
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Cytométrie en flux pour les protocoles surface et intracellulaire Antigen analyses des types cellulaires de neurones
Dans cet article
Résumé
We provide a detailed description of a protocol for flow cytometric analysis of surface antigens and/or intracellular antigens in neural cell types. Critical aspects of experimental planning, step-by-step methodological procedures, and fundamental principles of flow cytometry are explained in order to enable neurobiologists to exploit this powerful technology.
Résumé
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Introduction
La cytométrie en flux a été largement exploitée dans l'immunologie, hématologie et en oncologie pour définir des populations de cellules via des propriétés intrinsèques de dispersion, l'expression d'antigènes de surface cellulaire, et d'autres paramètres de fluorescence 3.1. Nos connaissances dans le sang le développement de la lignée et la maladie sont le résultat d'un degré significatif de l'amélioration continue de cette méthodologie après sa mise en œuvre initiale de 4,5. Sensibilisation accrue du potentiel analytique quantitative et globale de cytométrie en flux a récemment encouragé son utilisation plus répandue dans la recherche sur les cellules souches et peut permettre des progrès similaire profonde dans un délai plus court 6. Toutefois, l'application de la cytométrie en flux pour analyser spécifiquement et isoler des populations de neurones a longtemps été considérée comme difficile. Contrairement aux cellules hématopoïétiques qui existent naturellement en suspension, des types de cellules neurales sont typiquement récoltées à partir de sources excessivement complexes qui peuvent comprendre des cellules gliales et divers outres cellules environnantes ainsi que d'un réseau complexe de neurones de processus portant. Par conséquent, la neurobiologie a encore mis en œuvre la polyvalence de cytométrie de flux à son potentiel complet dans les routines quotidiennes de recherche. Toutefois, aussi longtemps que la suspension de cellules viables unique peut être générée (et protocoles ont été conçus et optimisés à cet effet 7), la cytométrie en flux et les cellules activé par fluorescence (FACS) peut être considéré comme un élément précieux du répertoire analytique en neurobiologie 8-11.

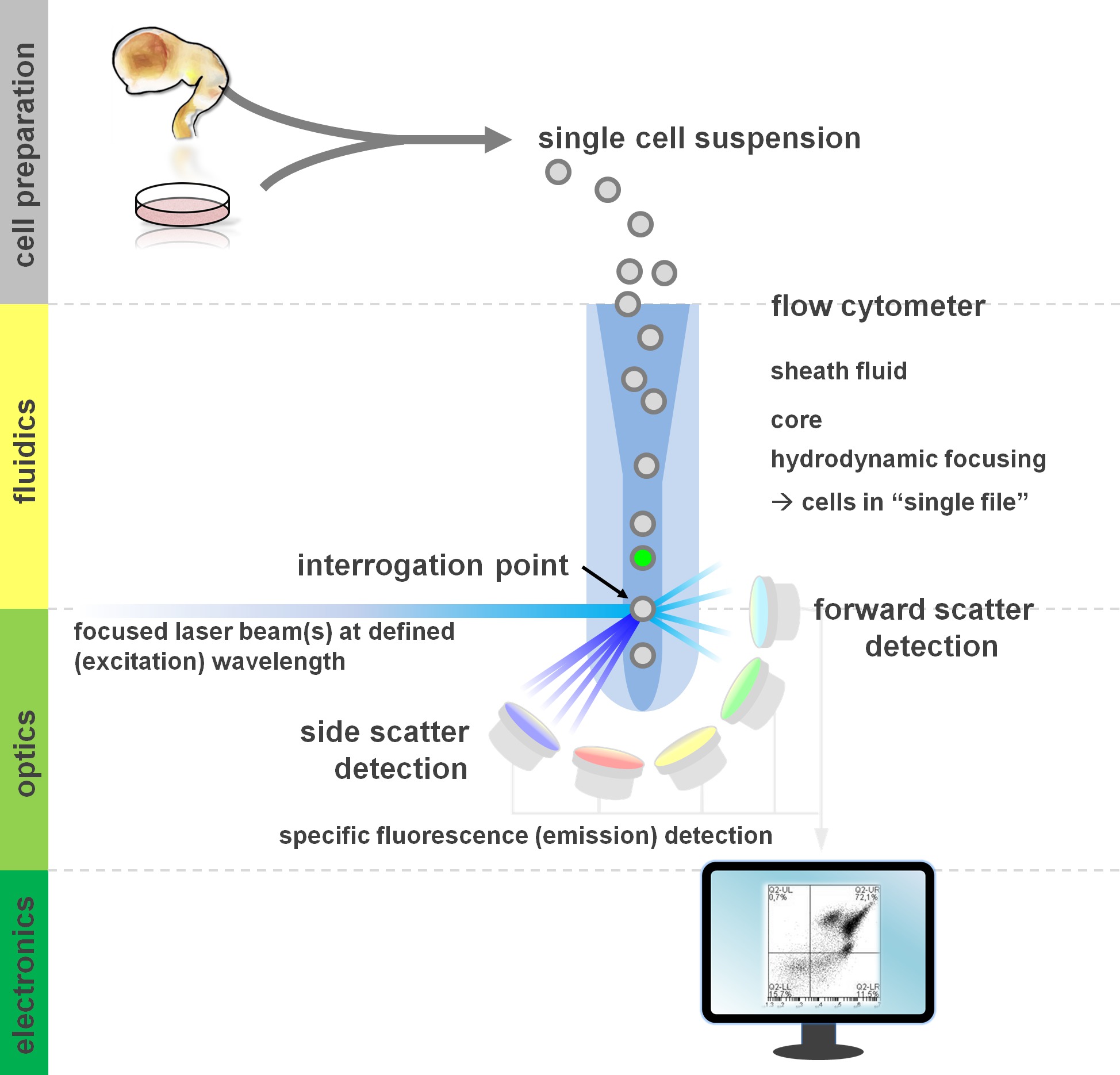
. Figure 1. Principe de l'analyse et de composants un cytomètre de flux cytométrie flux cytomètres de flux comprennent trois systèmes principaux: fluidiques, l'optique et l'électronique. Un écoulement laminaire de cellules en suspension (préparée à partir de tissu primaire ou en culture in vitro) est accomplie par la gaine de fluid via focalisation hydrodynamique, limitant l'échantillon à son noyau central. Les éléments optiques sont constitués de lasers qui illuminent le flux de cellules et de filtres optiques qui dirigent le signal de détecteurs appropriés. Les signaux lumineux détectés sont convertis en signaux électroniques, ensuite traitées par un ordinateur et visualisées sur un moniteur pour l'analyse des données et de déclenchement. Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Les utilisateurs de flux profit des méthodes de cytométrie d'au moins une compréhension de base des fondamentaux sous-jacents, y compris les blocs de construction d'un cytomètre (pour revue voir 12,13; voir aussi la figure 1). Un faisceau laser croise un flux fluide hydrodynamique concentré qui contient les cellules en suspension, qui à son tour passent par le faisceau laser dans "fichier unique" un après l'autre. Le interceptisur une cellule (ou tout autre particule, d'ailleurs) avec les résultats du laser dans la diffusion de la lumière à partir de ce point d'interrogation. La lumière diffusée peut être détectée dans la direction de poursuite laser (diffusion vers l'avant, associée à la taille de la particule), ainsi que perpendiculairement à sa direction (dispersion latérale; reflétant la granulosité de la particule / cellule). Ces propriétés de dispersion susmentionnés ne nécessitent pas de marquage spécifique, ce est pourquoi un échantillon non marqué (ou encore les débris cellulaires, des bulles d'air, etc.) va générer un signal (événement) sur la bivariée diffusion vers l'avant par rapport diagramme de dispersion latérale couramment utilisé pour déclenchement initial. En utilisant les lasers et des filtres spécifiques pour l'excitation et l'émission des spectres correspondant appropriées, une cellule peut être analysé pour déterminer sa positivité, le niveau d'intensité, ou l'absence de marqueurs fluorescents. La majorité des applications de cytométrie en flux ont porté sur la caractérisation par des antigènes de surface cellulaire. Contrairement à la LINEAG hématopoïétiquee, la lignée de neurones est resté moins largement définie selon les modèles 5 d'expression de l'épitope de surface. Un avantage de l'exploitation des antigènes de surface est que les cellules vivantes peuvent être soumis à des paradigmes de tri cellulaire telles que FACS. En revanche, l'antigène intracellulaire coloration nécessite fixation et perméabilisation étapes de médiation de l'interaction épitope-anticorps, ce qui empêche les applications en aval qui nécessitent des cellules viables. Fait à noter, ces approches permettent encore de nombreux essais quantitatifs 14 ainsi que des analyses en aval pour l'ARN et l'expression des protéines 15. Hématologie, l'immunologie et de l'oncologie ont souvent utilisé plus d'une douzaine de marqueurs en collaboration pour définir des sous-populations particulières 16. En outre, la cytométrie de masse CyTOF ou peuvent maintenant être utilisés pour analyser jusqu'à 30 paramètres simultanément 17,18.
Pour des applications de cellules souches neurales, ainsi que des cultures primaires 14,19,20 le hétérogénéité des cellules dansvitro est un phénomène commun 21-23. Les cellules qui ne représentent pas la population cible d'intérêt incarnent un facteur potentiellement confusion pour la lecture expérimentale 24,25. Idéalement, les différents sous-ensembles cellulaires présentes dans une suspension de cellules hétérogène portent les profils d'expression de l'antigène distinctes (connus ou encore à déchiffrer), qui peuvent être utilisés pour définir ces différentes populations. La cytométrie en flux peut donc jouer un rôle crucial dans la résolution de l'hétérogénéité cellulaire et, ainsi, faciliter les applications biomédicales (essais in vitro, thérapie cellulaire) et d'optimiser la lecture quantitative en se concentrant sur le sous-ensemble le plus pertinent 24,26. Diverses combinaisons d'antigènes de surface ont été identifiés au cours des dernières années afin de permettre la quantification et l'isolement de types spécifiques de cellules neurales. Cela comprend CD133 pour l'enrichissement de cellules souches neurales 27, une combinaison du CD15 / CD24 / CD29 des antigènes de surface pour l'isolement de NSC, differentiaTed neurone et les cellules de la crête neurale 28 ou CD15 / CD24 / CD44 / CD184 / CD271 pour isoler des sous-ensembles de neurones et cellules gliales 25, entre autres signatures 29,30. Au-delà de neurones comprennent des marqueurs gliaux A2B5 31, 25 CD44, NG2 32 et 33 GLAST. Une publication récente a exploité le mésencéphale platelage précurseur marqueur CORIN 34,35 pour enrichir précurseurs dopaminergiques dans la maladie de Parkinson transplantation de cellules paradigmes 36. CD molécules ne sont pas seulement des marqueurs, mais fonctionnellement pertinents de médiateurs interactions cellule-cellule et de la capacité d'une cellule à répondre à des signaux à partir de molécules de la matrice extracellulaire et des facteurs de croissance 37. Une stratégie de renforcer encore l'arsenal des antigènes CD combinatoires pour caractériser le développement de la lignée neuronale est d'utiliser des marqueurs intracellulaires connues pour cribler et définir des combinaisons d'antigènes de CD pour un type cellulaire particulier d'intérêt. Nous avons récemment exploité une telle approche et identifié CD49F - / CD200 des profils d'expression combinatoires élevées que une nouvelle approche pour enrichir des sous-ensembles de neurones différenciés neuronale pluripotentes induites systèmes de culture de cellules souches 38. Ici, nous incluons et discutons ce dernier protocole (et des variations de ceux-ci en option), dans lequel une coloration de surface et une coloration intracellulaire peuvent être utilisés simultanément pour définir des sous-populations de cellules neurales par cytométrie de flux.

Figure 2. Schéma du options de protocole expérimental. La figure illustre une représentation schématique des principales étapes dans le protocole. Les étapes facultatives (CFSE de colorants d'étiquetage ou d'antigène intracellulaire) sont indiquées par des cadres gris clair. Après la récolte, il est indispensable pour évaluer la viabilité cellulaire et nombre de suspensions de cellules neurales avant la coloration de la surface cellulaire. Positifainsi que des contrôles négatifs doivent être inclus, en plus des échantillons d'intérêt. Les échantillons peuvent être analysés par analyse de cytométrie en flux et / ou utilisés dans les paradigmes de triage des cellules. Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Bien que nous ayons précédemment utilisé anticorps primaire en combinaison avec un anticorps secondaire pour la coloration intracellulaire 38, nous introduisons maintenant étiquetage non covalente de l'anticorps primaire par l'intermédiaire de fragments Fab fluorescents (d'étiquetage Zenon) comme une légère variation, ce qui réduit les étapes de manipulation de la cellule 39. En outre, comme un autre exemple de la polyvalence du protocole, nous employons un étiquetage facultatif d'un sous-ensemble expérimentale par carboxyfluorescéine succinimidyl ester (CFSE) avant l'antigène de surface de coloration. Cette CFSE pré-étiquetage permet la comparaison directe immédiate de deux lignées cellulaires ou des conditions expérimentales (CFSE marqué vs. non marqué) dans un tube à échantillon unique, la réduction ou la variance des différences subtiles dans le temps d'incubation et l'enregistrement anticorps. CFSE est un colorant fluorescent mis en place qui est couramment utilisé pour le suivi de la cellule 40, dans la prolifération 41,42 43,44 expériences par code barre. Enfin, alors que des mesures réelles de tri (FACS, séparation immunomagnétique ou immunopanning) ne font pas partie de ce protocole, en principe, les procédures de récolte et d'étiquetage décrites ici ne échantillons de rendement qui peuvent être soumis à la surface l'antigène ou des applications intracellulaires base étiquetage-tri 15 25,28.
Avec cet article, nous visons à: résumer une surface viable protocole antigène coloration 25,28, résumer un protocole pour la détection de cibles intracellulaires ainsi que la surface combinée et intracellulaire analyse de l'antigène 38, présenter un CFSE étiquetage de teinture étape intracellulaire 41,45 comme un option expérimentale pour comcomparative analyse des populations de cellules neurales, et résumer les approches à l'écoulement analyse cytométrique (des contrôles appropriés 13,46, ouverture de porte stratégie et la présentation des données 47).
Access restricted. Please log in or start a trial to view this content.
Protocole
1. Neural cellulaire récolte
- Évaluation au microscope:
- Avant d'entreprendre une expérience, vérifier l'état de la culture avec champ lumineux ou microscopie à contraste de phase.
REMARQUE: Bien que le tissu neural primaire obtenu à partir dissections est, en principe, également ouvertes à couler analyse cytométrique 14,28, se il vous plaît noter que l'accent mis par le protocole est sur les cellules obtenues à partir de systèmes de cellules neurales in vitro.
- Avant d'entreprendre une expérience, vérifier l'état de la culture avec champ lumineux ou microscopie à contraste de phase.
- La récolte des cellules 7:
- Lavez délicatement le plat / flacon de cellules adhérentes avec Mg 2+ / Ca 2+ libre phosphate salin (PBS) à température ambiante (par exemple, 5 ml pour flacon T75, 3 ml par puits pour plaque de 6 puits, ou 10 ml boîte de 10 cm).
NOTE: Le PBS utilisé dans le protocole est Mg 2+ / Ca 2+ libre.- Pour l'exemple vidéo, utiliser un flacon T75 de cellules de neuroblastome SH-SY5Y à 80% de confluence. Appliquer étapes de lavage supplémentaires dans les cas oùconsidérable débris est présent dans la coupelle.
- Considérons lavages avec du sérum, Percoll, Ficoll ou 49 gradients de centrifugation 48 PBS contenant de l'albumine et / ou des billes disponibles dans le commerce 50,51. L'élimination de la myéline et d'autres lipides ou autres impuretés est essentiel, en particulier lorsque les sources de tissus adultes primaires sont utilisés.
- Ajouter préchauffé (37 ° C) remplacement de la trypsine à un volume approprié qui recouvre toute la surface du récipient de culture de tissus.
NOTE: Vous pouvez également envisager de Accutase ou d'autres options de digestion enzymatique. Cette étape critique peut affecter négativement l'expression de l'épitope de surface (voir 7). - Incuber le plat / fiole à 37 ° C pendant 2-5 min (selon le type cellulaire) pour permettre aux cellules de se détacher. Tapoter doucement le récipient de culture de tissu ou de niveau avec une pipette sérologique pour déloger les cellules. Évitez de trop digestion (car cela peut causer la perte de cellules et la coagulation à des stades ultérieurs).
- Quench le remplacement de la trypsine en ajoutant deux fois le volume de tampon d'écoulement (2% de FBS dans du PBS) et recueillir les cellules dans un tube conique de 15 ml.
- Triturer doucement la suspension de cellules en utilisant une pipette microlitres (100 - 1 000 pi) ou une pipette sérologique de 5 ml pour préparer une suspension cellulaire unique.
- Centrifuger les cellules à 220 xg pendant 5 min à 25 ° C. Aspirer soigneusement le surnageant laissant le culot derrière.
- Remettre en suspension le culot dans un volume approprié de tampon d'écoulement, en fonction de la taille de la pastille (par exemple, pour une confluence flacon T75 de cellules SH-SY5Y le rendement typique est d'au moins 10 x 10 6 cellules, dans ce cas, les cellules sont remis en suspension dans 5 ml de tampon de flux).
NOTE: Si plus gros morceaux ou coagulation sont observées, filtrer à travers un 30 - 100 um mesh.
- Lavez délicatement le plat / flacon de cellules adhérentes avec Mg 2+ / Ca 2+ libre phosphate salin (PBS) à température ambiante (par exemple, 5 ml pour flacon T75, 3 ml par puits pour plaque de 6 puits, ou 10 ml boîte de 10 cm).
- Le comptage des cellules 52:
- Transférer une petite portion aliquote de la suspension cellulaire dans un tube de microcentrifugation et diluer à un rapport défini dans un volume de trypan bleu ou un colorant viabilité alternatif avant le transfert à un hémocytomètre automatique ou système de comptage de cellules.
- Diluer la suspension cellulaire à une concentration de 1 x 10 6 cellules viables / ml en ajoutant le volume approprié de tampon d'écoulement ou du PBS avec 0,1% de BSA (si le traitement de l'étiquetage des CFSE).
NOTE: L'iodure de propidium, 7 D-aminoactinomycine, annexine V et kits de dosage de viabilité corrigeables disponibles dans le commerce représentent d'autres options afin d'évaluer la viabilité des cellules. En outre, des dosages d'apoptose en utilisant la caspase-3 fluorescence comme décrit précédemment 53 peuvent être utilisés. Canaux fluorescents seront "occupés" par ces réactifs qui peuvent limiter les options pour les étapes suivantes si inclus dans tous les échantillons.
2. intracellulaire Dye étiquetage Utilisation CFSE (Figure 3)
- Diluer le CFSE à une concentration de stock souhaité qui peut être facilement utilisé.
NOTE: Pour ces expériences, une concentration de l'action de0,01 mM a été utilisé. Déterminer la concentration de travail optimal de CFSE empiriquement. - Ajouter 10 ul d'une solution 0,01 mM de CFSE par ml de cellules (section 1.3.2, la concentration de 1 x 10 6 cellules / ml dans du PBS + BSA à 0,1%) pour une concentration finale de 0,1 uM. Brièvement vortex pour bien mélanger.
REMARQUE: concentrations CFSE utilisés ici sont environ dix fois plus faible que celles couramment appliquées dans les essais de prolifération, d'où la toxicité cellulaire du colorant est minime. Nous ne observons pas d'effets négatifs sur la viabilité des cellules. - Incuber pendant 5 min à température ambiante avec une agitation constante (200 rpm). Protéger de la lumière.
- Stopper la teinture en ajoutant 5 volumes de tampon d'écoulement dans les tubes. Centrifuger à 94 g pendant 5 min à température ambiante.
- Jeter le surnageant laissant le culot derrière. Remettre en suspension les cellules avec 5 volumes de tampon d'écoulement.
- Centrifuger à 94 g pendant 5 min à température ambiante. Jeter le surnageant et remettre en suspension les cellules dans du tampon de flux à une concentration de 1 x 10 6 cellules / ml.
- Ajouter un nombre égal de cellules non colorées présentant un intérêt pour la suspension de cellules colorées. Procéder à la surface protocole de coloration antigène (section 3).

Figure 3. Détection de l'expression différentielle de l'antigène de surface de CD entre deux lignées cellulaires par l'intermédiaire de cellules SH-SY5Y de neuroblastome CFSE colorant étiquetage. Sont pré-marquées avec CFSE pour identification ultérieure par rapport à des fibroblastes BJ non marqués. Co-coloration de l'échantillon mélangé (panneaux de droite) avec des marqueurs de surface CD24 ou CD54 (à la fois conjugué à APC) démontre que les lignées cellulaires sont facilement distinguables à cause de la coloration CFSE (flèches = SH-SY5Y, des pointes de flèches = fibroblastes BJ). La majorité des cellules SH-SY5Y expriment CD24 mais pas CD54 (ICAM-1). En revanche, les fibroblastes BJ (CFSE-négatif) sont positives pour CD54 mais largement négatif pour CD24.Cible "https://www.jove.com/files/ftp_upload/52241/52241fig3highres.jpg" = "_ blank"> Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
3. coloration de surface cellulaire
- tubes d'étiquetage y compris les échantillons et les contrôles critiques (voir le tableau 1).
| Tube pas. | Nom Sample | Antigen-fluorophore | Dilution |
| 1 | Cellules non colorées | - | |
| 2 | Teinté Simple | CD24-APC | 01h50 |
| 3 | Teinté Simple | TUJ1-Alexa Fluor 488 nm | 1: 2000 |
| 4 | Double teinté | CD24-APC | 01h50 |
| TUJ1-Alexa Fluor 488 nm | 1: 2000 | ||
| 5 | Teinté Simple | Secondaire seulement: Alexa fluor 488 nm | 1: 2000 |
Tableau 1. L ist de tubes à inclure dans un flux typique cytométrie expérience. Le tableau montre un ensemble minimal de tubes d'échantillon requis pour une expérience de co-coloration décrit dans cet article vidéo. Une expérience idéale doit inclure tous les contrôles nécessaires (contrôles négatifs, positifs de compensation ainsi que) pour l'interprétation précise des résultats obtenus.
- Ajouter 100 pi de suspension cellulaire (de la section 2.7 ou 1.3.2) à chaque tube de 1,5 ml.
NOTE: Assurez-vous un minimum de 0,1 x 10 6 cellules sont présents pour 100 ul de suspension cellulaire. - Ajouter fluorophore conjugué anticorps à l'échantillon à une dilution appropriée.
NOTE: Déterminer la dilution de travail pour chaque anticorps avant l'expérience. Voir le tableau 2 pour une liste of antigènes de surface de neurones.
| Antigène | Type de cellule | Référence |
| CD15 | Les cellules souches neurales | [28, 67] |
| CD24 | Cellules neuronales | [28, 68] |
| CD29 | Les cellules souches neurales | [28, 69, 70] |
| CD44 | Les cellules gliales | [25] |
| CD49f | Les cellules souches neurales | [38] |
| CD56 (NCAM) | Cellules neuronales | [71] |
| CD133 | Les cellules souches neurales | [27] |
| CD184 | Les cellules souches neurales et les cellules gliales | [25] |
| CD200 | Cellules neuronales | [38] |
| CD271 | Les cellules souches de la crête neurale | [25] |
| A2B5 | Les cellules gliales | [31] |
| CORIN | précurseurs dopaminergiques | [35, 36] |
| FORSE1 | Les cellules souches neurales (NSC) | [72] |
| GLAST | Les cellules gliales | [33] |
| NG2 | Les cellules gliales | [32] |
Tableau 2. Sélection des antigènes de surface de neurones. Ce tableau fournit une liste des épitopes de surface trouvés être exprimé par divers types de cellules neurales pour illustrer le panneau croissante des antigènes de surface utilisées pour caractériser la lignée de neurones. Notez que cette sélection est loin d'être complète et que la plupart de ces marqueurs sont également exprimée par une série d'autres cellules neurales et non neuronaux. Par conséquent, des combinaisons de plusieurs marqueurs seront nécessaires pour mieux définir et isoler les sous-ensembles de neurones indiqués.
- Incuber pendant 30 min sur un agitateur orbital (200 tpm) dans l'obscurité.
- Flux lavage tampon
- Ajouter 1 ml de tampon d'écoulement dans les tubes. Centrifuger à 380 g pendant 4 min à 4 ° C.
- Jeter le surnageant laissant le culot derrière.
- Répétez l'étape de lavage.
- Après le second lavage, décanter le surnageant et remettre en suspension les cellules dans du tampon de flux à un volume final de 100 ul.
- Utilisation échantillon pour l'analyse par cytométrie en flux. Sinon, passez à la section 4 et 5.
NOTE: Si les cellules doivent être triés et remis en culture post-FACS (c.-à-suspension de cellules viables sans fixation ou perméabilisation), appliquer les techniques d'asepsie pendant la récolte, la coloration et étapes analytiques.
4. Fixation et perméabilisation 38
- Fixation utilisant paraformaldéhyde (PFA):
- Préparer tampon de fixation contenant 2% de PFA dans du PBS.
NOTE: PFA est nocif pour les humains et l'environnement. Utiliser un équipement de protection individuelle approprié et jeter les déchets conformément à la réglementation locale. - Ajouter 500 ul de tampon de fixation à 100 ul de suspension cellulaire.
- Incuber les tubes à température ambiante pendant 15 min sur un agitateur orbital (100 tpm) dans l'obscurité.
- Préparer tampon de fixation contenant 2% de PFA dans du PBS.
- PBS lavage:
- Ajouter 1 ml de PBS au tube. Centrifuger à 380 g pendant 3 min à 4 ° C.
- Jeter / décanter le surnageant laissant environ 100 pi dans le tube.
- Perméabilisation avec du Tween-20:
- Préparer le tampon de perméabilisation contenant 0,7% de Tween-20 dans du PBS.
- Ajouter 500 ul de tampon de perméabilisation à 100 ul de suspension cellulaire.
- Incuber les tubes à température ambiante pendant 15 min sur un agitateur orbital (100 tpm) dans l'obscurité.
- Laver les cellules une fois avec du PBS (comme décrit à la section 4.2) et retirer le surnageant de la tubes completely, laissant seulement derrière le culot.
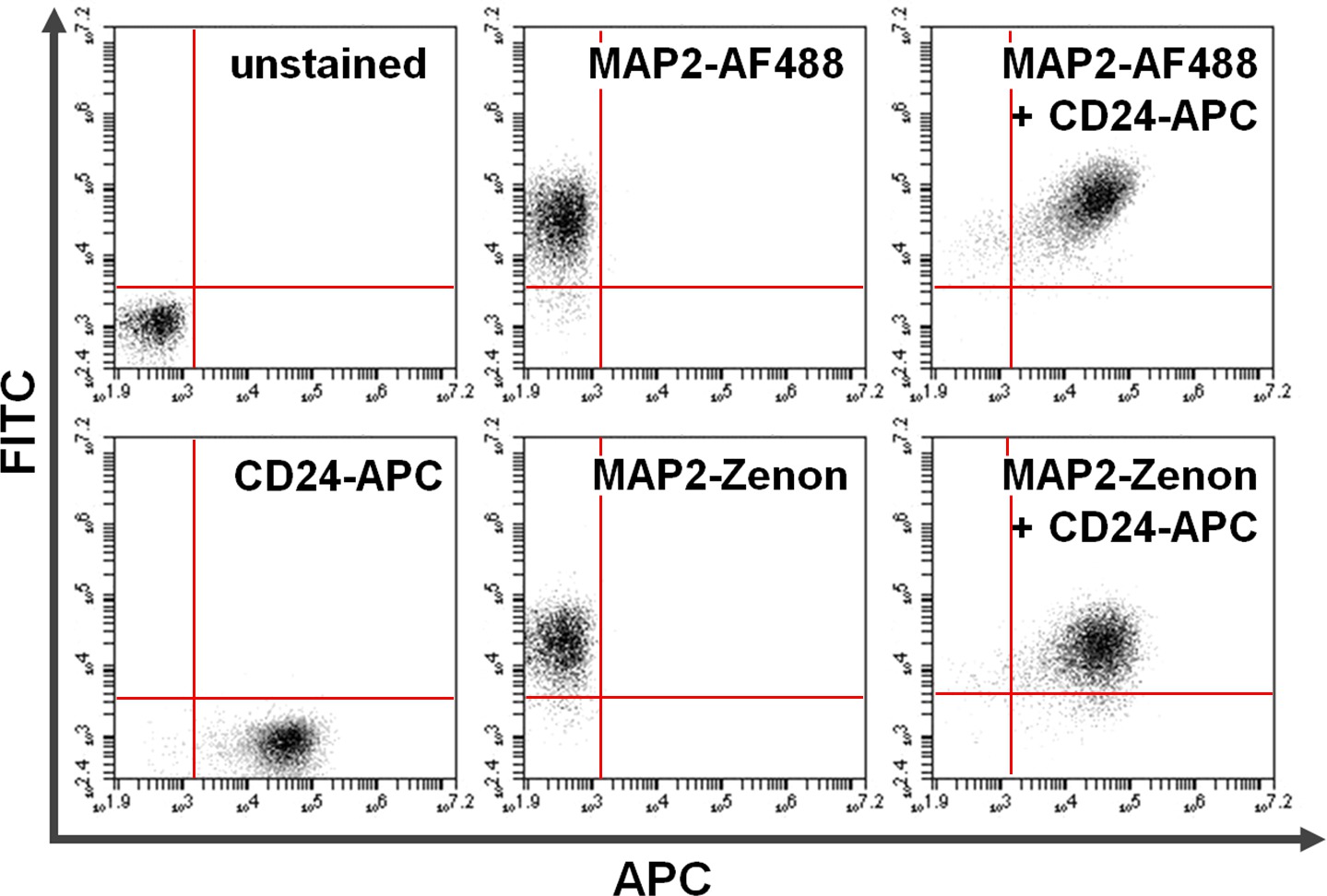
5. Antigène coloration intracellulaire 38 (Figure 4)
- Préparation des solutions d'anticorps primaires:
- Diluer les anticorps primaires dans du tampon de dilution contenant 1% d'albumine de sérum bovin, 10% de sérum (par exemple sérum de chèvre d'âne ou normal) et 0,5% de Tween-20 dans du PBS.
REMARQUE: Choisissez le sérum doit être utilisé selon les espèces les anticorps secondaires ont été soulevées dans. - Vous pouvez également utiliser l'étiquetage Zenon fluorescéine de l'anticorps primaire selon les instructions du fabricant.
- Préparer 1 pg de l'anticorps primaire dans du PBS à une dilution appropriée (volume total ≤ 20 ul).
- Ajouter 5 ul de fluorescéine Zenon réactif de marquage IgG (le composant A) à la solution d'anticorps.
- Incuber le mélange pendant 5 min à température ambiante.
- Ajouter 5 ul de réactif de blocage Zenon (composant B) au mélange de réaction.
- Incuberle mélange pendant 5 min à température ambiante. Appliquer l'anticorps à l'échantillon dans les 30 min.
- Diluer les anticorps primaires dans du tampon de dilution contenant 1% d'albumine de sérum bovin, 10% de sérum (par exemple sérum de chèvre d'âne ou normal) et 0,5% de Tween-20 dans du PBS.
- Coloration à l'anticorps primaire:
- Ajouter 100 ul de la solution d'anticorps primaire au culot cellulaire et triturer doucement pour mélanger.
- En variante, ajouter un anticorps marqué à la fluorescéine Zenon à la suspension cellulaire à la dilution appropriée.
- Incuber les tubes à température ambiante pendant 30 min sur un agitateur orbital (200 tpm), à l'abri de la lumière. Laver les cellules une fois avec du PBS (comme décrit à la section 4.2) et retirer le surnageant des tubes complètement, ne laissant que le culot derrière.
- Coloration d'anticorps secondaire (non requis pour les anticorps marqués Zenon fluorescéine):
- Diluer les anticorps secondaires dans du PBS à une concentration appropriée.
- Ajouter 100 ul de la solution d'anticorps secondaire au culot cellulaire et triturer doucement pour mélanger. Incuber les tubes à température ambiante pendant 30 min sur un agitateur (200 rpm) dans l'obscurité.
- Laver les échantillons deux fois avec du PBS (comme décrit à la section 4.2).
- Laver une fois avec le tampon de flux (voir section 3.5).
- Remettre en suspension les cellules dans environ 150 ul de tampon de flux et d'analyser sur le cytomètre de flux.

Figure 4. Co-coloration de la surface et des protéines intracellulaires. Cytométrie en flux de données illustre une comparaison entre repose-anticorps primaire + secondaire par rapport coloration intracellulaire base-fluorescéine Zenon en combinaison avec une coloration de surface. Positivité exclusive sur l'axe des y (quadrant supérieur gauche) et l'axe des x (quadrant inférieur droit) montre des cellules colorées pour MAP2 et CD24, respectivement. Après co-coloration, MAP2 partagée et d'expression CD24 peut être vu dans le quadrant supérieur droit (des panneaux de droite). Comparaison de l'utilisation deAlexa fluor 488. (En haut; AF488) contre Zenon fluorescéine (en bas) pour les rendements MAP2-en matière d'étiquetage des résultats similaires Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
6. Analyse par cytométrie en flux
- Effectuer une analyse par cytométrie en flux, immédiatement après l'achèvement du protocole de coloration à l'aide d'un cytomètre en flux avec des filtres appropriés pour la détection de signaux. Utilisez 488 nm bleu et 640 nm laser rouge avec FL-1 (533/30), FL-2 (585/40), et des filtres FL-4 (675/25) passe-bande.
- Mettre en place des portes primaires basées sur la diffusion vers l'avant et le côté excluant les débris et les cellules mortes.
- Set portes de fluorescence pour la surface et l'antigène intracellulaire ≤0.5% sur la base des échantillons non colorées et l'indemnisation des chevauchement spectral utilisant des contrôles simples colorés.
Access restricted. Please log in or start a trial to view this content.
Résultats
Le protocole présenté ici permet d'approches expérimentales polyvalent (figure 2). Dans sa version la plus courte (étapes 1, 3 et 6), il peut être considéré comme un guide pour les simples coloration des antigènes de surface. Dans sa forme la plus complexe, un certain nombre de paradigmes co-marquage avec une gamme d'antigènes intracellulaires peut être poursuivi (étapes facultatives 2 et / ou 4-5). E...
Access restricted. Please log in or start a trial to view this content.
Discussion
Le protocole présenté ici est bien établie pour les cultures de cellules neurales dérivées de cellules souches humaines, mais peut être appliqué également à d'autres sources de cellules neurales, y compris le tissu primaire ou des lignées de cellules neurales. En plus des sources embryonnaires, les cellules neuronales souches ou progénitrices peuvent être extraits à partir des régions neurogènes de cerveau adulte 27. En outre, la cytométrie en flux et FACS peuvent être exploitées à quan...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
The authors declare no potential conflicts of interest.
Remerciements
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| DMEM/F12 (1:1) (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) |  Life Technologies Life Technologies | 11330057 | |
| DPBS without Ca2+ Mg2+ | Life Technologies | 14190169 | |
| Fetal bovine serum, qualified, E.U.-approved, South America origin (FBS) | Life Technologies | 10270-106 | |
| MEM Non-essential amino acids (100x) | Life Technologies | 11140035 | |
| TrypLE Express | Life Technologies | 12604013 | |
| Trypan blue solution, 0.4% | Life Technologies | 15250061 | |
| Paraformaldehyde | Carl Roth | 335.3 | |
| Bovine serum albumin (BSA) Fraction V | PAA Laboratories, Coelbe | K41-001 | |
| Tween-20 Detergent | Calbiochem | 655205 | |
| Carboxyfluorescein succinimidyl ester (CFSE) | eBioscience | 65-0850-84 | |
| DMSO | AppliChem | A1584 | |
| Bottle top filters express plus 0.22 µm, 250 ml | Millipore | SCGPU02RE | |
| Cell culture treated flasks (T 25) | NUNC | 156367 | |
| Cell culture treated flasks (T 75) | NUNC | 156499 | |
| Conical tubes (15 ml) | Greiner Bio-One | 188271 | |
| Conical tubes (50 ml) | Greiner Bio-One | 227261 | |
| Pasteur pipet, glass (150 mm) | STEIN Labortechnik, Remchingen | S03710150 | |
| Pipet tips (0.1-10 µl) | Corning | 4125 | |
| Pipet tips (1-200 µl) | Corning | 4126 | |
| Pipet tips (100-1000 µl) | Corning | 4129 | |
| Serological pipets, 5 ml | Corning | 4051 | |
| Serological pipets, 10 ml | Corning | 4101 | |
| Serological pipets, 25 ml | Corning | 4251 | |
| Microcentrifuge tubes (0.5 ml) | Sarstedt | 72,699 | |
| Microcentrifuge tubes (1.5 ml) | Greiner Bio-One | 616201 | |
| Microcentrifuge tubes (2.0 ml) | Sarstedt | 72,695,500 | |
| Anti-Human CD24 APC monoclonal antibody | eBioscience | 17-0247-42 | Working dilution 1:50 |
| Anti-Human CD54 PE monoclonal antibody | eBioscience | 12-0549-42 | Working dilution 1:50 |
| Neuronal Class III β-Tubulin (Tuj1) polyclonal antibody | Covance | PRB-435P | Working dilution 1:2,000 |
| Alexa Fluor 488 Donkey anti Rabbit | Life Technologies | A21206 | Working dilution 1:2,000 |
| Zenon® Fluorescein Rabbit IgG Labeling Kit | Life Technologies | Z-25342 | |
| Neubauer-Improved counting chamber | Marienfeld | 0640010 | |
| Vortex | Scientific Industries | G560E | |
| Thermomixer comfort | Eppendorf | 5355 000.001 | |
| Accuri C6 flow cytometer | Becton Dickinson (BD) | 653118 | |
| Microcentrifuge refrigerated, PerfectSpin 24 R | Peqlab | 91-PSPIN-24R | |
| Orbital shaker, Unimax 1010 | Heidolph | 543-12310-00 | |
| Centrifuge refrigerated, Rotanta 96 RC | Hettich | 4480-50 | |
| Class II Biological safety cabinet Safe 2020 | Thermo Scientific | 51026640 | |
| CO2 Incubator, Heracell 240i | Thermo Scientific | 51026331 | |
| Vacuum system, Vacusafe comfort | Integra Biosciences | 158320 | |
| Microscope, Axiovert 40 CFL | Zeiss | 451212 | |
| Pipet controller, accu-jet pro | Brand | 26303 | |
| Micropipet, Pipetman neo P20N (2-20 µl) | Gilson | F144563 | |
| Micropipet, Pipetman neo P200N (20-200 µl) | Gilson | F144565 | |
| Micropipet, Pipetman neo P1000N (100-1000 µl) | Gilson | F144566 |
Références
- Herzenberg, L. A., et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 48 (10), 1819-1827 (2002).
- Chattopadhyay, P. K., Roederer, M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif). 57 (3), 251-258 (2012).
- Jaye, D. L., Bray, R. A., Gebel, H. M., Harris, W. A. C., Waller, E. K. Translational applications of flow cytometry in clinical practice). J. Immunol. 188 (10), 4715-4719 (2012).
- Henel, G., Schmitz, J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 38 (7), 428-436 (2007).
- Seita, J., Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2 (6), 640-653 (2010).
- Ulrich, H., Bocsi, J. Phenotypes of stem cells from diverse origin. Cytometry. A. 77 (1), 6-10 (2010).
- Panchision, D. M., et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 25 (6), 1560-1570 (2007).
- Meyer, R. A., Zaruba, M. E., McKhann, G. M. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 2 (1), 66-74 (1980).
- Junger, H., Junger, W. G. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 9 (16), 3749-3754 (1998).
- McLaren, F. H., Svendsen, C. N., Vander Meide, P., Joly, E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 112 (1-2), 35-46 (2001).
- Wang, S., Roy, N. S., Benraiss, A., Goldman, S. A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 22 (1-2), 167-176 (2000).
- Tanke, H. J., vander Keur, M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 11 (2), 55-62 (1993).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243 (1-2), 77-97 (2000).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 129 (1), 73-79 (2003).
- Ernst, A., et al. Neurogenesis in the striatum of the adult human brain. Cell. 156 (5), 1072-1083 (2014).
- Perfetto, S. P., Chattopadhyay, P. K., Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 4 (8), 648-655 (2004).
- Bandura, D. R., et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81 (16), 6813-6822 (2009).
- Bendall, S. C., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332 (6030), 687-696 (2011).
- Neveu, I., Rémy, S., Naveilhan, P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 113 (4), 767-777 (2002).
- Pruszak, J., Just, L., Isacson, O., Nikkhah, G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2 (Unit 2D.5), (2009).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99 (22), 14506-14511 (2002).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1-2), 18-29 (2003).
- Pruszak, J., Isacson, O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 651, 112-123 (2009).
- Carson, C. T., Aigner, S., Gage, F. H. Stem cells: the good, bad and barely in control. Nat. Med. 12 (11), 1237-1238 (2006).
- Yuan, S. H., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 6 (3), e17540(2011).
- Roy, N. S., Cleren, C., Singh, S. K., Yang, L., Beal, M. F., Goldman, S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 12 (11), 1259-1268 (2006).
- Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 97 (26), 14720-14725 (2000).
- Pruszak, J., Ludwig, W., Blak, A., Alavian, K., Isacson, O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 27 (12), 2928-2940 (2009).
- Peh, G. S. -L., Lang, R. J., Pera, M. F., Hawes, S. M. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 18 (2), 269-282 (2009).
- Golebiewska, A., Atkinson, S. P., Lako, M., Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 27 (6), 1298-1308 (2009).
- Dietrich, J., Noble, M., Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 40 (1), 65-77 (2002).
- Nishiyama, A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 14 (1), 77-82 (2001).
- Jungblut, M., et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 60 (6), 894-907 (2012).
- Ono, Y., et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 134 (17), 3213-3225 (2007).
- Chung, S., et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 108 (23), 9703-9708 (2011).
- Doi, D., et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2 (3), 337-350 (2014).
- Solozobova, V., Wyvekens, N., Pruszak, J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 8 (3), (2012).
- Turaç, G., et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 8 (6), e68519(2013).
- Buchwalow, I. B., Böcker, W. Chapter 2. Immunohistochemistry: Basics and Methods. , 9-17 (2010).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. (70), e4287(2012).
- Lyons, A. B., Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171 (1), 131-137 (1994).
- Hawkins, E. D., et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2 (9), 2057-2067 (2007).
- Quah, B. J. C., Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 44, (2010).
- Sukhdeo, K., et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 8 (1), e53015(2013).
- Jiang, L., et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 140, 90-99 (2014).
- Hulspas, R., et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 76 (6), 355-364 (2009).
- Moloney, M., Shreffler, W. G. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 101 (5), 544-549 (2008).
- Siebzehnrubl, F. A., et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 750, 61-77 (2011).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain). J. Neurosci. Methods. 203 (1), 10-18 (2012).
- Tham, C. -S., Lin, F. -F., Rao, T. S., Yu, N., Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 21 (8), 431-443 (2003).
- Nguyen, H. X., Beck, K. D., Anderson, A. J. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. (50), e2698(2011).
- Marchenko, S., Flanagan, L. Counting human neural stem cells. J. Vis. Exp. (7), 262(2007).
- Brunlid, G., Pruszak, J., Holmes, B., Isacson, O., Sonntag, K. C. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 25 (10), 2551-2558 (2007).
- Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 71 (2), 143-155 (1997).
- Cardona, A. E., Huang, D., Sasse, M. E., Ransohoff, R. M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1 (4), 1947-1951 (2006).
- Nielsen, J. A., Maric, D., Lau, P., Barker, J. L., Hudson, L. D. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 26 (39), 9881-9891 (2006).
- Daneman, R., et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 5 (10), e13741(2010).
- Gräbner, R., Till, U., Heller, R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 40 (3), 238-244 (2000).
- Quah, B. J. C., Parish, C. R. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 379 (1-2), 1-14 (2012).
- Lathia, J. D., et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6 (1), 117-129 (2014).
- Ganat, Y. M., et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 122 (8), 2928-2939 (2012).
- Hedlund, E., et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 26 (6), 1526-1536 (2008).
- Maroof, A. M., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12 (5), 559-572 (2013).
- Chivet, M., Hemming, F., Pernet-Gallay, K., Fraboulet, S., Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145(2012).
- Graner, M. W., et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23 (5), 1541-1557 (2009).
- Eldh, M., Lötvall, J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. (59), e3037(2012).
- Capela, A., Temple, S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291 (2), 300-313 (2006).
- Nieoullon, V., Belvindrah, R., Rougon, G., Chazal, G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 28 (3), 462-474 (2005).
- Nagato, M., et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 80 (4), 456-466 (2005).
- Hall, P. E., Lathia, J. D., Miller, N. G. A., Caldwell, M. A., French-Constant, C. Integrins are markers of human neural stem cells. Stem Cells. 24 (9), 2078-2084 (2006).
- Hargus, G., et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107 (36), 15921-15926 (2010).
- Elkabetz, Y., et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152-165 (2008).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.