
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Flow Cytometry Protokolle für Oberflächen- und intrazelluläre Antigen Analysen Neuronale Zelltypen
In diesem Artikel
Zusammenfassung
We provide a detailed description of a protocol for flow cytometric analysis of surface antigens and/or intracellular antigens in neural cell types. Critical aspects of experimental planning, step-by-step methodological procedures, and fundamental principles of flow cytometry are explained in order to enable neurobiologists to exploit this powerful technology.
Zusammenfassung
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Einleitung
Durchflusszytometrie wurde ausgiebig in der Immunologie, Hämatologie und Onkologie ausgenutzt, um Zellpopulationen über Eigenstreueigenschaften, Zelloberflächen-Antigen-Expression und andere Fluoreszenzparameter 1-3 definieren. Unsere Erkenntnisse über Blutslinie Entwicklung und Krankheit sind das Ergebnis in erheblichem Maße von der kontinuierlichen Verfeinerung dieser Methode nach ihrer erstmaligen Anwendung 4,5. Bewusstsein für die quantitative und die allgemeine analytische Potenzial der Durchflusszytometrie wurde vor kurzem aufgefordert seine weitere Verbreitung in der Stammzellforschung und können ähnlich tiefgreifende Fortschritte in einem kürzeren Zeitrahmen 6 zu ermöglichen. Allerdings ist die Anwendung der Durchflusszytometrie gezielt zu analysieren und zu isolieren, neuronale Populationen solange anspruchs wahrgenommen. Im Gegensatz zu den blutbildenden Zellen, die natürlicherweise in Suspension vorhanden sind, werden neuronalen Zelltypen in der Regel durch zu komplexe Quellen, die Glia und verschiedene o umfassen können geerntetther umgebenden Zellen sowie ein dichtes Netz von Prozesstragenden Neuronen. Folglich hat noch Neurobiologie, um die Vielseitigkeit der Durchflusszytometrie auf seine vollständige Potential im täglichen Forschungsroutinen implementieren. Jedoch solange eine tragfähige Einzelzellsuspension erzeugt werden können (und Protokollen entwickelt worden und zu diesem Zweck optimiert 7), Durchflusszytometrie und Fluoreszenz-aktivierte Zellsortierung (FACS) kann als wertvoller Bestandteil des analytischen Repertoire der Neurobiologie werden 8-11.

. Abbildung 1. Prinzip der Durchflusszytometrie und Komponenten von einem Durchflusszytometer Durchflusszytometer umfassen drei Hauptsysteme: Fluidtechnik, Optik und Elektronik. Eine stromlinienförmige Strömung der Zellen in Suspension wird durch die Hülle flui erreicht (von primärem Gewebe oder in vitro-Kultur hergestellt)d mittels hydrodynamischer Fokussierung, die Einschränkung der Probe zu seiner Mittelkern. Die optischen Teile sind von Lasern, die den Strom von Zellen und optische Filter, die das Signal an den entsprechenden Detektoren lenken beleuchten zusammensetzt. Die festgestellt werden, um elektronische Signale, anschließend von einem Computer verarbeitet und auf einem Monitor für die Datenanalyse und Gating visualisiert Lichtsignale umgewandelt. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
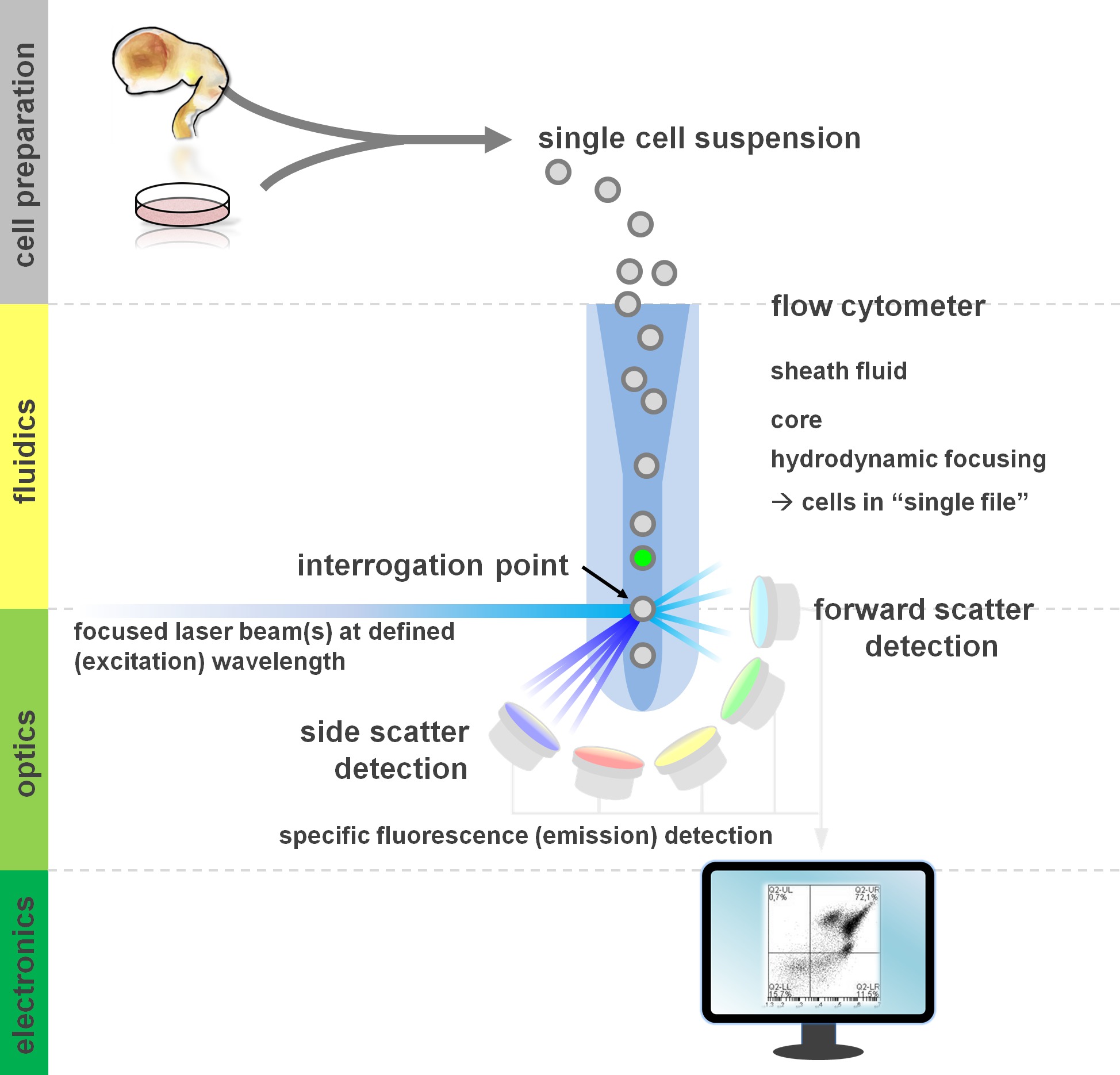{kind=link}
Benutzer von durchflusszytometrischen Methoden profitieren von mindestens einem grundlegenden Verständnis der zugrunde liegenden Fundamentaldaten einschließlich Bausteine eines Zytometer (für einen Überblick siehe 12,13; siehe auch Abbildung 1). Ein Laserstrahl schneidet mit einem hydrodynamisch fokussiert fluidischen Strom, der die Zellen in Suspension, die wiederum durch den Laserstrahl in 'Datei' eine nach der anderen vorbei enthält. Die interceptiauf einer Zelle (oder einer anderen Partikel, in diesem Fall) mit den Laser-Ergebnisse bei der Streuung von Licht, das von diesem Abfragepunkt. Streulicht kann in Weiterführung der Laserrichtung (Vorwärtsstreuung, wobei die Größe der Teilchen verbunden ist), als auch senkrecht zu seiner Verschieberichtung detektiert werden (side scatter; Reflektieren des granulosity der Partikel / Zelle). Diese zuvor genannten Streueigenschaften erfordern keine spezielle Kennzeichnung, weshalb ein unmarkierter Probe (oder auch Zelltrümmer, Luftblasen, etc.) ein Signal (event) auf der bivariate Vorwärtsstreuung im Vergleich zu Seitenstreudiagramm üblicherweise zur ersten Gating verwendet zu generieren. Unter Verwendung der entsprechenden Laser und die spezifisch für die entsprechende Anregungs- und Emissionsspektren Filter kann eine Zelle für seine Positivität, Intensität oder Abwesenheit von Fluoreszenzmarkern analysiert werden. Die Mehrheit der Durchflusszytometrie-Anwendungen haben sich auf die Charakterisierung über Zelloberflächenantigene gerichtet. Im Gegensatz zu den hämatopoetischen LineagE hat die neuronalen Linie nach Oberflächen Epitop Expressionsmuster 5 blieb weniger ausführlich definiert. Ein Vorteil der Nutzung von Oberflächenantigenen ist, dass lebende Zellen unterzogen, um das Sortieren Paradigmen, wie FACS-Zelle sein. Im Gegensatz dazu intrazelluläres Antigen-Färbung erfordert Fixierung und Permeabilisierung Schritte, um die Epitop-Antikörper-Wechselwirkung vermitteln, ausschließt nachgelagerten Anwendungen, die lebensfähige Zellen erfordern. Der Hinweis, derartige Ansätze noch für zahlreiche quantitative Assays 14 sowie Downstream-Analysen von RNA und Protein-Expression 15 ermöglichen. Hämatologie, Immunologie und Onkologie haben oft zusammen verwendet mehr als ein Dutzend Marker für bestimmte Subpopulationen 16 zu definieren. Darüber hinaus können Massen Zytometrie oder CyTOF jetzt zur Analyse von bis zu 30 Parameter gleichzeitig 17,18 werden.
Für neurale Stammzellen Anwendungen sowie Primärkulturen 14,19,20 die Heterogenität von Zellenvitro ist ein häufiges Phänomen 21-23. Die Zellen, die nicht die Zielgruppe von Interesse verkörpern eine potentiell verwirrenden Faktor für experimentelle Auslesen 24,25. Günstig sind die verschiedenen zellulären innerhalb einer heterogenen Zellsuspension vorliegenden Teilmengen tragen distinct (bekannt oder noch zu entschlüsseln) Antigen-Expressionsprofile, die verwendet werden können, um diese verschiedenen Populationen zu definieren. Durchflusszytometrie kann daher eine entscheidende Rolle bei der Lösung von zellulärer Heterogenität und damit zu erleichtern biomedizinische Anwendungen (in vitro-Assays, Zelltherapie) und optimieren quantitative Auslesen durch die Konzentration auf die wichtigsten Teilmenge 24,26. Verschiedene Oberflächenantigenkombinationen wurden in den letzten Jahren erkannt worden, die Quantifizierung und Isolierung von bestimmten neuronalen Zelltypen ermöglichen. Dies beinhaltet CD133 zur Anreicherung von neuralen Stammzellen 27, eine Kombination aus den CD15 / CD24 / CD29-Oberflächenantigene für die Isolierung von NSC, Differenzierungten Neurons und Neuralleistenzellen 28 oder CD15 / CD24 / CD44 / CD184 / CD271 auf neuronale und gliale Teilmengen 25, unter anderen Signaturen 29,30 isolieren. Jenseits Neuronen, Glia Marker umfassen A2B5 31, CD44 25, NG2 32 und GLAST 33. Eine aktuelle Publikation hat die Mittelhirn Bodenplatte Vorläufer Marker CORIN 34,35 für dopaminerge Vorstufen in Parkinson Zelltransplantation bereichern genutzt Paradigmen 36. CD-Moleküle sind nicht nur Marker, sondern funktionell relevante Mediatoren von Zell-Zell-Wechselwirkungen und die Fähigkeit einer Zelle, auf Hinweise von extrazellulären Matrixmolekülen reagieren und Wachstumsfaktoren 37. Eine Strategie, die weitere Verbesserung der Arsenal von kombinatorischen CD-Antigene zu charakterisieren neuronalen Linie Entwicklung ist es, bekannte intrazelluläre Marker verwenden, um für einen bestimmten Zelltyp von Interesse zu screenen und zu definieren CD Antigenkombinationen. Vor kurzem haben wir ausgenutzt solchen Ansatz und identifiziert CD49f - / CD200 hochkombiExpressionsMuster als neuer Ansatz zur Anreicherung von neuronalen Teilmengen von neural differenzierten induzierten pluripotenten Stammzellen Zellkultursystemen 38. Hier sind und die letztere Protokoll (und optionalen Varianten davon), in der Oberflächenfärbung und intrazelluläre Färbung gleichzeitig zur Definition neuronaler Zellpopulationen mittels Durchflusszytometrie verwendet werden diskutieren wir.

Abbildung 2: Flussdiagramm des Versuchsprotokoll-Optionen. Die Figur zeigt eine schematische Darstellung der wichtigsten Schritte im Protokoll beteiligt. Optionale Schritte (CFSE Farbstoff oder intrazelluläres Antigen Kennzeichnung) sind je nach hellgrau-Boxen angezeigt. Nach der Ernte wird es notwendig, um die Lebensfähigkeit und Zellzahl von neuronalen Zellsuspensionen vor der Zelloberflächenfärbung beurteilt. Positive wiesowie negative Kontrollen müssen zusätzlich zu den Proben von Interesse sein sollte. Die Proben können durch Durchflusszytometrie analysiert und / oder in Zellsortierung Paradigmen verwendet werden. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}
Während wir zuvor verwendeten primären Antikörper in Kombination mit sekundären Antikörper für die intrazelluläre Färbung 38 führen wir nun nicht kovalente Markierung des primären Antikörpers durch Fluoreszenz-Fab-Fragmente (Zenon Kennzeichnung) als geringfügige Veränderung, wodurch die Schritte der Bearbeitung der Zellen 39 verringert wird. Außerdem ist als weiteres Beispiel für das Protokoll der Vielseitigkeit, nutzen wir ein optional Kennzeichnung einer Versuchsmenge von Carboxyfluoresceinsuccinimidylester (CFSE) vor der Oberflächenantigen-Färbung. Solche CFSE vorge Kennzeichnung ermöglicht die sofortige direkten Vergleich zweier Zelllinien oder experimentellen Bedingungen (Vs CFSE-markierten. unmarkiert) innerhalb eines einzelnen Probenröhrchen, die Verringerung Varianz oder feine Unterschiede in der Inkubationszeit und Speichern Antikörper. CFSE ist ein etablierter Fluoreszenz-Farbstoff, der üblicherweise für Zellverfolgung 40 verwendet wird, bei der Proliferation 41,42 und Strichcodes Experimenten 43,44. Schließlich, während eigentliche Sortierschritte (FACS, immunomagnetischen Zelltrennung oder Immunopanning) sind nicht Teil des Protokolls im Prinzip die hier beschriebenen Ernte und Kennzeichnungsverfahren liefern zwar Proben, ausgesetzt sind, können an die Oberfläche Antigen oder die intrazelluläre Markierung basierte Sortieranwendungen werden 15 25,28.
Mit diesem Artikel wollen wir: Zusammenfassend eine tragfähige Oberflächenantigen Färbeprotokoll 25,28 fassen ein Protokoll zum Nachweis von intrazellulären Ziele sowie kombinierte Oberflächen- und intrazelluläres Antigen Analyse 38, präsentieren eine intrazelluläre Farbstoff CFSE Markierungsschritt 41,45 als experimentelle Option für comvergleichende Analysen von neuronalen Zellpopulationen, und fassen Ansätze zur zytometrische Analyse (geeignete Kontrollen 13,46, Gating-Strategie und Datendarstellung 47) fließen.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Neuronale Zellernte
- Bewertung durch Mikroskop:
- Vor Beginn eines Experiments, überprüfen Sie den Status der Kultur mit Hellfeld oder Phasenkontrastmikroskopie.
HINWEIS: Während primäre von Dissektionen erhalten Nervengewebe ist prinzipiell gleichermaßen zugänglich zytometrische Analyse 14,28 fließen, beachten Sie bitte, dass der Fokus des Protokolls ist auf Zellen, die aus in vitro neuronale Zellsystemen erhalten.
- Vor Beginn eines Experiments, überprüfen Sie den Status der Kultur mit Hellfeld oder Phasenkontrastmikroskopie.
- Ernten der Zellen 7:
- Die Schale / Kolben von adhärenten Zellen mit Mg 2+ schonend waschen / Ca 2+ freiem Phosphat-gepufferter Salzlösung (PBS) bei Raumtemperatur (beispielsweise 5 ml für T75-Kolben wurden 3 ml pro Vertiefung für 6-Well-Platte oder 10 ml für 10 cm Schale).
HINWEIS: Der PBS im gesamten Protokoll ist Mg 2+ / Ca 2+ frei.- Für das Video beispielsweise mit einem T75-Kolben von SH-SY5Y Neuroblastomzellen bei 80% Konfluenz. Übernehmen zusätzliche Waschschritte für den Fall,erhebliche Rückstände ist in der Schale vorhanden.
- Betrachten Sie Waschungen mit Serumalbumin enthaltenden PBS 48, Percoll oder Ficoll 49 Zentrifugation Steigungen und / oder handelsübliche Perlen 50,51. Entfernen von Myelin und andere Lipid oder anderen Verunreinigungen kritisch ist, insbesondere, wenn erwachsene primären Gewebequellen verwendet werden.
- Hinzufügen vorgewärmten (37 ° C) Trypsin Austausch an einem geeigneten Volumen, der die gesamte Fläche der Gewebekulturbehälter umfasst.
HINWEIS: Sie betrachten Accutase oder andere enzymatische Verdauung Optionen. Dieser entscheidende Schritt kann sich negativ auf Oberflächen Epitop Ausdruck (siehe 7). - Die Schale / Kolben bei 37 ° C für 2 inkubieren - 5 min (in Abhängigkeit vom Zelltyp), um Zellen zu trennen. Klopfen Sie leicht die Zellkulturgefäß oder bündig mit einer serologischen Pipette, um die Zellen zu entfernen. Vermeiden Sie zu der Verdauung (da dies zu Zellverlust und Gerinnung bei späteren Schritten verursachen).
- Quench das Trypsin Ersatz durch Zugabe des doppelten Volumens von Strömungspuffer (2% FBS in PBS) und sammle die Zellen in einem 15 ml konischen Röhrchen.
- Verreiben Sie vorsichtig die Zellsuspension mit Hilfe einer Mikroliterpipette (100 - 1000 ul) oder ein 5 ml serologische Pipette, um eine Einzelzellsuspension herzustellen.
- Zentrifugiere die Zellen bei 220 g für 5 min bei 25 ° C. Vorsichtig den Überstand aspirieren Verlassen des Pellet hinter.
- Re-suspendieren des Pellets in einem geeigneten Volumen von Strömungspuffer in Abhängigkeit von der Größe des Pellets (beispielsweise für einen konfluenten T75-Flasche von SH-SY5Y-Zellen die typische Ausbeute von mindestens 10 x 10 6 Zellen, wobei die Zellen werden in 5 ml-Fluß-Puffer) resuspendiert.
HINWEIS: Wenn größere Brocken oder Gerinnungseingehalten werden, durch ein 30-Filter - um Maschenweite 100.
- Die Schale / Kolben von adhärenten Zellen mit Mg 2+ schonend waschen / Ca 2+ freiem Phosphat-gepufferter Salzlösung (PBS) bei Raumtemperatur (beispielsweise 5 ml für T75-Kolben wurden 3 ml pro Vertiefung für 6-Well-Platte oder 10 ml für 10 cm Schale).
- Zellzählung 52:
- Übertragen einer kleinen Aliquot der Zellsuspension in ein Mikrozentrifugenröhrchen und verdünnt in einem festen Verhältnis in einem Volumen von trypan blau oder eine Alternative Lebensfähigkeit Farbstoff vor der Übertragung auf einem Hämozytometer oder automatisierte Zellzählung System.
- Verdünne die Zellsuspension auf eine Konzentration von 1 × 10 6 lebensfähigen Zellen / ml durch Zugabe des geeigneten Strömungsvolumen Puffer oder PBS mit 0,1% BSA (wenn mit CFSE Markierungsverfahrens).
HINWEIS: Propidiumiodid, 7-Aminoactinomycin, Annexin V und im Handel erhältliche fixierbar Lebensfähigkeit Assay-Kits stellen alternative Möglichkeiten, um die Lebensfähigkeit von Zellen zu beurteilen. Auch Apoptose-Assays unter Verwendung von Caspase-3-Fluoreszenz wie zuvor beschrieben 53 verwendet werden. Fluorescent Kanäle durch diese Reagenzien, die die Optionen für den nachfolgenden Schritten begrenzen können, wenn in allen Proben enthalten "besetzt" werden.
2. Intrazelluläre Farbstoffmarkierung Verwendung CFSE (Figur 3)
- Verdünne die CFSE zu einer gewünschten Ausgangskonzentration, die leicht verwendet werden kann.
HINWEIS: Für diese Experimente eine Stammkonzentration von0,01 mm verwendet. Bestimmen Sie die optimale Arbeitskonzentration von CFSE empirisch. - Zugabe von 10 ul 0,01 mM CFSE-Lösung pro ml Zellen (Abschnitt 1.3.2, der Konzentration von 1 x 10 6 Zellen / ml in PBS + 0,1% BSA) für eine Endkonzentration von 0,1 uM. Vortexen gut mischen.
HINWEIS: hier verwendeten CFSE Konzentrationen etwa zehnfach geringer als die, die üblicherweise in Proliferationstests angewendet, damit Zelltoxizität des Farbstoffs ist minimal. Wir haben nicht zu beobachten negative Auswirkungen auf die Lebensfähigkeit der Zellen. - Inkubation für 5 min bei RT unter konstantem Schütteln (200 rpm). Vor Licht schützen.
- Quenche die Farbstoff durch Zugabe von 5 Volumina Flusspuffer zu den Rohren. Zentrifugieren bei 94 × g für 5 min bei RT.
- Überstand verwerfen Verlassen des Pellet hinter. Resuspendieren der Zellen mit 5 Volumina Flusspuffer.
- Zentrifugieren bei 94 × g für 5 min bei RT. Verwerfen des Überstandes und Resuspendieren der Zellen in Fließpuffer bei einer Konzentration von 1 x 10 6 Zellen / ml.
- Fügen Sie die gleiche Anzahl von ungefärbten Zellen von Interesse für die Zellsuspension gefärbt. Fahren Sie mit der Oberfläche Antigen-Färbung Protokoll (Abschnitt 3).

Abbildung 3. Nachweis der Differenz CD-Oberflächen-Antigen-Expression zwischen zwei Zelllinien mittels CFSE Farbstoffmarkierung. SH-SY5Y Neuroblastomzellen sind bereits beschriftet mit CFSE zur späteren Identifizierung im Vergleich zum unmarkierten BJ Fibroblasten. Co-Färbung der Mischprobe (rechte Felder) mit Oberflächenmarker CD24 oder CD54 (beide konjugiert an APC) zeigt, dass Zelllinien sind leicht zu unterscheiden durch den CFSE-Färbung (Pfeile = SH-SY5Y, Pfeilspitzen = BJ Fibroblasten). Die Mehrheit der SH-SY5Y-Zellen exprimieren CD24 nicht aber CD54 (ICAM-1). Im Gegensatz dazu BJ Fibroblasten (CFSE-negativ) sind positiv für CD54, aber weitgehend negativ für CD24."Https://www.jove.com/files/ftp_upload/52241/52241fig3highres.jpg" target = "_ blank"> Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
3. Zelloberflächenfärbung
- Beschriften Sie die Röhrchen mit Proben und kritische Kontrollgruppe (siehe Tabelle 1).
| Rohr-Nr. | Beispielnamen | Antigen-Fluorophor | Verdünnung |
| 1 | Ungefärbte Zellen | - | |
| 2 | Einzelbunt | CD24-APC | 1.50 |
| 3 | Einzelbunt | TuJ1-Alexa Fluor 488 nm | 1: 2.000 |
| 4 | Doppelbunt | CD24-APC | 1.50 |
| TuJ1-Alexa Fluor 488 nm | 1: 2.000 | ||
| 5 | Einzelbunt | Sekundäre nur: Alexa Fluor 488 nm | 1: 2.000 |
Tabelle 1 L ist von Rohren in einer typischen Durchflusszytometrie Experiment einbezogen werden. Die Tabelle zeigt nur eine minimale Anzahl von Probenröhrchen für eine in diesem Video-Artikel beschriebenen Co-Färbung Experiment erforderlich. Ein ideales Experiment muss alle erforderlichen Kontrollen (negativ, positiv sowie eine Entschädigung Kontrollen) für eine genaue Interpretation der erzielten Ergebnisse umfassen.
- 100 l Zellsuspension (von Abschnitt 2.7 oder 1.3.2) zu jeder 1,5 ml Mikrozentrifugenröhrchen.
HINWEIS: Achten Sie darauf, ein Minimum von 0,1 x 10 6 Zellen pro 100 ul der Zellsuspension vorhanden sind. - Hinzufügen Fluorophor konjugiert Antikörper an die Probe bei einer geeigneten Verdünnung.
HINWEIS: Bestimmen Sie die Arbeitslösung für jeden Antikörper vor dem Experiment. Siehe Tabelle 2 für eine Liste of neuronalen Oberflächenantigene.
| Antigen | Zelltyp | Hinweis |
| CD15 | Neurale Stammzellen | [28, 67] |
| CD24 | Neuronalen Zellen | [28, 68] |
| CD29 | Neurale Stammzellen | [28, 69, 70] |
| CD44 | Gliazellen | [25] |
| CD49f | Neurale Stammzellen | [38] |
| CD56 (NCAM) | Neuronalen Zellen | [71] |
| CD133 | Neurale Stammzellen | [27] |
| CD184 | Neurale Stammzellen und Gliazellen | [25] |
| CD200 | Neuronalen Zellen | [38] |
| CD271 | Neuralleiste Stammzellen | [25] |
| A2B5 | Gliazellen | [31] |
| CORIN | Dopaminergen Vorläufer | [35, 36] |
| FORSE1 | Neurale Stammzellen (NSC) | [72] |
| GLAST | Gliazellen | [33] |
| NG2 | Gliazellen | [32] |
Tabelle 2. Auswahl der neuronalen Oberflächenantigene. Diese Tabelle enthält eine Liste von Oberflächenepitopen gefunden, die von verschiedenen neuronalen Zelltypen exprimiert wird, um die wachsende Gruppe von Oberflächenantigenen verwendet werden, um die neuronale Linie kenn exemplifizieren werden. Man beachte, dass diese Auswahl ist bei weitem nicht vollständig ist und die meisten von diesen Markierungen werden auch durch eine Reihe von anderen neuronalen und nicht-neuronalen Zellen exprimiert wird. Folglich werden Kombinationen mehrerer Marker erforderlich, um besser zu definieren und zu isolieren, die die angegebenen neuronalen Subsets werden.
- Inkubieren für 30 min auf einem Horizontalschüttler (200 rpm) in der Dunkelheit.
- Flusspuffer Wasch
- 1 ml der Pufferströmung an den Rohren. Zentrifuge bei 380 × g für 4 min bei 4 ° C.
- Überstand verwerfen Verlassen des Pellet hinter.
- Wiederholen Sie den Waschvorgang noch einmal.
- Nach dem zweiten Waschen, Dekantieren des Überstandes und Resuspendieren der Zellen in Fließpuffer auf ein Endvolumen von 100 ul.
- Verwenden Probe für die Durchflusszytometrie. Alternativ fahren Sie mit Abschnitt 4 und 5.
HINWEIS: Wenn Zellen sortiert werden und wieder in Kultur nach der FACS sind (dh lebensfähige Zellsuspension ohne Fixierung oder Permeabilisierung) gelten aseptische Technik bei der Ernte, Färbung und Analyseschritte.
4. Fixierung und Permeabilisierung 38
- Fixierung mit Paraformaldehyd (PFA):
- Bereiten Fixierungspuffer, der 2% PFA in PBS.
Anmerkung: PFA ist schädlich für Mensch und Umwelt. Verwenden Sie geeignete persönliche Schutzausrüstung und entsorgen Abfälle in Übereinstimmung mit den örtlichen Vorschriften. - Gib 500 ul Fixierungspuffer auf 100 & mgr; l Zellsuspension.
- Die Röhrchen bei RT für 15 min inkubieren, auf einem Orbitalschüttler (100 Upm) im Dunkeln.
- Bereiten Fixierungspuffer, der 2% PFA in PBS.
- PBS-Wasch:
- 1 ml PBS, um das Rohr. Zentrifuge bei 380 × g für 3 min bei 4 ° C.
- Entsorgen / Dekantieren des Überstandes verlassen etwa 100 ul in die Röhre.
- Permeabilisierung mit Tween-20:
- Bereiten Permeabilisierung Puffer mit 0,7% Tween-20 in PBS.
- Gib 500 ul Permeabilisierung Puffer auf 100 & mgr; l Zellsuspension.
- Die Röhrchen bei RT für 15 min inkubieren, auf einem Orbitalschüttler (100 Upm) im Dunkeln.
- Mit PBS (wie in Abschnitt 4.2 beschrieben) Waschen Sie die Zellen einmal und Überstand aus dem Röhrchen Completely, so dass nur das Pellet hinter.
5. Intrazelluläre Antigen Staining 38 (Figur 4)
- Herstellung von primären Antikörperlösungen:
- Verdünne die primären Antikörper in Verdünnungspuffer, der 1% Rinderserumalbumin, 10% Serum (zB normale Esel oder Ziegenserum) und 0,5% Tween-20 in PBS.
HINWEIS: Wählen Sie das Serum je nach Art der Sekundärantikörper wurden angehoben, um verwendet werden. - Alternativ verwenden Zenon Fluorescein Markierung des primären Antikörpers nach den Anweisungen des Herstellers.
- Herstellung von 1 & mgr; g des primären Antikörpers in PBS bei einer geeigneten Verdünnung (Gesamtvolumen ≤ 20 ul).
- Mit 5 ul Zenon Fluorescein IgG Markierungsreagenz (Komponente A) zu der Antikörperlösung.
- Inkubieren der Mischung für 5 min bei RT.
- Mit 5 ul Zenon Blockierungsreagenz (Komponente B) zu dem Reaktionsgemisch.
- Bebrütendie Mischung für 5 min bei RT. Anwenden des Antikörpers an die Probe innerhalb von 30 min.
- Verdünne die primären Antikörper in Verdünnungspuffer, der 1% Rinderserumalbumin, 10% Serum (zB normale Esel oder Ziegenserum) und 0,5% Tween-20 in PBS.
- Primäre Antikörper-Färbung:
- Füge 100 & mgr; l des primären Antikörperlösung zu dem Zellpellet und sanft verrieben zu mischen.
- Alternativ hinzuzufügen Zenon Fluorescein markierten Antikörper zu der Zellsuspension bei der geeigneten Verdünnung.
- Die Röhrchen bei Raumtemperatur 30 min Inkubieren auf einem Schüttler (200 rpm), vor Licht geschützt. Mit PBS Waschen Sie die Zellen einmal (wie beschrieben in Abschnitt 4.2) und Überstand aus den Röhrchen vollständig, so dass nur das Pellet hinter.
- Sekundäre Antikörper-Färbung (nicht für den Zenon Fluorescein markierten Antikörpern erforderlich):
- Verdünne die sekundären Antikörper in PBS in einer geeigneten Konzentration.
- Füge 100 & mgr; l des sekundären Antikörperlösung zu dem Zellpellet und sanft verrieben zu mischen. Die Röhrchen bei Raumtemperatur 30 min auf einem Schüttler (200 rpm) im Dunkeln inkubiert.
- Zweimal mit PBS waschen Sie die Proben (wie in Abschnitt 4.2 beschrieben).
- Einmal mit Fließpuffer waschen (siehe Abschnitt 3.5).
- Wieder die Zellen in ungefähr 150 & mgr; l Fließpuffer und von der Strömungs analysieren Cytometer.

Abbildung 4. Co-Färbung der Oberfläche und intrazellulären Proteinen. Durchflusszytometrie Daten veranschaulicht einen Vergleich zwischen primären + Sekundärantikörperbasis gegen Zenon Fluorescein basierende intrazelluläre Färbung in Kombination mit Oberflächenfärbung. Exclusive Positivität auf der y-Achse (obere linke Quadrant) und der x-Achse (untere rechte Quadrant) zeigt Zellen, die für MAP2 und CD24, angefärbt. Nach Co-Färbung kann geteilt MAP2 und CD24 Expression in der oberen rechten Quadranten (rechte Bilder) zu sehen. Vergleich mitAlexa Fluor 488. (Oben; AF488) gegenüber Zenon Fluorescein (unten) für MAP2-Markierungsausbeuten ähnliche Ergebnisse Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
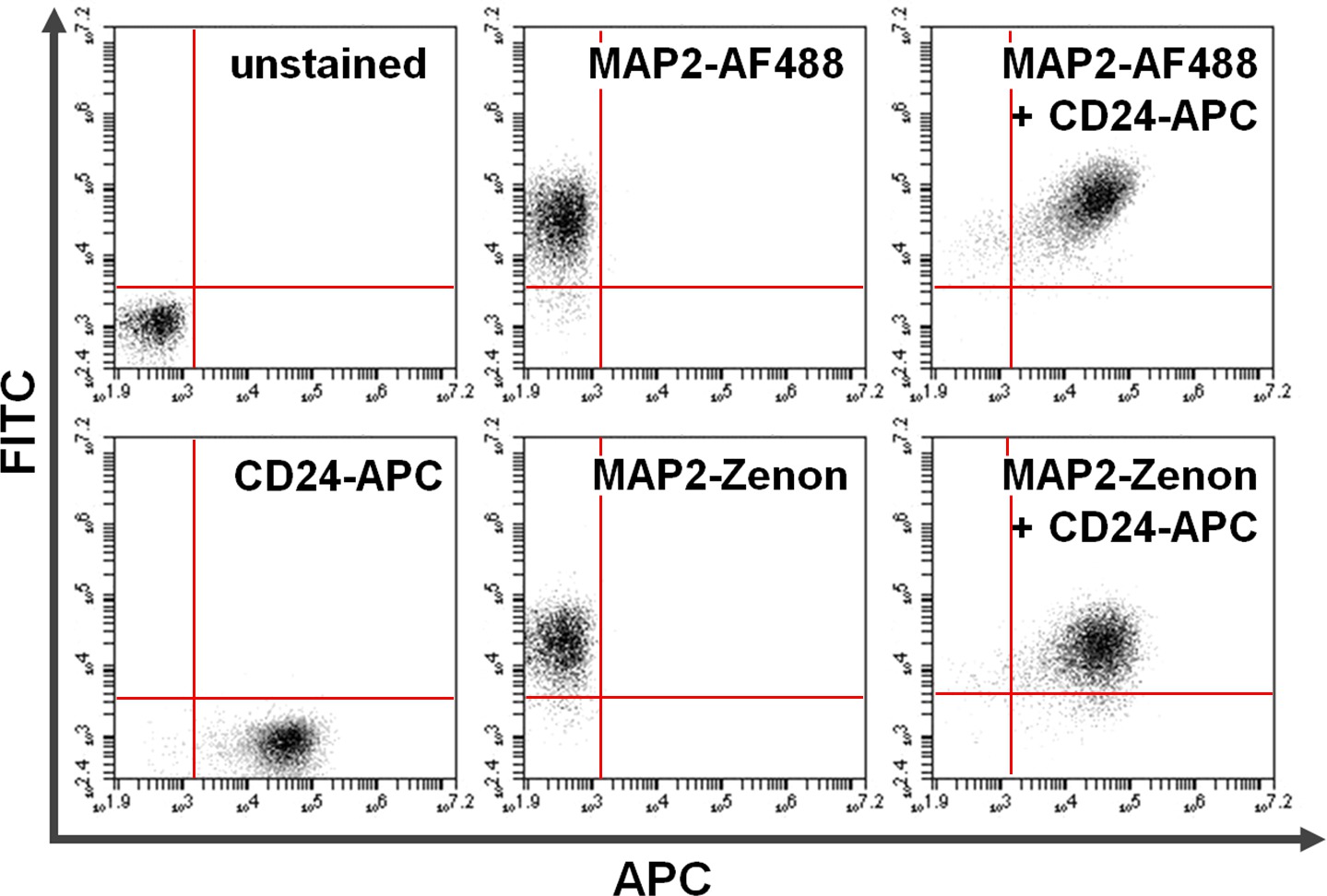{kind=link}
6. Analyse über Durchflusszytometrie
- Zuführen durchflusszytometrische Analyse unmittelbar nach der Beendigung des Färbeprotokoll mit einem Durchflusszytometer mit entsprechenden Filtern zur Signalerkennung. Verwenden 488 nm blau und rot 640 nm Laser mit FL-1 (533/30), FL-2 (585/40) und FL-4 (675/25) Bandpassfiltern.
- Up Haupttore auf der Grundlage der nach vorn und Seitenstreuung ohne Schmutz und abgestorbene Zellen ein.
- Stellen Fluoreszenz Tore für Oberflächen- und intrazelluläre Antigen ≤0.5% bezogen auf den nicht markierten Proben und die Entschädigung für spektrale Überlappung mit einzelnen Bunt Kontrollen.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Die hier vorgestellte Protokoll erlaubt vielseitige experimentelle Ansätze (Abbildung 2). In seiner kürzesten Ausführung (Schritte 1, 3 und 6), kann es als eine Führung für einfache Färbung von Oberflächenantigenen ist. In seiner komplexeren Form kann eine Reihe von Co-Kennzeichnung Paradigmen mit einer Reihe von intrazellulären Antigenen (optional Schritte 2 und / oder 4 bis 5) fortgesetzt. Darüber hinaus ist ...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Die hier vorgestellte Protokoll ist für neuronale Zellkulturen aus menschlichen Stammzellen gegründet, kann aber auch für andere neurale Zellquellen angewendet werden, einschließlich primärer Gewebe oder neuronalen Zelllinien. Neben embryonalen Quellen kann neuralen Stamm- oder Vorläuferzellen aus den neurogenen Regionen des erwachsenen Gehirns 27 extrahiert werden. Außerdem Durchflusszytometrie und FACS kann genutzt werden, um zu quantifizieren, zu analysieren und zu isolieren verschiedenen Zellpopula...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
The authors declare no potential conflicts of interest.
Danksagungen
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| DMEM/F12 (1:1) (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) |  Life Technologies Life Technologies | 11330057 | |
| DPBS without Ca2+ Mg2+ | Life Technologies | 14190169 | |
| Fetal bovine serum, qualified, E.U.-approved, South America origin (FBS) | Life Technologies | 10270-106 | |
| MEM Non-essential amino acids (100x) | Life Technologies | 11140035 | |
| TrypLE Express | Life Technologies | 12604013 | |
| Trypan blue solution, 0.4% | Life Technologies | 15250061 | |
| Paraformaldehyde | Carl Roth | 335.3 | |
| Bovine serum albumin (BSA) Fraction V | PAA Laboratories, Coelbe | K41-001 | |
| Tween-20 Detergent | Calbiochem | 655205 | |
| Carboxyfluorescein succinimidyl ester (CFSE) | eBioscience | 65-0850-84 | |
| DMSO | AppliChem | A1584 | |
| Bottle top filters express plus 0.22 µm, 250 ml | Millipore | SCGPU02RE | |
| Cell culture treated flasks (T 25) | NUNC | 156367 | |
| Cell culture treated flasks (T 75) | NUNC | 156499 | |
| Conical tubes (15 ml) | Greiner Bio-One | 188271 | |
| Conical tubes (50 ml) | Greiner Bio-One | 227261 | |
| Pasteur pipet, glass (150 mm) | STEIN Labortechnik, Remchingen | S03710150 | |
| Pipet tips (0.1-10 µl) | Corning | 4125 | |
| Pipet tips (1-200 µl) | Corning | 4126 | |
| Pipet tips (100-1000 µl) | Corning | 4129 | |
| Serological pipets, 5 ml | Corning | 4051 | |
| Serological pipets, 10 ml | Corning | 4101 | |
| Serological pipets, 25 ml | Corning | 4251 | |
| Microcentrifuge tubes (0.5 ml) | Sarstedt | 72,699 | |
| Microcentrifuge tubes (1.5 ml) | Greiner Bio-One | 616201 | |
| Microcentrifuge tubes (2.0 ml) | Sarstedt | 72,695,500 | |
| Anti-Human CD24 APC monoclonal antibody | eBioscience | 17-0247-42 | Working dilution 1:50 |
| Anti-Human CD54 PE monoclonal antibody | eBioscience | 12-0549-42 | Working dilution 1:50 |
| Neuronal Class III β-Tubulin (Tuj1) polyclonal antibody | Covance | PRB-435P | Working dilution 1:2,000 |
| Alexa Fluor 488 Donkey anti Rabbit | Life Technologies | A21206 | Working dilution 1:2,000 |
| Zenon® Fluorescein Rabbit IgG Labeling Kit | Life Technologies | Z-25342 | |
| Neubauer-Improved counting chamber | Marienfeld | 0640010 | |
| Vortex | Scientific Industries | G560E | |
| Thermomixer comfort | Eppendorf | 5355 000.001 | |
| Accuri C6 flow cytometer | Becton Dickinson (BD) | 653118 | |
| Microcentrifuge refrigerated, PerfectSpin 24 R | Peqlab | 91-PSPIN-24R | |
| Orbital shaker, Unimax 1010 | Heidolph | 543-12310-00 | |
| Centrifuge refrigerated, Rotanta 96 RC | Hettich | 4480-50 | |
| Class II Biological safety cabinet Safe 2020 | Thermo Scientific | 51026640 | |
| CO2 Incubator, Heracell 240i | Thermo Scientific | 51026331 | |
| Vacuum system, Vacusafe comfort | Integra Biosciences | 158320 | |
| Microscope, Axiovert 40 CFL | Zeiss | 451212 | |
| Pipet controller, accu-jet pro | Brand | 26303 | |
| Micropipet, Pipetman neo P20N (2-20 µl) | Gilson | F144563 | |
| Micropipet, Pipetman neo P200N (20-200 µl) | Gilson | F144565 | |
| Micropipet, Pipetman neo P1000N (100-1000 µl) | Gilson | F144566 |
Referenzen
- Herzenberg, L. A., et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 48 (10), 1819-1827 (2002).
- Chattopadhyay, P. K., Roederer, M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif). 57 (3), 251-258 (2012).
- Jaye, D. L., Bray, R. A., Gebel, H. M., Harris, W. A. C., Waller, E. K. Translational applications of flow cytometry in clinical practice). J. Immunol. 188 (10), 4715-4719 (2012).
- Henel, G., Schmitz, J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 38 (7), 428-436 (2007).
- Seita, J., Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2 (6), 640-653 (2010).
- Ulrich, H., Bocsi, J. Phenotypes of stem cells from diverse origin. Cytometry. A. 77 (1), 6-10 (2010).
- Panchision, D. M., et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 25 (6), 1560-1570 (2007).
- Meyer, R. A., Zaruba, M. E., McKhann, G. M. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 2 (1), 66-74 (1980).
- Junger, H., Junger, W. G. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 9 (16), 3749-3754 (1998).
- McLaren, F. H., Svendsen, C. N., Vander Meide, P., Joly, E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 112 (1-2), 35-46 (2001).
- Wang, S., Roy, N. S., Benraiss, A., Goldman, S. A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 22 (1-2), 167-176 (2000).
- Tanke, H. J., vander Keur, M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 11 (2), 55-62 (1993).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243 (1-2), 77-97 (2000).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 129 (1), 73-79 (2003).
- Ernst, A., et al. Neurogenesis in the striatum of the adult human brain. Cell. 156 (5), 1072-1083 (2014).
- Perfetto, S. P., Chattopadhyay, P. K., Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 4 (8), 648-655 (2004).
- Bandura, D. R., et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81 (16), 6813-6822 (2009).
- Bendall, S. C., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332 (6030), 687-696 (2011).
- Neveu, I., Rémy, S., Naveilhan, P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 113 (4), 767-777 (2002).
- Pruszak, J., Just, L., Isacson, O., Nikkhah, G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2 (Unit 2D.5), (2009).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99 (22), 14506-14511 (2002).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1-2), 18-29 (2003).
- Pruszak, J., Isacson, O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 651, 112-123 (2009).
- Carson, C. T., Aigner, S., Gage, F. H. Stem cells: the good, bad and barely in control. Nat. Med. 12 (11), 1237-1238 (2006).
- Yuan, S. H., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 6 (3), e17540(2011).
- Roy, N. S., Cleren, C., Singh, S. K., Yang, L., Beal, M. F., Goldman, S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 12 (11), 1259-1268 (2006).
- Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 97 (26), 14720-14725 (2000).
- Pruszak, J., Ludwig, W., Blak, A., Alavian, K., Isacson, O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 27 (12), 2928-2940 (2009).
- Peh, G. S. -L., Lang, R. J., Pera, M. F., Hawes, S. M. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 18 (2), 269-282 (2009).
- Golebiewska, A., Atkinson, S. P., Lako, M., Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 27 (6), 1298-1308 (2009).
- Dietrich, J., Noble, M., Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 40 (1), 65-77 (2002).
- Nishiyama, A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 14 (1), 77-82 (2001).
- Jungblut, M., et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 60 (6), 894-907 (2012).
- Ono, Y., et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 134 (17), 3213-3225 (2007).
- Chung, S., et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 108 (23), 9703-9708 (2011).
- Doi, D., et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2 (3), 337-350 (2014).
- Solozobova, V., Wyvekens, N., Pruszak, J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 8 (3), (2012).
- Turaç, G., et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 8 (6), e68519(2013).
- Buchwalow, I. B., Böcker, W. Chapter 2. Immunohistochemistry: Basics and Methods. , 9-17 (2010).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. (70), e4287(2012).
- Lyons, A. B., Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171 (1), 131-137 (1994).
- Hawkins, E. D., et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2 (9), 2057-2067 (2007).
- Quah, B. J. C., Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 44, (2010).
- Sukhdeo, K., et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 8 (1), e53015(2013).
- Jiang, L., et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 140, 90-99 (2014).
- Hulspas, R., et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 76 (6), 355-364 (2009).
- Moloney, M., Shreffler, W. G. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 101 (5), 544-549 (2008).
- Siebzehnrubl, F. A., et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 750, 61-77 (2011).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain). J. Neurosci. Methods. 203 (1), 10-18 (2012).
- Tham, C. -S., Lin, F. -F., Rao, T. S., Yu, N., Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 21 (8), 431-443 (2003).
- Nguyen, H. X., Beck, K. D., Anderson, A. J. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. (50), e2698(2011).
- Marchenko, S., Flanagan, L. Counting human neural stem cells. J. Vis. Exp. (7), 262(2007).
- Brunlid, G., Pruszak, J., Holmes, B., Isacson, O., Sonntag, K. C. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 25 (10), 2551-2558 (2007).
- Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 71 (2), 143-155 (1997).
- Cardona, A. E., Huang, D., Sasse, M. E., Ransohoff, R. M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1 (4), 1947-1951 (2006).
- Nielsen, J. A., Maric, D., Lau, P., Barker, J. L., Hudson, L. D. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 26 (39), 9881-9891 (2006).
- Daneman, R., et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 5 (10), e13741(2010).
- Gräbner, R., Till, U., Heller, R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 40 (3), 238-244 (2000).
- Quah, B. J. C., Parish, C. R. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 379 (1-2), 1-14 (2012).
- Lathia, J. D., et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6 (1), 117-129 (2014).
- Ganat, Y. M., et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 122 (8), 2928-2939 (2012).
- Hedlund, E., et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 26 (6), 1526-1536 (2008).
- Maroof, A. M., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12 (5), 559-572 (2013).
- Chivet, M., Hemming, F., Pernet-Gallay, K., Fraboulet, S., Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145(2012).
- Graner, M. W., et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23 (5), 1541-1557 (2009).
- Eldh, M., Lötvall, J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. (59), e3037(2012).
- Capela, A., Temple, S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291 (2), 300-313 (2006).
- Nieoullon, V., Belvindrah, R., Rougon, G., Chazal, G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 28 (3), 462-474 (2005).
- Nagato, M., et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 80 (4), 456-466 (2005).
- Hall, P. E., Lathia, J. D., Miller, N. G. A., Caldwell, M. A., French-Constant, C. Integrins are markers of human neural stem cells. Stem Cells. 24 (9), 2078-2084 (2006).
- Hargus, G., et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107 (36), 15921-15926 (2010).
- Elkabetz, Y., et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152-165 (2008).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten