celular paradigmas tais como FACS. Em contraste, a coloração do antigénio intracelular requer fixação e permeabilização passos para mediar a interacção epitopo-anticorpo, o que impede a jusante aplicações que exigem células viáveis. Digno de nota, tais abordagens ainda permitem inúmeros ensaios quantitativos 14, bem como análises a jusante para RNA e proteína expressão 15. Hematologia, imunologia e oncologia, muitas vezes utilizado mais de uma dúzia de marcadores em conjunto para definir determinadas subpopulações 16. Além disso, citometria de massa ou CyTOF pode agora ser utilizado para analisar se a 30 parâmetros simultaneamente 17,18.
Para aplicações de células-tronco neurais, bem como culturas primárias 14,19,20 A heterogeneidade das célulasvitro é um fenômeno comum 21-23. As células não representam a população alvo de interesse encarnar um fator potencialmente confusão para leitura experimental 24,25. Convenientemente, os diferentes tipos celulares presentes dentro de uma suspensão celular heterogénea suportar perfis de expressão de antigénio distintas (conhecidas ou ainda a ser decifrados), que podem ser utilizados para definir estes diferentes populações. A citometria de fluxo pode, portanto, desempenhar um papel crucial na resolução de heterogeneidade celular e, assim, facilitar aplicações biomédicas (ensaios in vitro, de terapia celular) e otimizar a leitura quantitativa, concentrando-se no subconjunto mais relevantes 24,26. Várias combinações de antigénios de superfície foram identificados nos últimos anos para permitir o isolamento e quantificação de tipos de células neuronais específicas. Isto inclui CD133 para o enriquecimento de células estaminais neurais 27, uma combinação dos / CD24 / CD29 antigénios de superfície CD15 para o isolamento da NSC, Differentiated neurônio e células da crista neural 28 ou CD15 / CD24 / CD44 / CD184 / CD271 para isolar neural e subconjuntos gliais 25, entre outras assinaturas 29,30. Além neurônios, marcadores gliais incluem A2B5 31, CD44 25, NG2 32 e GLAST 33. Uma publicação recente tem explorado o mesencéfalo marcador precursor floorplate CORIN 34,35 para enriquecer precursores dopaminérgicos no transplante de células Parkinson paradigmas 36. Moléculas de CD não são apenas marcadores, mas mediadores funcionalmente relevantes de interações célula-célula e da capacidade de uma célula para responder a estímulos a partir de moléculas de matriz extracelular e fatores de crescimento 37. Uma estratégia de reforçar ainda mais o arsenal de antigénios CD combinatórias para caracterizar desenvolvimento da linhagem neural é a utilização de marcadores intracelulares conhecidos para rastrear e definir as combinações de antigénios CD para um tipo particular de célula de interesse. Nós exploramos recentemente essa abordagem e identificaram CD49F - / CD200 padrões de expressão combinatórias altas como uma nova abordagem para o enriquecimento de subconjuntos de neurônios de pluripotentes induzidas sistemas de cultura de células-tronco diferenciadas neurally 38. Aqui, nós incluímos e discutir o último protocolo (e variações opcionais dos mesmos), em que a coloração da superfície e coloração intracelular pode ser utilizado simultaneamente para definir sub-populações de células neurais, por citometria de fluxo.

Figura 2. Diagrama do sistema de opções de protocolos experimentais. A figura ilustra uma representação esquemática dos principais passos envolvidos no protocolo. Passos opcionais (corante CFSE ou rotulagem antígeno intracelular) são indicados por caixas de cinza claro. Após a colheita, é essencial avaliar o número e a viabilidade de células de suspensões de células neuronais antes da coloração da superfície celular. Positivo comobem como os controlos negativos devem ser incluídos em adição às amostras de interesse. As amostras podem ser analisadas por análise de citometria de fluxo e / ou utilizadas em paradigmas de separação de células. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Embora se tenha usado previamente anticorpos primários em combinação com anticorpo secundário por 38 coloração intracelular, que agora introduzir a rotulagem não-covalente do anticorpo primário através de fragmentos Fab fluorescentes (rotulagem Zenon) como uma ligeira variação, reduzindo, assim, os passos de manipulação de células 39. Além disso, como mais um exemplo da versatilidade do protocolo, que empregam uma etiquetagem opcional de um subconjunto experimental por carboxifluoresceína éster de succinimidilo (CFSE) antes da coloração de antigénio de superfície. Essa pré-rotulagem CFSE permite a comparação direta imediata de duas linhas celulares ou condições experimentais (CFSE marcado vs. sem rótulo) dentro de um único tubo de amostra, reduzindo variação ou diferenças sutis no tempo de incubação e de poupança de anticorpos. CFSE é um corante fluorescente estabelecido que é comumente usado para rastreamento de células 40, na proliferação 41,42 e barcoding experiências 43,44. Finalmente, embora passos reais (FACS, de separação de células imunomagn�ica ou immunopanning) não fazem parte deste protocolo, em princípio, os procedimentos de colheita e de rotulagem descritos aqui fazer amostras de rendimento que podem ser submetidos a superfície antígeno ou aplicações de classificação baseada em rotulagem intracelulares 15 25,28.
Com este artigo, pretendemos: resumir um protocolo de coloração antígeno de superfície viável 25,28, resumir um protocolo para detecção de alvos intracelulares, bem como superfície combinada e análise do antígeno intracelular 38, apresentar uma CFSE marcação com corante passo intracelular 41,45 como um opção experimental para comanalisa comparativo de populações de células neurais, e resumir abordagens para análise de citometria de fluxo (controles apropriados 13,46, gating estratégia e dados apresentação 47).
Protocolo
1. Neural colheita celular
- Avaliação por microscópio:
- Antes de iniciar um experimento, verificar o estado da cultura com brilhante-campo ou microscopia de contraste de fase.
NOTA: Enquanto o tecido neural primário obtido a partir de dissecações é, em princípio, igualmente passível de análise por citometria de fluxo 14,28, por favor, note que o foco do protocolo está em células obtidas de sistemas in vitro de células neurais.
- Antes de iniciar um experimento, verificar o estado da cultura com brilhante-campo ou microscopia de contraste de fase.
- Colheita de células 7:
- Lavar suavemente o prato / frasco de células aderentes com Mg2 + / Ca2 + livre salina tamponada com fosfato (PBS) à temperatura ambiente (por exemplo, 5 ml para o frasco T75, 3 ml por poço de placa de 6 poços, ou 10 ml de prato de 10 cm).
NOTA: O PBS usado durante todo o protocolo é Mg2 + / Ca2 + livre.- Para o exemplo de vídeo, utilizar um frasco T75 de células de neuroblastoma SH-SY5Y em 80% de confluência. Aplicar etapas de lavagem suplementar nos casos emdetritos considerável está presente no prato.
- Considere 48 lavagens com PBS, contendo albumina de Percoll, ou de centrifugação de Ficoll 49 gradientes de soro e / ou grânulos comercialmente disponíveis 50,51. A remoção da mielina e outros lípidos ou de outros contaminantes é crítica, especialmente quando as fontes de tecidos adultos primários estão a ser utilizados.
- Adicionar pré-aquecido (37 ° C) substituição de tripsina a um volume apropriado que cobre toda a superfície do recipiente de cultura de tecidos.
NOTA: Como alternativa, considere Accutase ou outras opções de digestão enzimática. Este passo crítico pode afetar negativamente a expressão epitopo superfície (ver 7). - Incubar o prato / frasco a 37 ° C durante 2-5 minutos (dependendo do tipo de célula) para permitir que as células a separar. Bata levemente o vaso de cultura de tecidos ou flush com uma pipeta sorológica para desalojar as células. Evitar o excesso de digestão (pois isso pode causar a perda de células e coagulação em etapas posteriores).
- Quench a substituição da tripsina por adição de duas vezes o volume de tampão de fluxo (2% de FBS em PBS) e recolher as células num tubo de 15 ml.
- Triturar suavemente a suspensão de células utilizando uma pipeta de microlitro (100 - 1000 uL) ou uma pipeta de 5 ml serológico para preparar uma suspensão de célula única.
- Centrifuga-se as células a 220 xg durante 5 min a 25 ° C. Aspirar cuidadosamente o sobrenadante deixando um pelete de trás.
- Re-suspender o sedimento num volume apropriado de tampão de fluxo, dependendo do tamanho da pastilha (por exemplo, para um frasco T75 confluente de células SH-SY5Y o rendimento típico é de pelo menos 10 x 10 6 células, caso em que as células são ressuspensas em 5 ml de tampão de fluxo).
NOTA: Se forem observados pedaços maiores ou coagulação, filtrar através de um 30 - malha 100 mm.
- Lavar suavemente o prato / frasco de células aderentes com Mg2 + / Ca2 + livre salina tamponada com fosfato (PBS) à temperatura ambiente (por exemplo, 5 ml para o frasco T75, 3 ml por poço de placa de 6 poços, ou 10 ml de prato de 10 cm).
- A contagem das células 52:
- Transferir uma pequena alíquota da suspensão de células para um tubo de microcentrífuga e diluir em uma proporção definida em um volume de trypan azul ou um corante de viabilidade alternativa antes da transferência para um hemocitômetro ou sistema de contagem de células automatizado.
- Dilui-se a suspensão de células para uma concentração de 1 x 10 6 células viáveis / ml através da adição do volume apropriado de tampão de fluxo ou PBS com 0,1% de BSA (se prosseguir com a rotulagem CFSE).
NOTA: iodeto de propídio, D-7 aminoactinomycin, anexina V e comercialmente disponíveis kits de ensaio viabilidade solucionáveis representam opções alternativas, a fim de avaliar a viabilidade das células. Além disso, ensaios de apoptose caspase-3 utilizando a fluorescência como anteriormente descrito 53 pode ser usado. Canais fluorescentes serão "ocupado" por estes reagentes que podem limitar as opções para passos subsequentes, se incluído em todas as amostras.
2. Corante intracelular Etiquetagem Usando CFSE (Figura 3)
- Dilui-se a CFSE para uma concentração do material desejado que pode ser facilmente utilizado.
NOTA: Para estas experiências a concentração de ações de0,01 mM foi usada. Determinar a concentração de trabalho ideal de CFSE empiricamente. - Adicionam-se 10 ul de uma solução 0,01 mM de CFSE por ml de células (Secção 1.3.2, concentração de 1 x 10 6 células / ml em PBS + BSA a 0,1%) para uma concentração final de 0,1 uM. Resumidamente vortex para misturar bem.
NOTA: As concentrações de CFSE usados aqui são cerca de dez vezes menor do que os normalmente aplicados em ensaios de proliferação, por conseguinte, a toxicidade celular do corante é mínima. Não observamos efeitos negativos sobre a viabilidade celular. - Incubar durante 5 min à temperatura ambiente com agitação constante (200 rpm). Proteger da luz.
- Extingue-se o corante por adição de 5 volumes de tampão de fluxo para os tubos. Centrifuga-se a 94 x g durante 5 min à TA.
- Descartar o sobrenadante deixando um pelete de trás. Re-suspender as células com 5 volumes de tampão de fluxo.
- Centrifuga-se a 94 x g durante 5 min à TA. Descartar o sobrenadante e ressuspender as células em tampão de fluxo a uma concentração de 1 x 10 6 células / ml.
- Adicionar um número igual de células não coradas de interesse para a suspensão de células coradas. Proceder à tona protocolo de coloração antígeno (Seção 3).

Figura 3. Detecção de expressão diferencial entre o antigénio de superfície CD duas linhas de células por células SH-SY5Y neuroblastoma CFSE corante rotulagem. São pré-marcado com CFSE, para posterior identificação em comparação com fibroblastos BJ não marcados. Co-coloração da amostra mista (painéis à direita) com marcadores de superfície CD24 ou CD54 (ambos conjugados com APC) demonstra que as linhas celulares são facilmente distinguíveis devido à coloração CFSE (Setas = SH-SY5Y; setas = fibroblastos BJ). A maioria das células SH-SY5Y expressam CD24, mas não CD54 (ICAM-1). Em contraste, os fibroblastos BJ (CFSE-negativo) são positivas para CD54, mas amplamente negativo para CD24.Target "https://www.jove.com/files/ftp_upload/52241/52241fig3highres.jpg" = "_ blank"> Clique aqui para ver uma versão maior desta figura.
3. celular coloração da superfície
- Etiqueta de tubos, incluindo amostras e controlos críticos (ver Tabela 1).
| Tubo não. | Nome da amostra | Antigen-fluorophore | Diluição |
| 1 | Células não coradas | - | |
| 2 | Manchado Individual | CD24-APC | 01:50 |
| 3 | Manchado Individual | TUJ1-Alexa 488nm fluor | 1: 2000 |
| 4 | Duplo manchado | CD24-APC | 01:50 |
| TUJ1-Alexa 488nm fluor | 1: 2000 | ||
| 5 | Manchado Individual | Secundária só: Alexa 488nm fluor | 1: 2000 |
Tabela 1. L ist de tubos para ser incluído em um fluxo típico citometria experimento. A tabela mostra um conjunto mínimo de tubos de amostra necessários para uma experiência de co-coloração descrito neste artigo de vídeo. Um experimento ideal deve incluir todos os controles necessários (e positivas, bem como controles negativos de compensação) para a interpretação precisa dos resultados obtidos.
- Adicionar 100 ul de suspensão de células (a partir da Secção 2.7 ou 1.3.2) a cada tubo de microcentrífuga de 1,5 ml.
NOTA: Certifique-se de um mínimo de 0,1 x 10 6 células estão presentes por 100 mL de suspensão de células. - Adicionar fluoróforo anticorpo conjugado com a amostra a uma diluição adequada.
NOTA: Determinar a diluição de trabalho de cada um dos anticorpos, antes da experiência. Ver Tabela 2 para uma lista of antigénios de superfície neurais.
| Antígeno | Tipo celular | Referência |
| CD15 | As células estaminais neurais | [28, 67] |
| CD24 | Células neuronais | [28, 68] |
| CD29 | As células estaminais neurais | [28, 69, 70] |
| CD44 | As células da glia | [25] |
| CD49f | As células estaminais neurais | [38] |
| CD56 (NCAM) | Células neuronais | [71] |
| CD133 | As células estaminais neurais | [27] |
| CD184 | Células-tronco neurais e células gliais | [25] |
| CD200 | Células neuronais | [38] |
| CD271 | Células-tronco da crista neural | [25] |
| A2B5 | As células da glia | [31] |
| CORIN | Precursores dopaminérgicos | [35, 36] |
| FORSE1 | Células-tronco neurais (NSC) | [72] |
| GLAST | As células da glia | [33] |
| NG2 | As células da glia | [32] |
Tabela 2. Selecção de antigénios de superfície neurais. Esta tabela fornece uma lista de epítopos de superfície que se verificou ser expresso por diversos tipos de células neurais para exemplificar o aumento painel de antigénios de superfície utilizados para caracterizar a linhagem neural. Note-se que esta selecção está longe de ser completa e que a maioria destes marcadores são também expressos por uma variedade de outras células neurais e não neurais. Por conseguinte, as combinações de vários marcadores será necessária para melhor definir e isolar os subconjuntos neurais indicados.
- Incubar durante 30 min num agitador orbital (200 rpm) no escuro.
- Tampão de lavagem de fluxo
- Adicionar 1 ml de tampão de fluxo para os tubos. Centrifugar a 380 g durante 4 min a 4 ° C.
- Descartar o sobrenadante deixando um pelete de trás.
- Repita o passo de lavagem de novo.
- Após a segunda lavagem, decantar o sobrenadante e ressuspender as células em tampão de fluxo para um volume final de 100 ul.
- Use amostra para análise de citometria de fluxo. Como alternativa, proceder com a Seção 4 e 5.
NOTA: Se as células estão a ser classificado e colocado de volta na cultura pós-FACS (ou seja, a suspensão de células viáveis sem fixação ou permeabilização), aplicar técnicas de assepsia durante a colheita, a coloração e as etapas de análise.
4. Fixação e permeabilização 38
- Fixação com paraformaldeído (PFA):
- Preparar tampão de fixação contendo 2% de PFA em PBS.
NOTA: PFA é prejudicial aos seres humanos e ao meio ambiente. Usar equipamento de protecção individual adequado e descarte de resíduos, de acordo com os regulamentos locais. - Adicionar 500 ul de tampão de fixação de 100 ul de suspensão de células.
- Incubar os tubos à temperatura ambiente durante 15 min num agitador orbital (100 rpm) no escuro.
- Preparar tampão de fixação contendo 2% de PFA em PBS.
- PBS lavagem:
- Adicionar 1 ml de PBS ao tubo. Centrifuga-se a 380 xg durante 3 min a 4 ° C.
- Descartar / decanta-se o sobrenadante deixando aproximadamente 100 ul do tubo.
- Permeabilização com Tween-20:
- Preparar tampão de permeabilização que contém 0,7% de Tween-20 em PBS.
- Adicionar 500 ul de tampão de permeabilização de 100 ul de suspensão de células.
- Incubar os tubos à temperatura ambiente durante 15 min num agitador orbital (100 rpm) no escuro.
- Lavar as células uma vez com PBS (como descrito na Secção 4.2) e remover o sobrenadante do co tubosmpletely, deixando apenas o sedimento atrás.
5. Coloração Intracelular Antigen 38 (Figura 4)
- Preparação das soluções de anticorpos primários:
- Dilui-se os anticorpos primários em tampão de diluição contendo 1% de albumina de soro bovino, 10% de soro (por exemplo, burro ou soro de cabra normal) e 0,5% de Tween-20 em PBS.
NOTA: Escolha o soro a ser utilizado, dependendo da espécie os anticorpos secundários foram levantados no. - Alternativamente, usar Zenon rotulagem de fluoresceína o anticorpo primário de acordo com as instruções do fabricante.
- Prepare 1 jig de anticorpo primário em PBS a uma diluição adequada (volume total ≤ 20 ul).
- Adiciona-se 5 ul de reagente de marcação Zenon fluoresceína IgG (Componente A) à solução de anticorpo.
- Incubar a mistura durante 5 minutos à temperatura ambiente.
- Adiciona-se 5 ul de reagente de bloqueio Zenon (Componente B) à mistura de reacção.
- Incubara mistura durante 5 minutos à temperatura ambiente. Aplicar o anticorpo com a amostra em 30 min.
- Dilui-se os anticorpos primários em tampão de diluição contendo 1% de albumina de soro bovino, 10% de soro (por exemplo, burro ou soro de cabra normal) e 0,5% de Tween-20 em PBS.
- Coloração de anticorpo primário:
- Adicionar 100 ul da solução de anticorpo primário para a pelete de células e tritura-se suavemente para misturar.
- Alternativamente, adicionar Zenon anticorpo marcado com fluoresceína para a suspensão de células na diluição apropriada.
- Incubar os tubos à temperatura ambiente durante 30 min num agitador orbital (200 rpm), protegida da luz. Lavar as células uma vez com PBS (como descreve na Secção 4.2) e remover o sobrenadante dos tubos completamente, deixando apenas o sedimento atrás.
- Coloração do anticorpo secundário (não necessária para os anticorpos Zenon fluoresceína marcados):
- Dilui-se os anticorpos secundários em PBS a uma concentração apropriada.
- Adicionar 100 ul da solução de anticorpo secundário para a pelete de células e tritura-se suavemente para misturar. Incubar os tubos à temperatura ambiente durante 30 min num agitador (200 rpm) no escuro.
- Lavam-se as amostras duas vezes com PBS (como descrito na Secção 4.2).
- Lavar uma vez com tampão de fluxo (ver secção 3.5).
- Re-suspender as células em cerca de 150 ul de tampão de fluxo e analisar no citómetro de fluxo.

Figura 4. Co-coloração de superfície e proteínas intracelulares. A citometria de fluxo de dados exemplifica uma comparação entre baseada em anticorpo primário + secundário contra a coloração intracelular com base em fluoresceína Zenon em combinação com coloração de superfície. Positividade exclusiva no eixo y (quadrante superior esquerdo) e o eixo x (quadrante inferior direito) mostra células coradas para MAP2 e CD24, respectivamente. Após co-coloração, MAP2 partilhada e expressão de CD24 pode ser visto no quadrante superior direito (painéis da direita). Comparação do usoAlexa Fluor 488. (Top; AF488) versus Zenon fluoresceína (em baixo) para os rendimentos de rotulagem MAP2 resultados semelhantes por favor, clique aqui para ver uma versão maior desta figura.
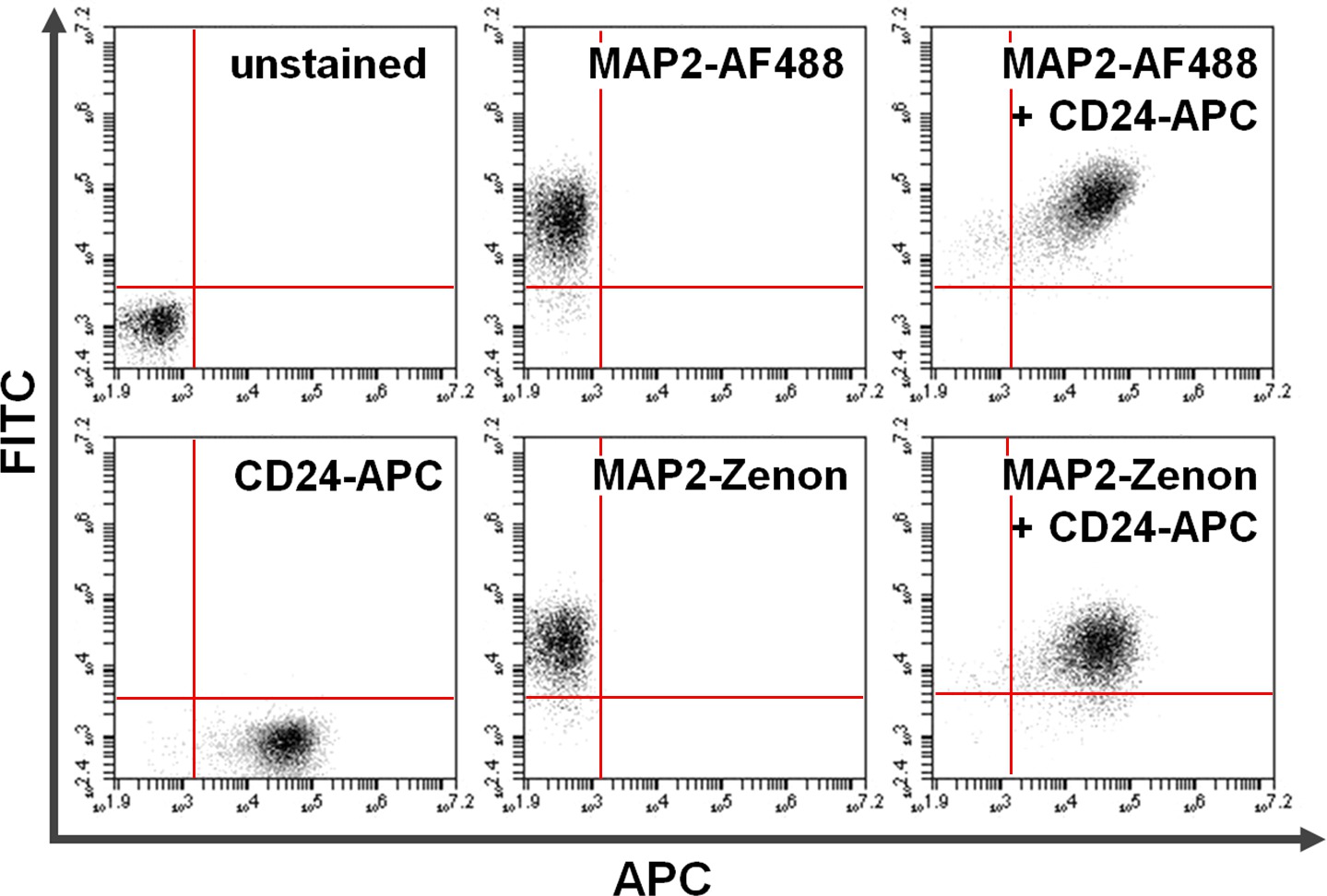{kind=link}
6. Fluxo de análise de citometria
- Realizar uma análise de citometria de fluxo, imediatamente após a conclusão do protocolo de coloração utilizando um citómetro de fluxo com filtros adequados para a detecção de sinal. Use 488 nm laser vermelho nm azul e 640 com FL-1 (533/30), FL-2 (585/40), e filtros FL-4 (675/25) de banda.
- Configure portas primárias com base na dispersão frontal e lateral excluindo detritos e células mortas.
- Definir portões fluorescência para superfície e antígeno intracelular para ≤0.5% com base nas amostras não coradas e indemnizações por sobreposição espectral usando controles manchadas de solteiro.
Resultados
O protocolo aqui apresentado permite abordagens para versátil experimentais (Figura 2). Na sua versão mais curta (passos 1, 3 e 6), ele pode ser considerado como um guia para a coloração simples de antigénios de superfície. Na sua forma mais complexa, um número de paradigmas de co-marcação com uma gama de antigénios intracelulares pode ser prosseguido passos opcionais (2 e / ou 4 a 5). Além d...
Discussão
O protocolo aqui apresentado é bem estabelecido para as culturas de células neurais derivadas de células estaminais humanas, mas pode ser igualmente aplicado a outras fontes de células neurais, incluindo tecido primário ou linhas de células neuronais. Para além das fontes embrionárias, células estaminais ou progenitoras neurais pode ser extraído a partir das regiões do cérebro adulto neurogénicos 27. Além disso, a citometria de fluxo e FACS pode ser explorada para quantificar, analisar e isolar ...
Divulgações
The authors declare no potential conflicts of interest.
Agradecimentos
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
Materiais
| Name | Company | Catalog Number | Comments |
| DMEM/F12 (1:1) (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) |  Life Technologies Life Technologies | 11330057 | |
| DPBS without Ca2+ Mg2+ | Life Technologies | 14190169 | |
| Fetal bovine serum, qualified, E.U.-approved, South America origin (FBS) | Life Technologies | 10270-106 | |
| MEM Non-essential amino acids (100x) | Life Technologies | 11140035 | |
| TrypLE Express | Life Technologies | 12604013 | |
| Trypan blue solution, 0.4% | Life Technologies | 15250061 | |
| Paraformaldehyde | Carl Roth | 335.3 | |
| Bovine serum albumin (BSA) Fraction V | PAA Laboratories, Coelbe | K41-001 | |
| Tween-20 Detergent | Calbiochem | 655205 | |
| Carboxyfluorescein succinimidyl ester (CFSE) | eBioscience | 65-0850-84 | |
| DMSO | AppliChem | A1584 | |
| Bottle top filters express plus 0.22 µm, 250 ml | Millipore | SCGPU02RE | |
| Cell culture treated flasks (T 25) | NUNC | 156367 | |
| Cell culture treated flasks (T 75) | NUNC | 156499 | |
| Conical tubes (15 ml) | Greiner Bio-One | 188271 | |
| Conical tubes (50 ml) | Greiner Bio-One | 227261 | |
| Pasteur pipet, glass (150 mm) | STEIN Labortechnik, Remchingen | S03710150 | |
| Pipet tips (0.1-10 µl) | Corning | 4125 | |
| Pipet tips (1-200 µl) | Corning | 4126 | |
| Pipet tips (100-1000 µl) | Corning | 4129 | |
| Serological pipets, 5 ml | Corning | 4051 | |
| Serological pipets, 10 ml | Corning | 4101 | |
| Serological pipets, 25 ml | Corning | 4251 | |
| Microcentrifuge tubes (0.5 ml) | Sarstedt | 72,699 | |
| Microcentrifuge tubes (1.5 ml) | Greiner Bio-One | 616201 | |
| Microcentrifuge tubes (2.0 ml) | Sarstedt | 72,695,500 | |
| Anti-Human CD24 APC monoclonal antibody | eBioscience | 17-0247-42 | Working dilution 1:50 |
| Anti-Human CD54 PE monoclonal antibody | eBioscience | 12-0549-42 | Working dilution 1:50 |
| Neuronal Class III β-Tubulin (Tuj1) polyclonal antibody | Covance | PRB-435P | Working dilution 1:2,000 |
| Alexa Fluor 488 Donkey anti Rabbit | Life Technologies | A21206 | Working dilution 1:2,000 |
| Zenon® Fluorescein Rabbit IgG Labeling Kit | Life Technologies | Z-25342 | |
| Neubauer-Improved counting chamber | Marienfeld | 0640010 | |
| Vortex | Scientific Industries | G560E | |
| Thermomixer comfort | Eppendorf | 5355 000.001 | |
| Accuri C6 flow cytometer | Becton Dickinson (BD) | 653118 | |
| Microcentrifuge refrigerated, PerfectSpin 24 R | Peqlab | 91-PSPIN-24R | |
| Orbital shaker, Unimax 1010 | Heidolph | 543-12310-00 | |
| Centrifuge refrigerated, Rotanta 96 RC | Hettich | 4480-50 | |
| Class II Biological safety cabinet Safe 2020 | Thermo Scientific | 51026640 | |
| CO2 Incubator, Heracell 240i | Thermo Scientific | 51026331 | |
| Vacuum system, Vacusafe comfort | Integra Biosciences | 158320 | |
| Microscope, Axiovert 40 CFL | Zeiss | 451212 | |
| Pipet controller, accu-jet pro | Brand | 26303 | |
| Micropipet, Pipetman neo P20N (2-20 µl) | Gilson | F144563 | |
| Micropipet, Pipetman neo P200N (20-200 µl) | Gilson | F144565 | |
| Micropipet, Pipetman neo P1000N (100-1000 µl) | Gilson | F144566 |
Referências
- Herzenberg, L. A., et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 48 (10), 1819-1827 (2002).
- Chattopadhyay, P. K., Roederer, M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif). 57 (3), 251-258 (2012).
- Jaye, D. L., Bray, R. A., Gebel, H. M., Harris, W. A. C., Waller, E. K. Translational applications of flow cytometry in clinical practice). J. Immunol. 188 (10), 4715-4719 (2012).
- Henel, G., Schmitz, J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 38 (7), 428-436 (2007).
- Seita, J., Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2 (6), 640-653 (2010).
- Ulrich, H., Bocsi, J. Phenotypes of stem cells from diverse origin. Cytometry. A. 77 (1), 6-10 (2010).
- Panchision, D. M., et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 25 (6), 1560-1570 (2007).
- Meyer, R. A., Zaruba, M. E., McKhann, G. M. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 2 (1), 66-74 (1980).
- Junger, H., Junger, W. G. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 9 (16), 3749-3754 (1998).
- McLaren, F. H., Svendsen, C. N., Vander Meide, P., Joly, E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 112 (1-2), 35-46 (2001).
- Wang, S., Roy, N. S., Benraiss, A., Goldman, S. A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 22 (1-2), 167-176 (2000).
- Tanke, H. J., vander Keur, M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 11 (2), 55-62 (1993).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243 (1-2), 77-97 (2000).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 129 (1), 73-79 (2003).
- Ernst, A., et al. Neurogenesis in the striatum of the adult human brain. Cell. 156 (5), 1072-1083 (2014).
- Perfetto, S. P., Chattopadhyay, P. K., Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 4 (8), 648-655 (2004).
- Bandura, D. R., et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81 (16), 6813-6822 (2009).
- Bendall, S. C., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332 (6030), 687-696 (2011).
- Neveu, I., Rémy, S., Naveilhan, P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 113 (4), 767-777 (2002).
- Pruszak, J., Just, L., Isacson, O., Nikkhah, G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2 (Unit 2D.5), (2009).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99 (22), 14506-14511 (2002).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1-2), 18-29 (2003).
- Pruszak, J., Isacson, O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 651, 112-123 (2009).
- Carson, C. T., Aigner, S., Gage, F. H. Stem cells: the good, bad and barely in control. Nat. Med. 12 (11), 1237-1238 (2006).
- Yuan, S. H., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 6 (3), e17540 (2011).
- Roy, N. S., Cleren, C., Singh, S. K., Yang, L., Beal, M. F., Goldman, S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 12 (11), 1259-1268 (2006).
- Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 97 (26), 14720-14725 (2000).
- Pruszak, J., Ludwig, W., Blak, A., Alavian, K., Isacson, O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 27 (12), 2928-2940 (2009).
- Peh, G. S. -. L., Lang, R. J., Pera, M. F., Hawes, S. M. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 18 (2), 269-282 (2009).
- Golebiewska, A., Atkinson, S. P., Lako, M., Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 27 (6), 1298-1308 (2009).
- Dietrich, J., Noble, M., Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 40 (1), 65-77 (2002).
- Nishiyama, A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 14 (1), 77-82 (2001).
- Jungblut, M., et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 60 (6), 894-907 (2012).
- Ono, Y., et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 134 (17), 3213-3225 (2007).
- Chung, S., et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 108 (23), 9703-9708 (2011).
- Doi, D., et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2 (3), 337-350 (2014).
- Solozobova, V., Wyvekens, N., Pruszak, J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 8 (3), (2012).
- Turaç, G., et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 8 (6), e68519 (2013).
- Buchwalow, I. B., Böcker, W. Chapter 2. Immunohistochemistry: Basics and Methods. , 9-17 (2010).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. (70), e4287 (2012).
- Lyons, A. B., Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171 (1), 131-137 (1994).
- Hawkins, E. D., et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2 (9), 2057-2067 (2007).
- Quah, B. J. C., Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 44, (2010).
- Sukhdeo, K., et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 8 (1), e53015 (2013).
- Jiang, L., et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 140, 90-99 (2014).
- Hulspas, R., et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 76 (6), 355-364 (2009).
- Moloney, M., Shreffler, W. G. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 101 (5), 544-549 (2008).
- Siebzehnrubl, F. A., et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 750, 61-77 (2011).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain). J. Neurosci. Methods. 203 (1), 10-18 (2012).
- Tham, C. -. S., Lin, F. -. F., Rao, T. S., Yu, N., Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 21 (8), 431-443 (2003).
- Nguyen, H. X., Beck, K. D., Anderson, A. J. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. (50), e2698 (2011).
- Marchenko, S., Flanagan, L. Counting human neural stem cells. J. Vis. Exp. (7), 262 (2007).
- Brunlid, G., Pruszak, J., Holmes, B., Isacson, O., Sonntag, K. C. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 25 (10), 2551-2558 (2007).
- Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 71 (2), 143-155 (1997).
- Cardona, A. E., Huang, D., Sasse, M. E., Ransohoff, R. M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1 (4), 1947-1951 (2006).
- Nielsen, J. A., Maric, D., Lau, P., Barker, J. L., Hudson, L. D. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 26 (39), 9881-9891 (2006).
- Daneman, R., et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 5 (10), e13741 (2010).
- Gräbner, R., Till, U., Heller, R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 40 (3), 238-244 (2000).
- Quah, B. J. C., Parish, C. R. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 379 (1-2), 1-14 (2012).
- Lathia, J. D., et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6 (1), 117-129 (2014).
- Ganat, Y. M., et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 122 (8), 2928-2939 (2012).
- Hedlund, E., et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 26 (6), 1526-1536 (2008).
- Maroof, A. M., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12 (5), 559-572 (2013).
- Chivet, M., Hemming, F., Pernet-Gallay, K., Fraboulet, S., Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145 (2012).
- Graner, M. W., et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23 (5), 1541-1557 (2009).
- Eldh, M., Lötvall, J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. (59), e3037 (2012).
- Capela, A., Temple, S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291 (2), 300-313 (2006).
- Nieoullon, V., Belvindrah, R., Rougon, G., Chazal, G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 28 (3), 462-474 (2005).
- Nagato, M., et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 80 (4), 456-466 (2005).
- Hall, P. E., Lathia, J. D., Miller, N. G. A., Caldwell, M. A., French-Constant, C. Integrins are markers of human neural stem cells. Stem Cells. 24 (9), 2078-2084 (2006).
- Hargus, G., et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107 (36), 15921-15926 (2010).
- Elkabetz, Y., et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152-165 (2008).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados