
Method Article
Promotor captura Hola-C: Perfiles de alta resolución, genoma de interacciones promotor
* Estos autores han contribuido por igual
En este artículo
Resumen
Elementos reguladores del ADN, como potenciadores, controlan la expresión génica físicamente en contacto con promotores de genes de la blanco, a menudo a través de interacciones cromosómicas largo alcance que abarca grandes distancias genómicas. Promotor de captura Hi-C (PCHi-C) identifica interacciones significativas entre promotores y regiones distales, permitiendo la asignación de posibles secuencias reguladoras a sus genes diana.
Resumen
La organización tridimensional del genoma está ligada a su función. Por ejemplo, elementos reguladores como potenciadores transcripcionales controlan de la expresión espacio-temporal de sus genes diana a través del contacto físico, a menudo salvar distancias genómica considerable (en algunos casos cientos de kilobases) y pasando por alto genes cercanos. El genoma humano alberga un estimado potenciadores 1 millón, la mayoría de los cuales tiene desconocido objetivos gen. Así es crucial para entender el control de la expresión de gene asignar distales regiones reguladoras de sus genes diana. Hemos desarrollado promotor capturar Hi-C (PCHi-C) para permitir la detección de genoma de regiones de interacción promotor distales (PIR), para todos los promotores en un solo experimento. PCHi-c, altamente complejo Hi-C bibliotecas son enriquecidas específicamente para secuencias de promotor a través de la selección de híbridos en solución con miles de cebos biotinilado RNA complementarios a los extremos de los fragmentos de restricción que contiene el promotor. El objetivo es secuencias de promotor entonces hacia abajo y sus compañeros de interacción frecuente como potenciadores y otros posibles elementos reguladores. Después de la secuenciación de alto rendimiento extremo apareado, se aplica una prueba estadística a cada fragmento de restricción ligada promotor identificar PIRs significativas a nivel de fragmento de restricción. Hemos utilizado PCHi-C para generar un atlas de interacciones de largo alcance promotor en decenas de humanos y tipos de células de ratón. Estos mapas del interactoma promotor han contribuido a una mayor comprensión del control de la expresión de genes mamíferos asignando regiones reguladoras supuestas a sus genes diana y revela redes de interacción espacial preferencial promotor-promotora. Esta información también tiene gran importancia para entender la enfermedad genética humana y la identificación de genes de enfermedades potenciales, vinculando no codificante asociada a enfermedad secuencia variantes en o cerca de secuencias de control a sus genes diana.
Introducción
La acumulación de pruebas sugiere que la organización tridimensional del genoma juega un papel funcional importante en una variedad de procesos nucleares, incluyendo gene activación1,2,3, represión4 ,5,6,7,8, recombinación9,10, DNA repair11, DNA replication12,13, y senescencia celular14. Potenciadores del distantes se encuentran en estrecha proximidad espacial a los promotores regulan15,16,17, que es esencial para el control de la expresión de gene spatio-temporal adecuada. Eliminaciones de reforzador muestran que potenciadores distales son esenciales para el objetivo gene transcripción18,19,20,21,22y 'forzado cromatina bucles' demuestra que es suficiente para impulsar la activación transcripcional23ingeniería tethering entre un intensificador y su promotor de destino en el lugar geométrico de la Hbb . Además, los cambios del genoma que llevan genes bajo el control de reforzadores ectópicos pueden resultar en activación de gen inadecuado y enfermedad24,25,26. Juntos, estos ejemplos ilustran que las interacciones promotor enhancer son esenciales para el control del gen y requieren regulación apretada para expresión génica adecuada. El ser humano y los genomas del ratón cada uno se estima que alrededor 1 millón potenciadores del puerto. Para la gran mayoría de estos potenciadores, genes de la blanco son desconocidos, y las reglas de enfrentamiento entre promotores y potenciadores son poco conocidas. Asignar sus genes diana transcripcionales enhancers así sigue siendo un desafío importante en descifrar el control de la expresión de genes mamíferos.
Nuestra comprensión de la arquitectura tridimensional del genoma ha sido revolucionado por la introducción de 3C27 (captura de la conformación de cromosoma) y sus variantes28,29,30,31 . La más poderosa de estas técnicas, Hi-C (captura de conformación de cromosoma de alto rendimiento) está diseñada para identificar todo el conjunto de interacciones cromosómicas dentro de una población celular. Hola C bibliotecas, normalmente generadas por millones de células, son altamente complejas con un estimado 1011 productos de ligadura independientes entre fragmentos de ~ 4 kb en el genoma humano32. Como una consecuencia, identificación confiable y reproducible de las interacciones entre la restricción individual fragmentos (como los que contienen un promotor o potenciador) de datos Hi-C no están factibles a menos que Hi-C bibliotecas son objeto de secuenciación profunda ultra, que no es una solución económicamente viable para preparar bibliotecas Hi-C rutinariamente los laboratorios. Para evitar esta carencia, hemos desarrollado promotor capturar Hi-C específicamente enriquecer productos de ligadura que contienen el promotor de Hi-C bibliotecas. Nos enfocamos en promotores por dos razones. En primer lugar, contactos promotor enhancer han demostrado ser cruciales para los niveles de expresión génica adecuada en numerosos estudios (ver referencias anteriores), y en segundo lugar, como promotores en gran medida invariantes entre tipos celulares, el mismo sistema de captura de cebo puede utilizarse para interrogar el circuito regulador en múltiples tipos celulares y condiciones. Nuestro enfoque se basa en la solución en hibridación de bibliotecas Hi-C con decenas de miles de 120mers biotinilado RNA complementario a los productos que contienen el promotor Hi-C ligadura y posterior captura de bolas magnéticas recubiertas de estreptavidina. Esto se traduce en bibliotecas PCHi-C con mucho menor complejidad en comparación con la original biblioteca de HI-c, centrándose sólo en la identificación de los fragmentos que se unen a los promotores a significativamente altas frecuencias.
Hemos utilizado PCHi-C en un número de humanos y tipos de células de ratón para contribuir a una mejor comprensión del control de expresión del gene por descubrir regiones interacción promotor distal largo alcance con supuesta función reguladora, así como no-al azar contactos de promotor-promotora en el espacio tridimensional del núcleo. Los estudios han asignado cientos de miles de contactos promotor potenciador a través de numerosas células tipos33,34,35,36,37,38, 39, identificado organización mediado Polycomb represivo complejo espacial genoma en células madre embrionarias de ratón7, demostró gran cableado de interactomes promotor durante la diferenciación celular37, 38 , 39y vinculado no codificante asociada a enfermedad de la secuencia de variantes de genes promotores35.
PCHi-C es un método ideal para mapear el genoma conjunto de secuencias de ADN interaccionando con los promotores. Enfoques relacionados, tales como captura de HI-c de regiones genómicas continuadas (ver discusión) son el método de elección para obtener perfiles de interacción de alta resolución para las regiones genómicas. PCHi-C y capturar Hi-C son muy similares desde el punto de vista experimental (la única diferencia es la elección del sistema de captura), para que los consejos y directrices que le ofrecemos son aplicables a ambos enfoques. Aquí, presentamos una descripción detallada de PCHi-C. Esquema de la lógica y el diseño de un experimento de PCHi-C, proporcionar un protocolo de generación de biblioteca PCHi-C paso a paso e ilustran cómo se puede supervisar la calidad de bibliotecas PCHi-C en varios pasos en el protocolo para producir datos de alta calidad.
Protocolo
1. formaldehído fijación
-
Preparación de la célula: iniciar con un mínimo de 2 x 107 células por experimento.
- Para las células cultivadas en cultura, resuspender las células en medio de cultivo. De células ex vivo , resuspender en 1 x de Dulbecco modificado Eagle Medium (DMEM), suplementado con suero bovino fetal (FBS) de 10% (vol/vol).
- Para las células adherentes, eliminar el medio de cultivo y añadir 30,625 mL de medio fresco con 10% (vol/vol) FBS a temperatura ambiente (RT; 20 – 25 ° C).
- Para las células de suspensión, recoger y centrifugar las células a 400 x g y 20 ° C por 3 minutos eliminar sobrenadante y Resuspenda el sedimento celulares en 30,625 mL de medio con 10% (vol/vol) FBS a TA.
- Para los tejidos sólidos, use tripsina (concentración final de 0.05% a 2.5%, dependiendo del tipo de la célula) o dounce de homogeneización para obtener una suspensión unicelular. Después de este paso adicional, tratar células como las células de suspensión.
-
Añadir 4,375 mL de paraformaldehído al 16% libre de metanol (ampolla abierta justo antes de usar) a una concentración final de 2% (vol/vol). Fijar por 10 min a temperatura ambiente con mezcla suave en un eje de balancín.
PRECAUCIÓN: Paraformaldehido es un producto químico peligroso. Siga las regulaciones de salud y seguridad apropiadas. - Calmar la reacción agregando 5 mL de glicina helado de 1 M recién preparado. Mezclar durante 5 min con suave balanceo en RT y luego incubar en hielo durante 15 min con inversión ocasional.
-
Lavar y recoger las células fijas.
- Para las células adherentes, eliminar el sobrenadante, añadir 10 mL de helado 1 x pH 7.4 de PBS en la placa de pared y retírela. Añadir 1 mL de helado 1 x pH 7.4 de PBS, recoger las células usando un raspador celular y transferencia en un tubo de 50 mL. Repetir dos veces para recoger tantas células como sea posible. Añadir PBS helado hasta volumen final de 50 mL.
- Para las células de suspensión, centrifugar células x 760 g y 4 ° C por 5 min, retirar el sobrenadante y Resuspenda el sedimento celulares en 50 mL de pH PBS helado 7.4.
- Centrifugar las células a 400 x g y 4 ° C durante 10 minutos y retire con cuidado el sobrenadante. El precipitado de células puede ser complemento congelado en nitrógeno líquido y posteriormente almacenados a-80 ° C durante varios meses.
2. lisis de la célula
- Resuspenda el precipitado de células en 50 mL de tampón de lisis helado recién preparado (pH de 10 mM Tris-HCl 8, de 0.2% (vol/vol) Igepal CA-630, NaCl, 10 mM y cóctel de inhibidor de la proteasa de una tableta) y mezclar. Incubar en hielo durante 30 minutos, de vez en cuando mezclar por inversión del tubo. Centrifugue los núcleos en 760 x g y 4 ° C por 5 min y retirar el sobrenadante.
3. hindIII digestión
- Lave el núcleo celular con el tampón de restricción de 1,25 x 2. Resuspenda el sedimento en 1 mL de tampón de restricción helada x 1.25 2 celulares y la transferencia en un tubo de 1,5 mL. Spin de los núcleos en 760 x g y 4 ° C por 5 min y retirar el sobrenadante.
- Resuspenda el sedimento celulares en 1790 μl de tampón de restricción de 1,25 x 2. Tomar alícuotas de 5, cada una con 5 millones de células en 358 μl de 1.25 x tampón de restricción 2.
- Añadir 11 μl de 10% (peso/vol) SDS alícuota y agitar a 950 revoluciones por minuto (rpm) durante 30 min a 37 ° C en un Termomezcladores. Si aparecen grumos de células, disociar mediante pipeteo, evitando burbujas.
- Añadir 75 μl de 10% Tritón X-100 (vol/vol) por la alícuota y agitar a 950 rpm y 37 ° C durante 15 min en un Termomezcladores. Si aparecen grumos de células, disociar mediante pipeteo, evitando burbujas.
-
Añadir 12 μl de 100 U/μl HindIII 100 (1.200 unidades en total) por la alícuota e incubar a 37 ° C durante la noche (O/N) agitando a 950 rpm en un Termomezcladores.
- Para el control de la digestión, 25 μl de muestra (5 μl de cada alícuota) en un nuevo tubo de transferencia antes de agregar la enzima (control sin digerir) y repita el mismo procedimiento después de agregar la enzima (control digerida). Incubar ambos tubos en la misma forma que la biblioteca Hi-C.
- A la mañana siguiente, añadir 5 μl de 100 U/μl HindIII (500 unidades en total) por alícuota e incubar a 37 ° C por 2 h agitando a 950 rpm en un Termomezcladores.
-
Control de la digestión: para los controles digeridos y no digeridos (ver 3.5.1), realizar la reversión de la reticulación (paso 6), extracción de fenol: cloroformo y precipitación de DNA (paso 7).
- Diseño de un par de primers que abarcan un sitio de dIII Hin. En la misma región, diseñar otro par de iniciadores que no abarcan un sitio de dIII Hin. Diseño de primers para PCR cuantitativa (Q-PCR) usando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) y los siguientes parámetros:
Tamaño cartilla: óptima 20 (min.: máximo 18: 27); Cartilla Tm: Óptima 60 (mín.: 57, máx.: 63); % De contenido de primer CG: mín.: 20, máx.: 80; Tamaño de amplicón: RT-PCR ~ 100 bp (para PCR convencional ~ 300 bp); Mispriming biblioteca: humanos (cartillas humanas) o roedor y simple (cartillas de ratón). - Realizar la Q-PCR para obtener 4 Cts media (ciclo umbral): Ct [D; H], Obtenido de la muestra digerida [D] con el par de primers que abarcan un HindIII sitio [H]; CT [D;-], obtenida de la muestra digerida [D] con el par de iniciadores que no abarcan un HindIII sitio web [-]; CT [U; H], Obtenido de la muestra sin digerir [U] con el par de primers que abarcan un sitio de dIII Hin; CT [U;-], obtenida de la muestra sin digerir [U] con el par de iniciadores que no abarcan un HindIII sitio web [-]. Calcular el porcentaje de digestión como: digestión % = 100-100/2(Ct[D,H]-Ct[D,-]) - (Ct[U,H]-Ct[U,-]).
- Diseño de un par de primers que abarcan un sitio de dIII Hin. En la misma región, diseñar otro par de iniciadores que no abarcan un sitio de dIII Hin. Diseño de primers para PCR cuantitativa (Q-PCR) usando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) y los siguientes parámetros:
4. Biotinilación de voladizos de fragmento de restricción
- Preparar mezcla maestro Biotinilación: 30,6 μl de 10 x tampón de restricción 2, 10.2 μl de H2O (grado de biología molecular), 7,65 μl de 10 mM dCTP, 7.65 μl de 10 mM dGTP, 7.65 μl de dTTP 10 mM, 191.25 μl de 0.4m m biotina-14-dATP y 51 μl de 5.000 U/mL ADN polimerasa I (grandes Fragmento de Klenow).
- Añadir 60 μL de mezcla principal Biotinilación por alícuota, mezcla e incubar a 37 ° C por 1 h sacudiendo a 700 rpm (Termomezcladores) durante 5 s, cada 30 s. Después de 1 h, colocar alícuotas en el hielo.
5. en el núcleo la ligadura
- Preparar mezcla maestro ligadura: 510 μl de 10 x buffer de T4 ADN ligasa, 51 μl de 10 mg/mL de albúmina sérica bovina (BSA 100 x), 1754.4 μL de agua (grado de biología molecular) y 127,5 μl de 1 U/μl T4 ADN ligasa (véase Tabla de materiales).
- Añadir 479 μl de la mezcla principal ligadura por mezclar alícuota e incubar a 16 ° C durante 4 horas sacude a 700 rpm durante 5 seg cada 2 min en un Termomezcladores.
- Incubar 30 min a TA.
6. reticulación revocación
- Combinar todas las alícuotas en un tubo de centrífuga de 50 mL (conveniente para la centrifugación de alta velocidad).
- Añadir 62.5 μl de Rnasa A, mix, de 10 mg/mL e incubar durante 30 min a 37 ° C.
- Añadir 300 μL de 10 mg/mL proteinasa K, mezcla e incubar por 30 min a 37 ° C.
- Incubar la reacción O/N (o por lo menos 4 h) a 65 ° C. A la mañana siguiente, añadir 300 μL de 10 mg/mL proteinasa K, mezcla e incubar 1 h a 65 ° C.
7. purificación de ADN
- Añadir 4337.5 μl de buffer TLE (10 mM Tris-HCl pH 8.0; 0.1 mM EDTA pH 8.0) y mezclar.
- Agregue 1 volumen (10 mL) fenol pH 8.0, vortex de 10 s y centrífuga en RT y 20.000 x g durante 3 min transferencia de 9 mL de la fase superior (acuosa) a un nuevo tubo de 50 mL.
PRECAUCIÓN: El fenol es un producto químico peligroso. Siga las regulaciones de salud y seguridad apropiadas. - Añadir 2 mL de tampón de TLE la fase acuosa restante, vortex de 10 s y centrifugar a RT y 20.000 x g durante 3 minutos transferir 2.5 mL de la fase acuosa en el nuevo tubo de paso 7.2, haciendo que el volumen final mL 11,5. Deseche el tubo conteniendo la fase inferior (orgánica).
- Agregar 1 volumen (11,5 mL) de alcohol fenol: cloroformo: isoamílico (25:24:1), vortex de 10 s y centrífuga en RT y 20.000 x g durante 3 minutos transferir 11 mL de la fase superior (acuosa) a un nuevo tubo de 50 mL. Repetir el punto 7.3. El volumen total de la muestra será 13,5 mL.
- Añadir 1,35 mL de 3 M sodio acetato pH 5.2 y 33.75 mL de hielo frío 100% etanol, mezcla e incubar a-80 ° C por 45 min o alternativamente durante la noche a-20 ° C.
- Centrifugar a 4 ° C y 20.000 x g durante 10 min, eliminar el sobrenadante, vuelva a suspender el sedimento en 1 mL de etanol (vol/vol) al 70% recién preparado y transferir a un tubo nuevo.
- Centrifugar a 4 ° C y a toda velocidad por 3 min en una centrífuga de sobremesa y luego retire el sobrenadante.
- Vuelva a suspender el sedimento en 1 mL de hielo frío 70% (vol/vol) de etanol y repita el paso 7.7. Seca a la pelotilla a 37 ° C por 10 min y resuspender en 650 μl de tampón de TLE. Determinar el rendimiento de ADN utilizando un ensayo de fluorescencia para cuantificar ADN de doble hebra.
Nota: El protocolo se puede pausarse aquí por presión, congelación y almacenamiento de la muestra a-80 ° C durante varios meses o a-20 ° C por un período más corto de tiempo.
8. controles de calidad
- Monitor biblioteca integridad y ligadura por electroforesis de ADN. Ejecución 200 ng de biblioteca en un 0,8% agarosa/1 x TBE gel. El ADN debe funcionar como una banda más de 10 kb.
- Detectar el tipo de célula conocido invariante corto largo alcance y las interacciones mediante PCR convencional. Uso 100 ng de plantilla de DNA por la reacción de PCR. Diseño de los cebadores PCR cerca y hacia los sitios de restricción siguiendo las instrucciones de arriba (ver 3.7.1). Secuencias del primer control de calidad de ratón y humanas Hi-C bibliotecas se enumeran en la tabla 1.
-
Control de relleno y la ligadura: cortar las bandas de gel que contiene los amplicones de control 8.2, gel-Extracto de ADN y usar la DNA como plantilla para 4 reacciones de PCR individuales con combinaciones de cartilla idéntica.
- Purificar mediante un kit de purificación PCR amplicones y cuantificar la concentración de ADN.
- Preparar cuatro digestión reacciones (HindIII [a], NheI [b], HindIII + NheI [c] y no hay enzima [d]) para cada amplicón en un volumen final de 15 μl: 500 ng de amplicones, 1,5 μl de 10 x buffer de restricción 2.1, 0,15 μl de 10 mg/mL de albúmina sérica bovina (BSA x 100) y 0,1 μl (10 unidades) de la enzima (HindIII [a], NheI [b], HindIII + NheI [c] o agua [d]).
- Digerir durante 1 h a 37 ° C, y luego ejecute reacciones de digestión en gel de x TBE de agarosa/1 1.5% (peso/vol).
9. fragmentación del ADN
- Transferencia 50,5 μg de muestra en un tubo nuevo y añadir tampón TLE a un volumen final de 655 μl. Split muestra en 5 frascos de sonicación (véase Tabla de materiales) mediante la adición de 130 μl de biblioteca (10 μg) en cada frasco. Corte a un tamaño de ~ 400 bp en un ultra-sonicador (véase Tabla de materiales) usando los siguientes parámetros: factor de servicio: 10%; pico de potencia incidente (w): 140; ciclos por explosión: 200; tiempo: 55 s.
- Recoger muestra de sonicación en un nuevo tubo 2 mL.
10. selección de tamaño de grano de SPRI doble cara
- Solución de grano mezcla SPRI (inmovilización Reversible de fase sólida) bien por inversión del tubo, transferencia 1,85 mL de solución de grano a un tubo nuevo y traer a RT durante 15 minutos.
- Añadir 350 μl de agua (grado de biología molecular) a la muestra (volumen final 1 mL).
- Añadir 600 μl de solución de grano SPRI para la muestra (volumen total 1,6 mL; relación entre solución SPRI para ADN: 0.6 a 1), incubar por 5 min a temperatura ambiente y muestra en una centrífuga de mesa para 2-3 s recoger la muestra de la vuelta.
- Abra la tapa, coloque la muestra en el stand de separación magnética durante 5 min., transferencia claro sobrenadante en un tubo nuevo y descartar los granos.
- Concentrado de granos de la SPRI para la segunda etapa de selección de tamaño: 930 transferir μl de SPRI de perlas en un tubo nuevo, lugar en el stand de separación magnética para 5 minutos y descartar sobrenadante claro. Vuelva a suspender las cuentas en 310 μl de solución de grano SPRI.
- Añadir 300 μL de concentrado granos SPRI (paso 10.5) a la muestra (volumen total 1,9 mL; relación SPRI solución al ADN ahora es 0.9 a 1), incubar a temperatura ambiente durante 5 min y centrifugado de la muestra en una centrífuga de mesa para 2-3 s. Abra cuidadosamente la tapa , coloque el tubo en el soporte de separación magnética por 5 min y descarte el sobrenadante.
- Añadir 1 mL de etanol al 70% recién preparada (vol/vol) al tubo de muestras en el stand de separación magnética, incube durante 30 s y descartar sobrenadante. Repetir dos veces.
- Secar granos a 37 ° C en un Termomezcladores (tapa de tubo abierta) de no más de 5 minutos añadir 300 μL de tampón de ELT para la muestra, mezcla e incubar 10 min a temperatura ambiente.
- Girar la muestra en una centrífuga de mesa para 2-3 s, abra la tapa y el tubo de la separación magnética reposar 5 min transferencia claro sobrenadante en un tubo nuevo y descartar los granos.
11. biotina/estreptavidina Pull-down de ligadura productos
- Preparación de buffers: 1 x buffer TB (5mM Tris-HCl de pH 8.0, EDTA 0.5m, 1 M NaCl; 0,05% Tween 20); 2 x de buffer NTB (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 2 M de NaCl); 1 buffer x NTB (5 mM Tris-HCl de pH 8.0, EDTA 0.5m m; 1 M NaCl).
- Añadir 200 μL de granos magnéticos de estreptavidina acoplado (véase Tabla de materiales) en un tubo nuevo, colóquelo en el soporte de separación magnética para 1 minuto y eliminar el sobrenadante.
-
Lavar granos dos veces con 500 μl de tampón de TB 1 x.
- Para cada paso de lavado durante la biotina desplegable, reparación final y retiro de biotina en los extremos de DNA no ligada, dATP tizón y pasos de ligadura de adaptador, vuelva a suspender las cuentas en el búfer, rotar en RT y 15 rpm durante 3 minutos, girar el tubo en una centrífuga de sobremesa para 2-3 s, coloque el tubo en el soporte de separación magnética para 3 minutos y retirar el sobrenadante.
- Vuelva a suspender cuentas en 300 μL de tampón de x NTB 2. Mezcla de granos y la muestra (volumen total de 600 μL) e incube a TA por 15 min en un disco giratorio a 3 rpm.
- Recuperar los granos en el stand de separación magnética para 3 minutos y retirar el sobrenadante. Lavar granos dos veces en 500 μl de tampón de x NTB 1 primero y después en 200 μL de tampón de la ligadura de x 1. Vuelva a suspender las cuentas en 50 μl de tampón de la ligadura de x 10.
12. terminar la reparación y la eliminación de la biotina en los extremos de DNA no ligada
- La muestra (50 μL en total) se combinan con 50 μl de 2.5 mM dNTP mix (12,5 μl de 10 mM de cada dNTP), 18,1 μl de 3.000 U/mL T4 DNA polimerasa, 18,1 μl de 10.000 U/mL T4 PNK, 3.7 μl de 5.000 U/mL ADN polimerasa I grande (Klenow) fragmento y 360.1 μl de H2O.
- Mezclar e incubar a 20 ° C durante 1 hora, agitando 5 s en cada 2 min en un Termomezcladores de 700 rpm.
- Recuperar los granos en el stand de separación magnética, eliminar el sobrenadante y lavar granos dos veces en 500 μl de 1 x buffer TB.
- Lavar los granos en 500 μl de tampón de x NTB 1, seguido de un lavado en 500 μl de 1 x TLE.
- Recuperar los granos en el stand de separación magnética, eliminar el sobrenadante y Resuspenda granos en 415 μl de tampón de x TLE 1.
13. dATP de jales
- Muestra (415 ml) se combinan con 50 μl de 10 x tampón de restricción 2, 5 μl de 10 mM de dATP y 30 μl de 5 U/μl Klenow exo de menos.
- Mezclar e incubar a 37 ° C durante 30 min, agitando 5 s en cada 2 min en un Termomezcladores de 700 rpm.
- Recuperar los granos en el stand de separación magnética, eliminar el sobrenadante y lavar granos dos veces en 500 μl de 1 x buffer TB.
- Lavar los granos en 500 μl de tampón de x NTB 1.
14. adaptador ligadura
- Lavar los granos en 200 μL de 1 x buffer de reacción de la ligadura (véase Tabla de materiales).
- Vuelva a suspender granos en 200 μL de tampón de reacción de la ligadura de x 1. Añadir 4 μL de ADN ligasa (véase Tabla de materiales) y 16 μl de 15 μm, previamente recocido adaptadores de PE (pre-cocer los adaptadores PE mezclando volúmenes iguales de adaptador de PE 1 y PE adaptador 2 (ambos a 30 μm) y incubar durante unos minutos a temperatura ambiente). Incubar a temperatura ambiente durante 15 minutos.
- Recuperar los granos en el stand de separación magnética, eliminar el sobrenadante y lavar granos dos veces en 500 μl de 1 x buffer TB.
- Lavar los granos en 500 μl de tampón de x NTB 1. Luego, lave los granos en 100 μl de 1 x de tampón de restricción 2, vuelva a suspender cuentas en 50 μl de 1 x de tampón de restricción 2 y transferir a un tubo nuevo.
15. Hola-C biblioteca amplificación
- Preparar mezcla maestro PCR: 100 μl de buffer de x Phusion 5; 6 μl de la cartilla de PCR de PE de 25 μm 1.0; 6 μl de la cartilla de PCR de PE de 25 μm 2.0; 14 μl de mezcla de dNTP (10 mM cada uno); 6 μl de Phusion polimerasa; 318 μl de H2O.
- Mix master Mix PCR con los granos (500 μL en total), se dividen en 10 alícuotas de 50 μl y amplificar por PCR usando las siguientes condiciones:
años 30 a 98 ° C
7 ciclos de: 10 s a 98 ° C; 30 s a 65 ° C; 30 s a 72 ° C
7 minutos a 72 ° C - Recoger las reacciones de PCR en un tubo nuevo, recuperar cuentas en el stand de separación magnética y la transferencia flotante (500 μl) en un tubo nuevo.
-
Purificar el ADN de biblioteca con perlas de SPRI.
- Abalorios mezcla SPRI, transferir 460 μl de granos en un tubo nuevo, hacer RT para 15 minutos añadir 450 μl de granos de la SPRI para las reacciones de PCR (volumen final 950 μL), incubar por 5 min a temperatura ambiente y muestra en una centrífuga de mesa para 2-3 s recoger la muestra de la vuelta.
- Abra la tapa, coloque la muestra en el stand de separación magnética durante 5 min. y eliminar el sobrenadante.
- Mantener los granos en el stand de separación magnética, añadir 1 mL de etanol al 70% (vol/vol) al tubo de la muestra sobre un área libre de granos, dejar por 30 s y descartar sobrenadante.
- Repita el paso 15.4.3 dos veces más.
- Secar granos a 37 ° C en un Termomezcladores (tapa de tubo abierta) de no más de 5 minutos.
- Agregar 51 μl de tampón de ELT para la muestra, mezcla e incubar 10 min a 37 ° C, agitando a 950 rpm en un Termomezcladores.
- Girar la muestra en una centrífuga de mesa para 2-3 s, abra la tapa y el tubo de la separación magnética reposar 5 min transferencia claro sobrenadante en un tubo nuevo y descartar los granos.
- Cuantificar la concentración de la biblioteca Hi-C. Después de 7 rondas de amplificación por PCR, obtenemos rutinariamente 500 – 1.500 ng de biblioteca Hi-C.
16. híbridos en solución captura
Nota: Bloqueador y buffer (SHS1-4) a continuación las soluciones son de la SureSelect kit (véase Tabla de materiales).
- Transferencia 500 ng a 1 μg de biblioteca Hi-C en un tubo nuevo y se evapora la muestra en un concentrador de vacío (véase Tabla de materiales, 45 ° C, presión del vacío: nivel 30.0, rampa 5) hasta que se seque.
- Vuelva a suspender biblioteca Hi-C evaporada agregando 3.6 μl de H2O (grado de biología molecular), 2.5 μl de bloqueador de 1, 2.5 μl de blocker 2 y 0,6 μl de bloqueador personalizado.
- Transferir la muestra en un pozo de una nueva tira de tubo PCR, cierre con una tira de la tapa PCR y coloque en hielo. Etiqueta como "D" (para ADN-C).
- Preparar el tampón de hibridación: 12,5 μl de tampón de SHS1; 0,5 μl de tampón de SHS2; 5 μl de tampón de SHS3; 6.5 μl de tampón de SHS4.
- Incubar a 65 ° C por 5 min en un Termomezcladores. Transferir a un pozo de una nueva tira de tubo PCR, cierre con una tira de la tapa PCR y mantener en RT. etiqueta como "H" (para el tampón de hibridación).
- En un pozo de una nueva tira de tubo PCR, mezclar 5 μl de 100 ng/μl biotinilado sondas de RNA (almacenamiento a-80 ° C y descongelar en hielo antes de uso); 0,5 μl de SRNase B (inhibidor de la Rnasa) y 1,5 μl de H2O (grado de biología molecular).
- Cerrar la tira de tubo PCR con un lugar en el hielo y tira de la tapa PCR. Etiqueta como "R" (RNA).
- Configurar la máquina de la polimerización en cadena usando los siguientes parámetros:
5 min a 95 ° C; 25 h a 65 ° C; tapa calentada; Volumen 29 μl de la reacción de PCR.
Nota: Proceder tan rápidamente como sea posible durante todos los procedimientos en marcha la máquina PCR con el fin de evitar la evaporación de la muestra. - Coloque la tira de tubo "D" polimerización en cadena en la máquina de la polimerización en cadena y cierre la tapa de la máquina PCR, iniciar la reacción de PCR. Cuando el programa PCR alcanza los 65 ° C, abra la tapa de la máquina de la polimerización en cadena y coloque la tira de tubo "H" polimerización en cadena en la máquina de la polimerización en cadena. Cierre la tapa de la máquina PCR e incubar 3 minutos abre la tapa de la máquina PCR, lugar el tubo de PCR "R" de la tira en la máquina de la polimerización en cadena y cerca de la máquina de la polimerización en cadena.
- Después de 2 minutos, abra la tapa de la máquina de la polimerización en cadena y todas las tiras de tubo PCR. Transferir 13 μl del bien "H" en pozo "R", entonces todo el volumen del bien "D" a bien "R". Pipeta de arriba y abajo 3 veces para mezclar la reacción, cerrar la tira de tubos PCR, quitar la "H" y "D" PCR tubo tiras y cierren la tapa de la máquina PCR. Incubar la reacción a 65 ° C durante 24 h.
17. aislamiento de promotor que contiene fragmento ligadura productos
Nota: Los pasos siguientes se recomiendan hacerse con kit de adaptador SureSelect y biblioteca (ver Tabla de Materals).
- Caliente previamente 1,5 mL de tampón de lavado 2 por muestra a 65 ° C por adelantado.
- Añadir 60 μL de estreptavidina acoplado bolas magnéticas (véase Tabla de materiales) en un tubo nuevo, coloque en el soporte de separación magnética para 1 minuto y eliminar el sobrenadante.
- Lavar granos tres veces con 200 μL de 1 x de tampón de Unión.
Nota: Para cada etapa de lavado durante el aislamiento post-captura de productos que contienen el promotor de la ligadura, vuelva a suspender los granos en el búfer, gire durante 3 min a temperatura ambiente y 15 rpm en un disco giratorio, girar suavemente el tubo en una centrífuga de mesa para 2-3 s recoger muestra, lugar del tubo de la separación magnética reposar 3 minutos y eliminar sobrenadante. - Vuelva a suspender granos en 200 μL de 1 x de tampón de Unión. Abra la máquina de la polimerización en cadena y la Faja de tubo PCR (mientras sigue funcionando el programa PCR) y transferencia de la reacción de hibridación en el tubo con las bolas magnéticas. Incubar a temperatura ambiente por 30 min en un disco giratorio a 3 rpm.
- Recuperar los granos en el stand de separación magnética y eliminar el sobrenadante. Vuelva a suspender cuentas en 500 μl de tampón de lavado 1, mezcla e incubar durante 15 min a 20 ° C agitando a 950 rpm en un Termomezcladores.
- Recuperar los granos en el stand de separación magnética para 3 minutos y retirar el sobrenadante. Vuelva a suspender cuentas en 500 μl de tampón de lavado 2, mezclar e incubar 10 min a 65 ° C agitando a 950 rpm en un Termomezcladores. Repita el paso dos veces más de 17.5.
- Recuperar los granos en el stand de separación magnética, eliminar el sobrenadante y Resuspenda granos en 200 μL de 1 x de tampón de restricción 2. Recuperar granos en el stand de separación magnética, eliminar el sobrenadante y resuspender los granos en 30 μl de 1 x de tampón de restricción 2.
18. PCHi-C biblioteca amplificación
- Preparar mezcla maestro PCR: 60 μL de 5 x PCR buffer (tampón de Phusion), 3.6 μl de 25 μm PCR PE la cartilla 1.0 3.6 μl de 25 μm PCR PE la cartilla 2.0, 8.4 μL de mezcla de dNTP (10 mM), 3.6 μl de Phusion polimerasa y 190,8 μl de H2O.
- Mix master Mix PCR con los granos (μL 300 en total), se dividen en 6 alícuotas de 50 μl y amplificación de PCR utilizando las siguientes condiciones:
30 s a 98 ° C
4 ciclos de: 10 s a 98 ° C, 30 s a 65 ° C, 30 s a 72 ° C
7 minutos a 72 ° C - Recoger todas las reacciones de PCR en un tubo nuevo, recuperar los granos en el imán y transferir el sobrenadante (300 μL; contiene biblioteca de PCHi-C) en un tubo nuevo.
- Purificar la biblioteca PCHi-C con perlas de SPRI, siguiendo los pasos descritos anteriormente en 15.4.
- Cuantificar la concentración de la biblioteca de PCHi-C.
Resultados
Promotor de captura Hi-C se ha utilizado para enriquecer ratón7,34,36,39 y humano33,35,37,38 Hi-C bibliotecas para interacciones del promotor. Un protocolo similar (llamado HiCap) ha sido descrito por el grupo de Sandberg40. Figura 1A muestra el mismo flujo de trabajo para promotor de captura Hi-C. En el protocolo descrito aquí, Hi-C bibliotecas se generan utilizando ligadura en núcleo41, que se traduce en un número significativamente reducido de ligadura falsos productos42. PCHi-c, ratón muy complejo o humana Hi-C bibliotecas son objeto de solución en hibridación y capturan usando 39.021 ARN biotinilado complementario a 22.225 fragmentos de la restricción de Hinel dIII que contienen el promotor del ratón, o ARN biotinilado 37.608 dirige a 22.076 humanos que contienen el promotor HindIII fragmentos de restricción, respectivamente. Promotor que contiene fragmentos de restricción puede ser objetivo en uno o ambos extremos de individual biotinilado RNAs (figura 1B). Encontramos que captura de los dos termina mejor cobertura de promotores individuales (figura 1; Lee secuencia cruda) casi doble, como se esperaba. Por lo tanto, siempre que sea posible (es decir, en regiones no repetitivas), aconsejamos utilizar ARN biotinilado complementario a ambos extremos de un fragmento de la restricción para ser capturados.
Evaluar la calidad de biblioteca PCHi-C en una etapa temprana durante la preparación de la biblioteca, realizamos dos controles después de ligadura de ADN y purificación, como se describió anteriormente31. La primera es utilizar pares de imprimación específica para amplificar productos ligadura como 3 C27. Utilizamos pares de cartilla (tabla 1) para amplificar productos invariante ligadura de largo alcance de tipo de la célula, tales como entre el gene de Myc y sus conocidos potenciadores situado aproximadamente 2 Mb de distancia (figura 2A) o entre los genes de la (lugar geométrico) Hist1 separados por 1,5 Mb) y entre dos regiones ubicadas en las proximidades de lineal ('control' de corto alcance).
El segundo control de calidad se lleva a cabo para determinar la eficacia de la incorporación de biotina durante el relleno de Klenow-mediada de restricción sitio voladizos con biotina-dATP. Relleno de Klenow éxito y posterior ligadura de Roma-final resultados en la desaparición del sitio original de restricción entre las moléculas de ADN de un producto de la ligadura y en el caso de HindIII en la formación de un nuevo sitio de reconocimiento de NheI (figura 2B ). La proporción de HindIII al producto digerido ligadura de NheI es una lectura directa de la eficacia de incorporación de biotina. Una biblioteca de Hi-C mala calidad mostrará un alto nivel de HindIII digestión, mientras que las bibliotecas de alta calidad tienen digestión de NheI casi completa de los productos de ligadura (figura 2B).
Después de la preparación de biblioteca Hi-C (es decir, después de biotina-estreptavidina tire hacia abajo de tamaño seleccionado Hi-C ligadura productos, ligadura de adaptador y pre-captura de PCR), la distribución de la biblioteca de C de alta integridad y tamaño es evaluada por equipo Bioanalyzer (figura 2 C). el mismo control se realiza al final de la preparación de biblioteca PCHi-C (es decir, después de la captura de la hibridación de productos que contienen el promotor de la ligadura y post-captura de PCR). Comparación de los perfiles de HI-c y equipo Bioanalyzer PCHi-C muestra que como se esperaba, Hi-C bibliotecas son mucho más concentradas que las correspondientes bibliotecas de PCHi-C, pero la distribución de tamaño de las bibliotecas es muy similar, indicando que la captura en PCHi-C no introduce un sesgo de tamaño (figura 2, D).
Después de la secuencia final emparejado, se asigna el PCHi-C Lee, calidad controlada y había filtrada utilizando la tubería HiCUP del43. Bibliotecas de PCHi-C de alta calidad contienen entre 70-90% 'pares válidos' (es decir, la secuencia de final emparejado lee entre dos fragmentos de restricción que no son vecinos en el mapa genómico lineal; Figura 3A, B). La ligadura en núcleo protocolo41,42, el porcentaje de trans Lea pares (es decir, la secuencia de final emparejado lee entre dos fragmentos de restricción que se encuentran en diferentes cromosomas) son generalmente bajos, entre 5 y 25%, lo que refleja la existencia de territorios del cromosoma e indicando la calidad de la buena biblioteca. Comparación directa del porcentaje de pares de válido entre bibliotecas Hi-C y sus correspondientes de las bibliotecas de PCHi-C35, muestra que en todos los casos el porcentaje de pares válidos es mayor en las bibliotecas de PCHi-C (figura 3B). Esto se acompaña de una reducción en el porcentaje de lecturas no válidas 'mismo fragmento interno' en PCHi-C (figura 3). Se espera, como el paso de la captura no sólo enriquece para productos que contienen el promotor de la ligadura, sino también para los extremos del fragmento de restricción, debido a la posición de los oligos de captura en la restricción de los fragmentos (ver figura 1B).
Después HiCUP filtrado, determinamos la eficiencia de captura. PCHi-C bibliotecas contienen tres tipos de lecturas de secuencia válido después de filtrar HiCUP:
1.) promotor: genoma Lee (es decir, lecturas entre un fragmento promotor capturados y un fragmento de restricción HindIII no promotor en cualquier lugar del genoma)
2.) promotor: promotor Lee (lee entre dos fragmentos del promotor capturado)
3.) genoma: genoma Lee (productos de fondo Hi-C ligadura donde ninguno de los socios producto de la ligadura se asigna a un promotor capturado). Estos son desechados antes de análisis de aguas abajo.
Bibliotecas de PCHi-C de alta calidad tienen eficiencias de captura (suma de las categorías 1 y 2) entre 65-90% (figura 3D). Una comparación directa a bibliotecas Hi-C muestra que PCHi-C resulta en un ~ 15 dobleces enriquecimiento para promotor-productos que contienen la ligadura (figura 3D), en algunos casos 17-fold. Esto está cerca del máximo hipotético (19.6-fold) enriquecimiento PCHi-c, que es dependiente en el porcentaje de los fragmentos de restricción del genoma cubiertos por el sistema de captura. Mayor enriquecimiento puede lograrse mediante el diseño de sistemas de captura a menos restricción fragmentos44,45,46.
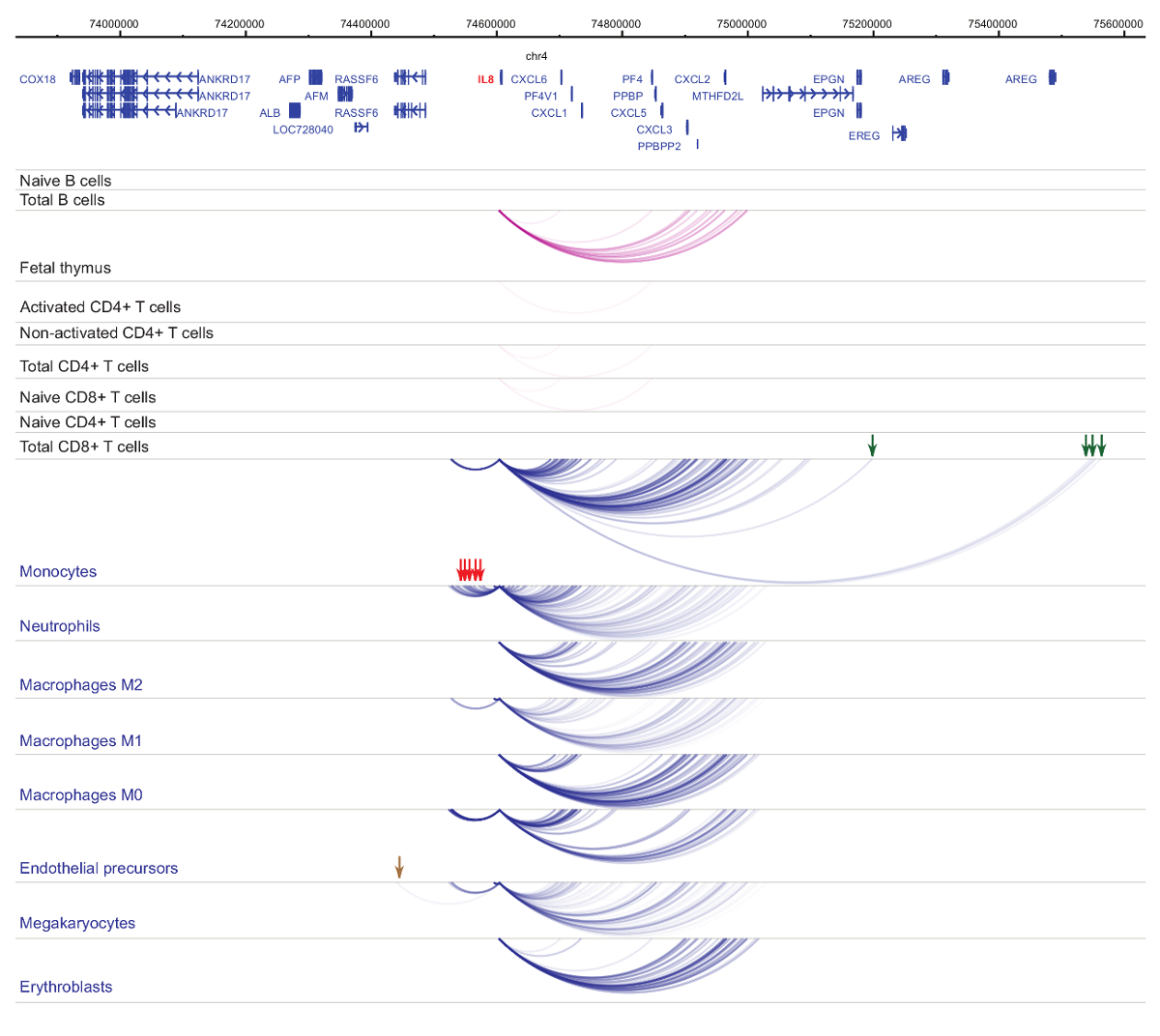
Análisis de interactomes promotor demuestra células tipo y linaje-especificidad33,34,35, con cambios pronunciados durante la diferenciación celular37,38,39 . Figuras 4 y 5 muestran ejemplos de dinámicas de especificidad y diferenciación de linaje en promotores específicos. Por ejemplo, ALAD se expresa constitutivamente en todas las células pero su expresión es upregulated en eritroblastos47. El promotor de la ALAD en contacto varios fragmentos distales en todas las células hematopoyéticas y se involucra en interacciones adicionales específicamente en eritroblastos (figura 4). IL-8 no muestra interacciones estadísticamente significativas en las células de B, muy pocas interacciones en las células de T, pero decenas de interacciones en las células del linaje mieloide, incluyendo interacciones específicas del tipo de células (neutrófilos, monocitos y megacariocitos Figura 5). Estos ejemplos demuestran cómo se puede utilizar PCHi-C desentrañar interactomes específicos del tipo de la célula e identificar regiones promotor interactuando con potencial regulatorio.

Figura 1 : Promotor capturar Hi-C justificación y captura cebo diseño. Flujo de trabajo esquemático (A) PCHi-c. Ligadura en núcleo Hi-C41,42 (I) es seguida por solución en hibridación con cebos biotinilado RNA (II) dirigidas a los fragmentos de restricción de todo humano (representado aquí) o ratón de promotores de genes (III). (B) diseño de cebo para PCHi-C. Cebos de la captura de ARN biotinilado (líneas curvas rojas) están diseñados contra los extremos de los fragmentos de restricción que contiene el promotor (gris, nota que las secuencias de promotor se (rojo) sólo son blanco de los cebos de captura RNA si están situados en la restricción extremos del fragmento). Productos de ligadura de promotor que contiene fragmentos de restricción (gris) y sus interacción fragmentos de restricción (amarillo y verde) son aislados mediante hibridación de secuencia-complementariedad entre cebo de RNA y DNA Diana y posterior biotina-estreptavidina desplegable, como se muestra en (C) comparación de PCHi-A.c. eficiencia de captura de fragmentos de restricción que contiene el promotor dirigido por un RNA cebo captura sonda vs dos sondas de captura de cebo RNA (ver diagrama b). Haga clic aquí para ver una versión más grande de esta figura.
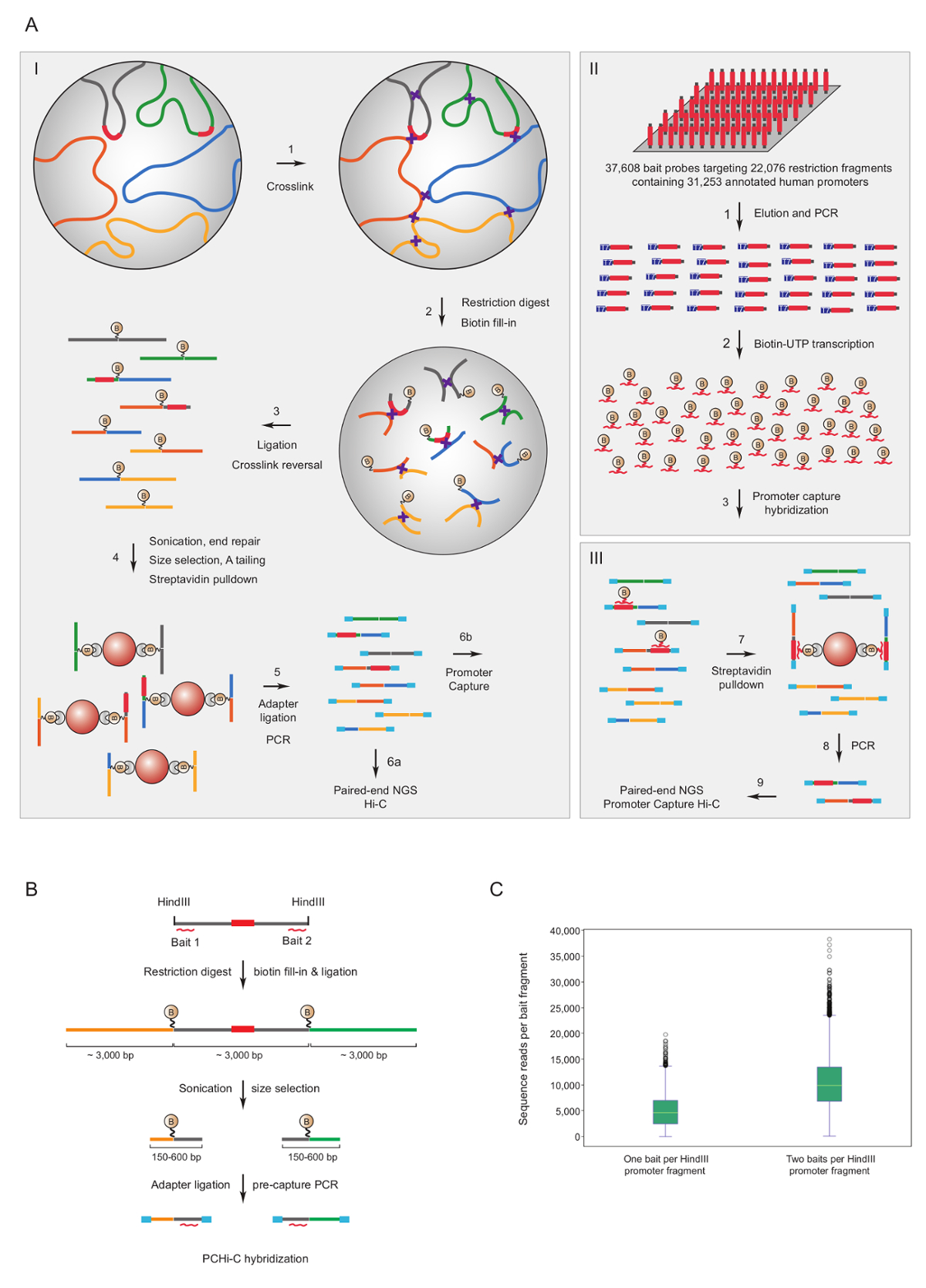{kind=link}

Figura 2 : Controles de calidad de la secuencias de PCHi-C. (A) a la izquierda, esquema de yuxtaposición espacial entre promotor y PIR, resultando en un producto de la ligadura de Hi-C que consiste en un fragmento de restricción que contiene el promotor (gris, secuencia de promotor en rojo) y un fragmento de la restricción de PIR (amarillo). Derecha, ADN gel electroforesis mostrando ejemplos de ligadura Hi-C los productos amplificados con pares de primer específico (como se muestra en el esquema de la izquierda). (B) a la izquierda, digiere los ejemplos representativos de restricción HindIII, NheI y NheI/HindIII de Hi-C ligadura (productos PCR se muestra en la A). A la derecha, esquema de ADN secuencia después de la ligadura de la Hi-C después de fracasado (arriba) o relleno de Klenow de dNTP exitosa (parte inferior) de las ensambladuras de la restricción y la ligadura subsecuente. (C) representante Hi-C biblioteca bioanalyzer el perfil (dilución al 1/5). (D) representante PCHi-C biblioteca bioanalyzer el perfil (sin dilución). Haga clic aquí para ver una versión más grande de esta figura.
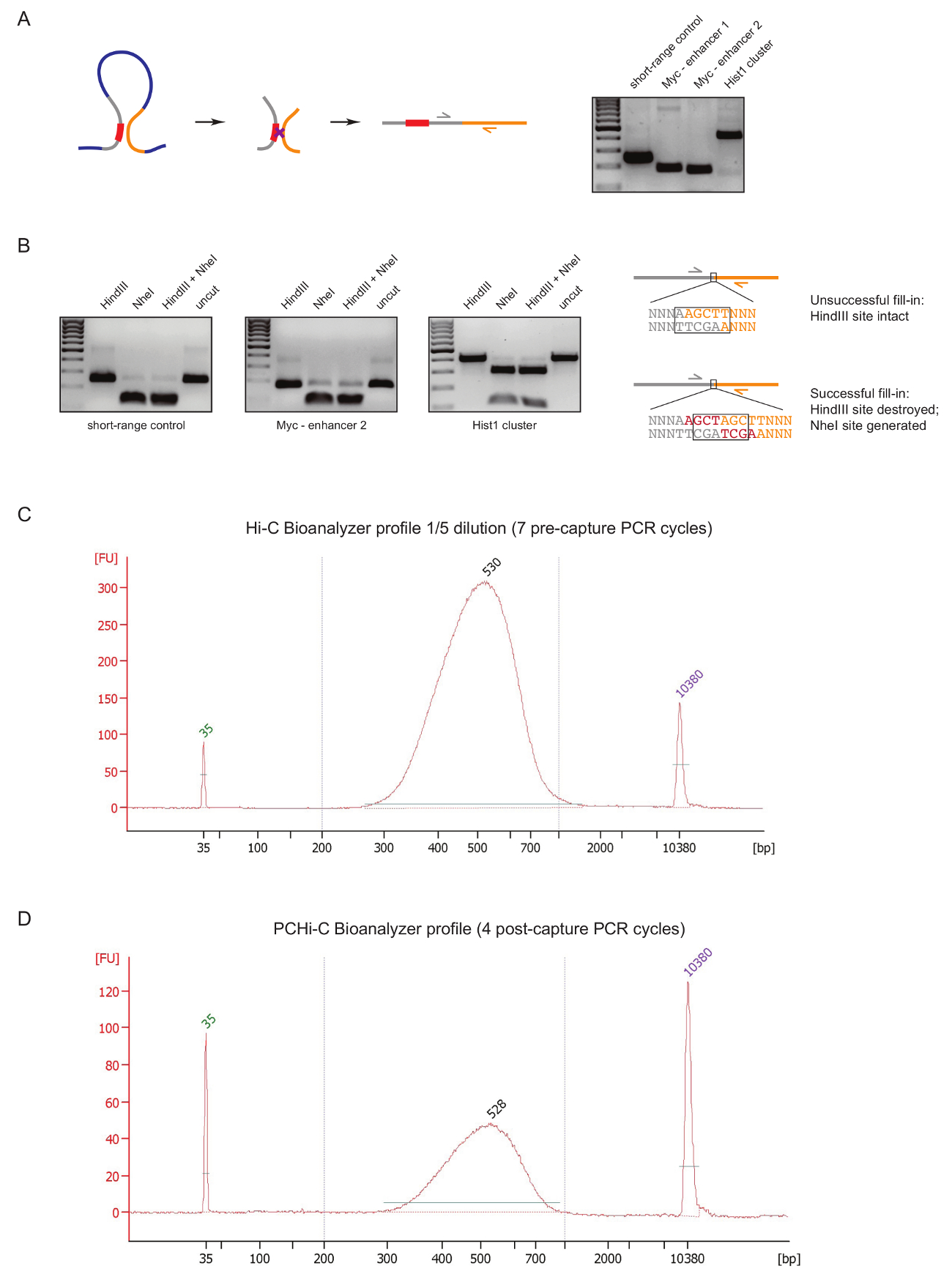{kind=link}

Figura 3 : Controles de calidad de la secuencias de PCHi-C. (A) comparación de la secuencia válida porcentaje Lee pares después HiCUP43 en PCHi-C vs correspondientes bibliotecas Hi-C (datos de Javierre et al., 201635). (B) representante HiCUP PCHi-C resultado mostrando válido Lee pares y otras categorías de secuencia que son desechados antes de análisis aguas abajo (datos de Javierre et al., 201635). (C) comparación de porcentaje 'mismo fragmento interno' Lee después HiCUP en PCHi-C vs correspondientes bibliotecas Hi-C (datos de Javierre et al., 201635). (D) comparación de la secuencia de porcentaje Lee con fragmentos del promotor con cebo (eficiencia de captura) en PCHi-C vs correspondientes bibliotecas Hi-C (datos de Javierre et al., 201635). Haga clic aquí para ver una versión más grande de esta figura.
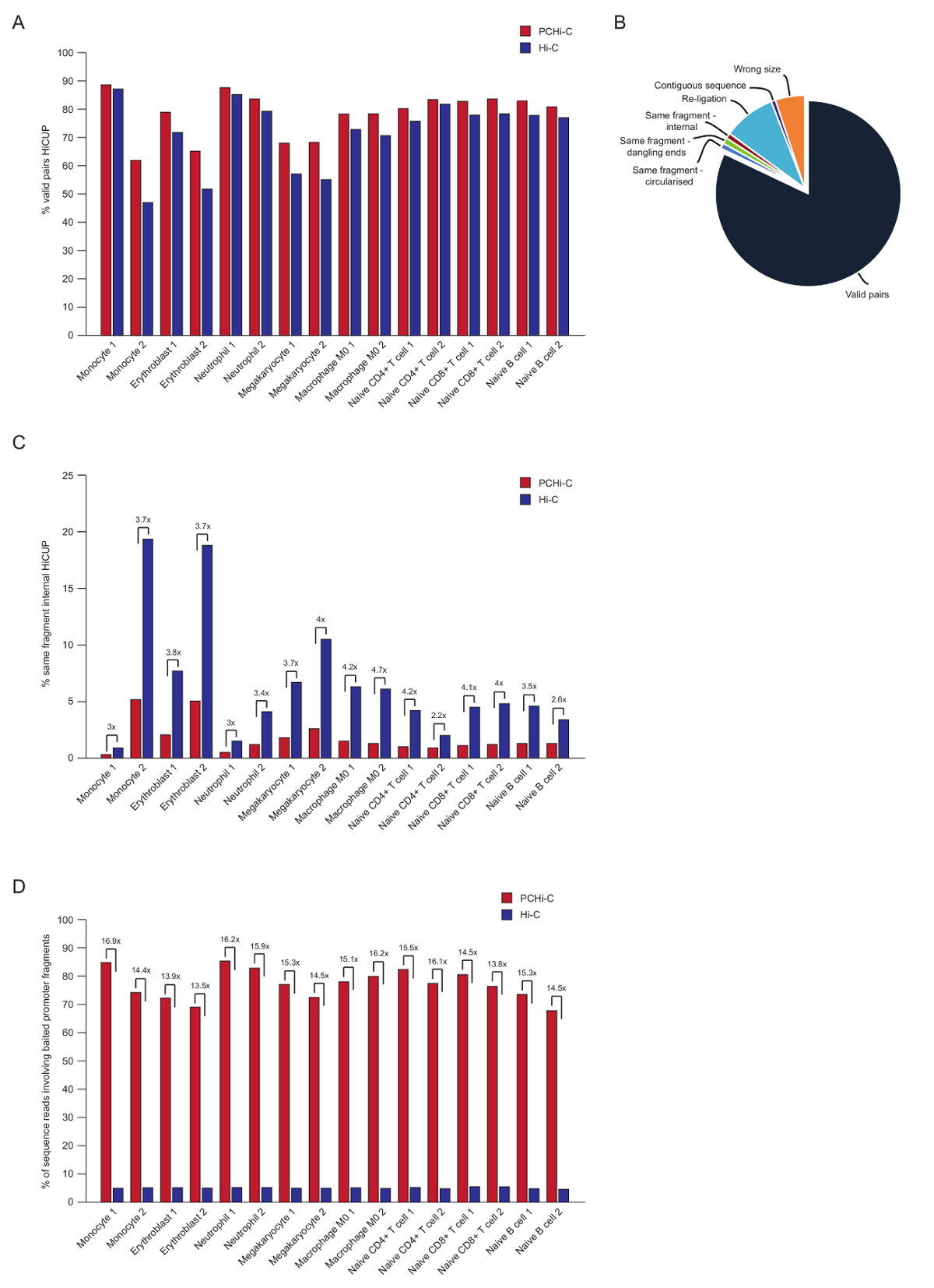{kind=link}

Figura 4: ALAD PCHi-C perfil en células hematopoyéticas humanas. Interacciones promotor de tipos de la célula mieloide se muestran como arcos azules, y las interacciones promotor de tipos de células linfoides se muestran como arcos púrpura. Las interacciones específicas del erythroblast se indican con flechas rojas (datos de Javierre et al., 201635). Haga clic aquí para ver una versión más grande de esta figura.
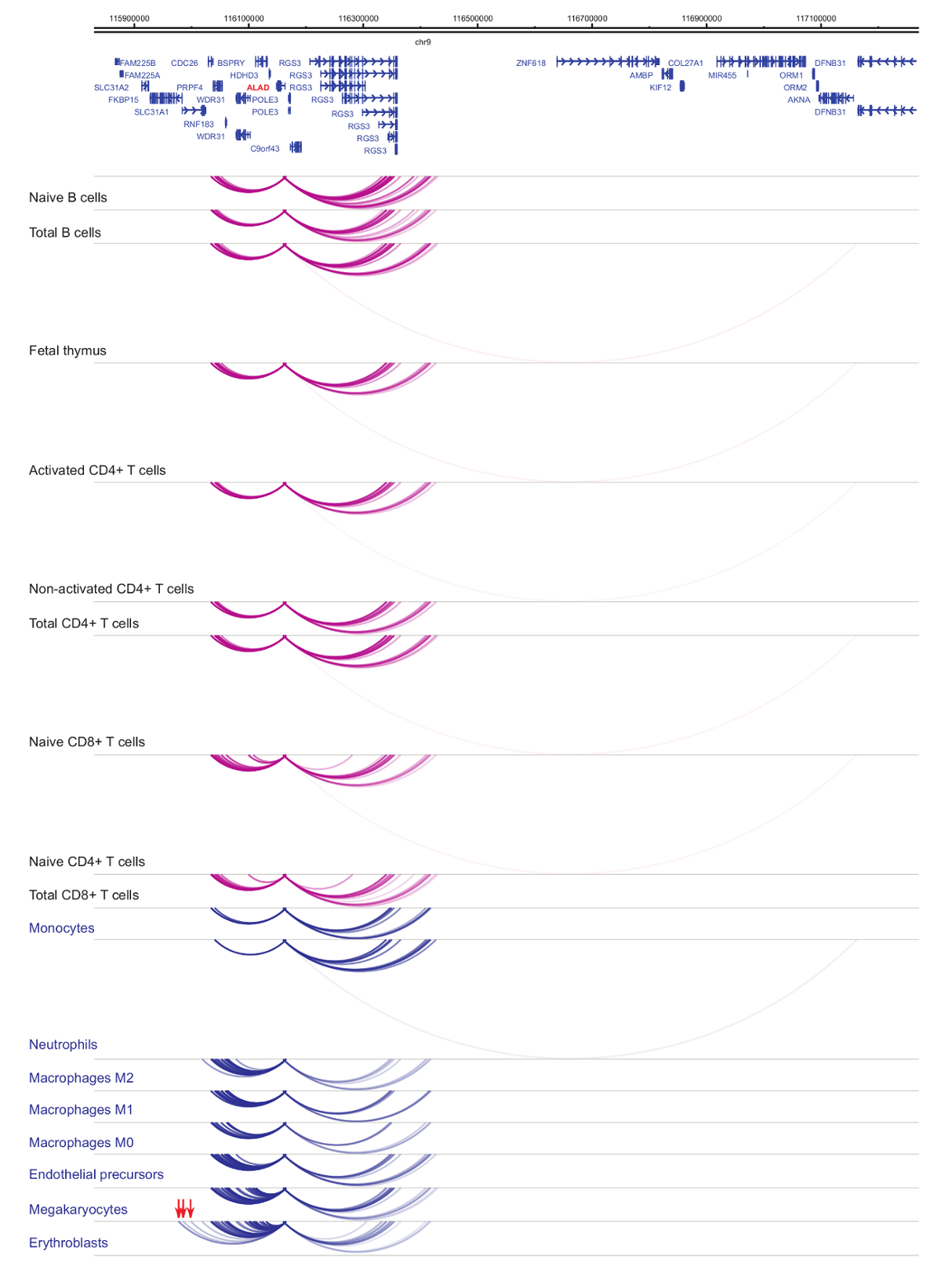{kind=link}

Figura 5 : IL8 Perfil de PCHi-C en las células hematopoyéticas humanas. Interacciones promotor de tipos de la célula mieloide se muestran como arcos azules, y las interacciones promotor de tipos de células linfoides se muestran como arcos púrpura. Interacciones específicas de monocitos están indicadas por las flechas verdes, interacciones neutrófilos específicos se indican con flechas rojas, y una interacción específica de megacariocitos está indicada por una flecha marrón (datos de Javierre et al., 201635). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Humano | ||||||||
| Nombre | Secuencia de | Cromosoma | Strand | Inicio GRCh38/hg38 | Final GRCh38/hg38 | Combinaciones de cartilla para probar las interacciones de 3C y biotina incorporación | ||
| Capítulo AHF64 Dekker | GCATGCATTAGCCTCTGCTGTTCTCTGAAATC | 11 | + | 116803960 | 116803991 | utilizar en combinación con hs AHF66 Dekker | ||
| Capítulo AHF66 Dekker | CTGTCCAAGTACATTCCTGTTCACAAACCC | 11 | + | 116810219 | 116810248 | utilizar en combinación con hs AHF64 Dekker | ||
| lugar geométrico MYC HS | GGAGAACCGGTAATGGCAAA | 8 | - | 127733814 | 127733833 | usar en combinación con hs MYC +1820 o capítulo MYC-538 | ||
| Capítulo MYC +1820 | AAAATGCCCATTTCCTTCTCC | 8 | + | 129554527 | 129554547 | usar en combinación con el locus MYC de capítulo | ||
| Capítulo MYC-538 | TGCCTGATGGATAGTGCTTTC | 8 | - | 127195696 | 127195716 | usar en combinación con el locus MYC de capítulo | ||
| Capítulo HIST1 F | AAGCAGGAAAAGGCATAGCA | 6 | + | 26207174 | 26207193 | utilizar en combinación con hs HIST1 R | ||
| Capítulo HIST1 R | TCTTGGGTTGTGGGACTTTC | 6 | + | 27771575 | 27771594 | utilizar en combinación con hs HIST1 F | ||
| Ratón | ||||||||
| Secuencia de | Cromosoma | Strand | Inicio GRCm38/mm10 | Final GRCm38/mm10 | Combinaciones de cartilla para probar las interacciones de 3C y biotina incorporación | |||
| TCATGAGTTCCCCACATCTTTG | 8 | + | 84841090 | 84841111 | usar en combinación con mm Calr2 | |||
| CTGTGGGCACCAGATGTGTAAAT | 8 | + | 84848519 | 84848541 | usar en combinación con mm Calr1 | |||
| TATCAAGGGTGCCCGTCACCTTCAGC | 6 | + | 125163098 | 125163123 | usar en combinación con Gapdh4 Dekker | |||
| GGGCTTTTATAGCACGGTTATAAAGT | 6 | + | 125163774 | 125163799 | usar en combinación con Gapdh3 Dekker | |||
| GGAGGAGGGAAAAGGAGTGATT | 6 | + | 52212829 | 52212850 | usar en combinación con mm Hoxa13 | |||
| CAGGCATTATTTGCTGAGAACG | 6 | - | 52253490 | 52253511 | usar en combinación con mm Hoxa7 | |||
| GGGTAATGGTGTCACTAACTGG | 13 | + | 23571284 | 23571305 | usar en combinación con m Hist1h3e o m Hist1h4i | |||
| GGGTTTGATGAGTTGGTGAAG | 13 | + | 23566541 | 23566561 | usar en combinación con mm Hist1h2ae | |||
| TTGGGCCAAAGCCTATATGA | 13 | + | 22043085 | 22043104 | usar en combinación con mm Hist1h2ae | |||
Tabla 1: Secuencias de Primer control de calidad de humano y ratón Hi-C bibliotecas.
Discusión
Diseño modular del promotor capturar Hi-C
Promotor de captura Hi-C está diseñado para específicamente enriquecer bibliotecas Hi-C para interacciones con promotores. Estas interacciones constituyen sólo un subconjunto de productos de ligadura en una biblioteca de C Hi.
Captura de Hi-C puede modificarse fácilmente para enriquecer bibliotecas Hi-C para cualquier región genómica o regiones de interés por cambiar el sistema de captura. Captura de regiones pueden ser continua de segmentos genomic44,45,46,48, potenciadores que se han identificado en PCHi-C (' inversa capturar Hi-C'35), o DNasa y sitios hipersensibles49 . El tamaño del sistema de captura puede ajustarse dependiendo del ámbito experimental. Por ejemplo, Dryden et al. blanco 519 fragmentos de cebo en tres desiertos de genes asociados con cáncer de mama44. El sistema de captura por Martin et al. objetivos de ambos segmentos genomic continuo ('Captura de región': 211 regiones genómicas en total; 2.131 fragmentos de restricción) y promotores (3.857 promotores de genes)45.
SureSelect bibliotecas están disponibles en diferentes tamaños: 1 kb a 499 kb (5.190-4.806), 500 kb a 2,9 Mb (5.190-4.816) y 3 Mb a Mb 5,9 (5.190-4.831). Como cada captura individual biotina-RNA 120 nucleótidos de largo, estos capturan sistemas de acomodar a un máximo de 4.158, 24.166 y 49.166 individual captura las puntas de prueba, respectivamente. Esto corresponde a 2.079, 12.083 y fragmentos de restricción específicas 24.583, respectivamente (nota que los números de fragmentos de restricción son límites inferiores basadas en el supuesto que dos sondas de captura individual pueden ser diseñadas para cada restricción fragmento — en realidad debido a secuencias repetitivas esto no será el caso para cada restricción del fragmento (véase también figura 1B, C), resultando en un mayor número de fragmentos de restricción targetable para un número constante de sondas de captura disponible ).
El protocolo aquí descrito se basa en el uso de una enzima de restricción con un sitio de reconocimiento de bp 6 para descubrir las interacciones de largo alcance. Usando una enzima de restricción con un sitio de reconocimiento de bp 4 para una mayor resolución de interacciones más proximales es también posible40,49.
Limitaciones de PCHi-C
Una limitación inherente de todo cromosoma conformación captura de ensayos es que su resolución está determinada por la enzima de restricción utilizada para la generación de la biblioteca. Las interacciones que se producen entre elementos de ADN situados en el mismo fragmento de restricción son invisibles para los ensayos de 'Tipo C'. Además, en PCHi-C, en algunos casos más de un sitio de inicio de transcripción puede encontrarse en el mismo fragmento de restricción que contiene el promotor y PIRs en algunos casos albergan ambas marcas histona activo y represivo, lo que es difícil de identificar que regulador elementos median las interacciones y para predecir la salida regulador de las interacciones del promotor. Usando enzimas de restricción con 4 sitios de reconocimiento de bp atenúa este problema pero viene a expensas de mucho mayor complejidad de biblioteca Hi-C (Hi-C bibliotecas generadas con enzimas de restricción de sitio de reconocimiento 4 de bp son al menos 100 veces más complejas que Hi-C bibliotecas generadas con enzimas de restricción de sitio de reconocimiento 6 de bp) y los costos asociados para la siguiente secuencia de generación.
Otra limitación es que el actual protocolo de PCHi-C requiere de millones de células como material, que el análisis de las interacciones de promotor en tipos raros de la célula de partida. Una versión modificada de PCHi-C para permitir que el interrogatorio de los contactos de promotor en poblaciones celulares con 10.000 a 100.000 células (por ejemplo durante el desarrollo embrionario o células madre hematopoyéticas) por lo tanto sería una adición valiosa a la captura Hola C caja de herramientas.
Por último, como todos los métodos que se basan en la fijación de formaldehído, PCHi-C sólo registra las interacciones que se 'congelan' en el momento de la fijación. Por lo tanto, para estudiar la cinética y dinámica de las interacciones del promotor, se requieren métodos como la microscopía de superresolución vivo de la célula junto con PCHi-C.
Métodos para disecar la organización espacial del cromosoma en alta resolución
La gran complejidad de las bibliotecas cromosómicas interacción prohíbe la identificación confiable de los productos de la interacción entre dos fragmentos de restricción específica con significación estadística. Para evitar este problema, captura de secuencia se ha utilizado para enriquecer Hi-C33,34,40,44 o50,de 3 C51 bibliotecas de interacciones específicas. La ventaja principal de usar bibliotecas de bibliotecas sobre 3C Hi-C para el paso de enriquecimiento es que Hi-C, a diferencia de C 3, incluye un paso de enriquecimiento de productos genuinos de la ligadura. Como consecuencia, el porcentaje de lecturas válidas en bibliotecas PCHi-C es aproximadamente 10 veces mayor que en captura-C Lee bibliotecas50, que contenía alrededor de 5 – 8% válido después de filtrar HiCUP. Sahlen et al han comparado directamente captura-C a HiCap, que como PCHi-C utiliza bibliotecas Hi-C para el enriquecimiento de la captura, en contraste con C de captura que utiliza bibliotecas C 3. De acuerdo con nuestros resultados, encontraron que las bibliotecas de captura de C se componen principalmente de fragmentos ligados no40. Además, las bibliotecas HiCap tenían una complejidad mayor de las bibliotecas de captura de C40.
Una variante de captura-C, llamado generación captura C52 NG captura-C utiliza un oligo por final de fragmento de restricción, como previamente establecidas en el PCHi-C33,34, en lugar de la superposición de las sondas utilizadas en el original Protocolo de captura de C50. Esto aumenta el porcentaje de lecturas válidas en comparación con C captura modestamente, pero NG captura-C emplea dos ciclos secuenciales de captura de enriquecimiento, y un número relativamente elevado de PCR ciclos (ciclos de 20 a 24 en total, en comparación con 11 ciclos típicamente PCHi-c), que inevitablemente resulta en un número de secuencia duplicados y menor complejidad de la biblioteca. En experimentos de prueba durante la optimización de PCHi-C, encontramos que el porcentaje de único (es decir, no duplicados) lee pares era solamente alrededor 15% cuando se utilizaron 19 ciclos PCR (13 ciclos de pre-capturan + 6 ciclos post capturan; datos no mostrados), sin embargo optimización a un menor número de ciclos de PCR, típicamente rinde 75 – 90% únicos pares de leer. Por lo tanto, reduce sustancialmente el número de ciclos PCR aumenta la cantidad de datos de la secuencia informativa.
Un método reciente combina ChIP con Hi-C para centrarse en las interacciones cromosómicas mediadas por una proteína específica de interés (HiChIP53). Comparado con el animal doméstico de ChIA54, que se basa en una lógica similar, HiChIP datos contienen un mayor número de lecturas de secuencia informativa, permitiendo mayor confianza interacción llamando a53. Sería muy interesante comparar directamente la correspondiente HiChIP y conjuntos de datos de captura Hi-C una vez que estén disponibles (por ejemplo HiChIP usando un anticuerpo contra la cohesina unidad Smc1a53 con captura de Hi-C para todos Smc1a obligado restricción fragmentos) lado a lado. Una diferencia inherente entre estos dos enfoques es que captura Hi-C no depende de inmunoprecipitación de cromatina y por lo tanto es capaz de interrogar a interacciones cromosómicas independientemente de la ocupación de la proteína. Esto permite la comparación de la organización del genoma 3D en la presencia o ausencia de atascamiento del factor específico, como se ha utilizado para identificar PRC1 como un regulador clave del ratón ESC genoma espacial arquitectura7.
PCHi-C y GWAS
Estudios de Asociación de genoma completo (GWAS) han revelado que más del 95% de enfermedad asociada a variantes de la secuencia se encuentran en regiones no codificantes del genoma, a menudo a grandes distancias para genes de codificación de la proteína55. Suelen ser variantes GWAS encontró en proximidad cercana a DNasa I sitios hipersensibles, que es una seña de identidad de secuencias con potencial actividad reguladora. PCHi-C y capturar Hi-C se han utilizado extensivamente para vincular promotores a loci de riesgo GWAS implicados en cáncer de mama44, cáncer colorrectal48y enfermedad autoinmune35,45,46. Un PCHi-C estudio de 17 células hematopoyéticas humano diferentes tipos encontrar SNPs asociados con enfermedad autoinmune se enriquecieron en el PIRs en células linfoides, mientras que variantes de la secuencia asociadas a rasgos específicos de plaquetas y glóbulos rojos se encontraron predominantemente en los macrófagos y eritroblastos, respectivamente35,56. Así, promotor específico tipo de tejido interactomes por PCHi-C puede ayudar a entender la función de no codificación asociada a enfermedad de variantes de secuencia e identificar nuevos genes de enfermedades potenciales para la intervención terapéutica.
Características de las regiones de interacción promotor
Varias líneas de evidencia enlace promotor interactomes al control de la expresión de genes. En primer lugar, varios estudios de PCHi-C han demostrado que regiones genómicas interactuando con los promotores de genes expresados (altamente) se enriquecen en marcas asociadas con actividad enhancer, como la acetilación de H3K27 y p300 enlace33,34 , 37. se encontró una correlación positiva entre el nivel de expresión génica y el número de reforzadores interactuantes, lo que sugiere que los efectos aditivos de resultado potenciadores en la expresión génica aumento niveles34,35. En segundo lugar, naturales loci de rasgos cuantitativos (eQTLs) están enriquecidos en PIRs que están conectados a los mismos genes cuya expresión se ve afectada por los eQTLs35de expresión. En tercer lugar, mediante la integración de viaje57 y PCHi-C datos, Cairns et al. encontrado que genes del reportero viaje a PIRs en ratón CES muestran a reportero más fuerte expresión génica de genes del reportero en los sitios de integración en las regiones de interacción de promotor no 58, lo que indica que los PIRs poseen actividad de regulación transcripcional. Juntos, estos resultados sugieren que promotor interactomes descubiertas por PCHi-C en varios tipos de células humanas y ratón incluyen módulos regulatorios claves para el control de la expresión de genes.
Cabe destacar que los Potenciadores representan sólo una pequeña fracción (~ 20%) de los PIRs por PCHi-C33,34. Otros PIRs podrían tener funciones estructurales o topológicas en lugar de funciones de regulación transcripcionales directas. Sin embargo, también hay evidencia que PCHi-C puede descubrir elementos de ADN con función reguladora que no albergan marcas clásica potenciador. En una línea de células linfoides humanas, el promotor BRD7 fue encontrado para interactuar con una región desprovista de marcas potenciador que fue demostrada para poseer actividad potenciador en reportero gene ensayos33. Elementos reguladores de similares características pueden ser más abundantes que actualmente apreciado. Por ejemplo, una pantalla basada en CRISPR para regulación ADN elementos identificados sin marcar elementos reguladores (UREs) que controlan la expresión génica pero están desprovistos de reforzador marca59.
En otros casos, han demostrado PIRs puerto marcas cromatina asociadas con la represión transcripcional. PIRs y promotores interactúan por PRC1 en ratón CES participaron en una extensa red espacial de genes reprimidos teniendo que el represivo Marcos H3K27me37. En células linfoblastoides humanas, un elemento distante interactuando con el promotor de BCL6 reprimido transgen reportero gene expresión33, sugiriendo que puede funcionar para reprimir la transcripción de BCL6 en su contexto nativo.
PIRs enriquecidas para la ocupación de la proteína del aislador de cromatina CTCF en CES y CNE humano37 puede representar otra clase de PIRs. Colectivamente, estos resultados sugieren que PIRs albergan una colección de actividades reglamentarias gene a caracterizarse funcionalmente.
Divulgaciones
Autores no tienen nada que revelar.
Agradecimientos
Agradecemos Valeriya Malysheva lectura crítica del manuscrito y ayuda de un experto con la figura 1. Este trabajo fue apoyado por el Consejo de investigación médica, Reino Unido (Señor/L007150/1) y el Reino Unido biotecnología y Consejo de investigación de ciencias biológicas, Reino Unido (BB/J004480/1).
Materiales
| Name | Company | Catalog Number | Comments |
| 16% (vol/vol) paraformaldehyde solution | Agar Scientific | R1026 | |
| Dulbecco's Modified Eagle Medium (DMEM) 1x | Life Technologies | 41965-039 | |
| Fetal bovine serum (FBS) sterile filtered | Sigma | F9665 | |
| Low-retention filter tips | Starlab | S1180-3810, S1180-1810, S1180-8810 and S1182-1830 | |
| 10x PBS pH 7.4 | Life Technologies | 70011-036 | |
| Molecular biology grade water | Sigma-Aldrich | W4502 | |
| 1 M Tris-HCl pH 8.0 | Life Technologies | 15568-025 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 5 M NaCl | Life Technologies | 24740-011 | |
| Protease inhibitor cocktail (EDTA-free) | Roche Diagnostics | 11873580001 | |
| Restriction buffer 2 (10x NEBuffer 2) | New England Biolabs | B7002 | |
| DNA LoBind tube, 1.5 mL | Eppendorf | 0030 108.051 | |
| DNA LoBind tube, 2 mL | Eppendorf | 30108078 | |
| 20% (wt/vol) SDS | Bio-Rad Laboratories | 161-0418 | |
| 20% (vol/vol) Triton X-100 | Sigma-Aldrich | T8787 | |
| HindIII, 100 U/uL | New England Biolabs | R0104 | |
| 10 mM dCTP | Life Technologies | 18253-013 | |
| 10 mM dGTP | Life Technologies | 18254-011 | |
| 10 mM dTTP | Life Technologies | 18255-018 | |
| 0.4 mM Biotin-14-dATP | Life Technologies | 19524-016 | |
| DNA polymerase I large (Klenow) fragment 5000 units/mL | New England Biolabs | M0210 | |
| 10x T4 DNA ligase reaction buffer | New England Biolabs | B0202 | |
| 100x 10mg/ml Bovine Serum Albumin | New England Biolabs | B9001 | |
| T4 DNA ligase, 1 U/μL | Invitrogen | 15224-025 | |
| RNase A | Roche | 10109142001 | |
| Proteinase K, recombinant, PCR grade | Roche | 3115836001 | |
| 20 000×g 50 ml centrifuge tube | VWR | 525-0156 | |
| 0.5 M EDTA pH 8.0 | Life Technologies | 15575-020 | |
| Phenol pH 8.0 | Sigma | P4557 | |
| Phenol: Chloroform: Isoamyl Alcohol 25:24:1 | Sigma | P3803 | |
| Sodium acetate pH 5.2 | Sigma | S7899 | |
| Quant-iT PicoGreen | Invitrogen | P7589 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| Restriction buffer 2.1 (10x NEBuffer 2.1) | New England Biolabs | B7202 | |
| NheI, 100U/uL | New England Biolabs | R0131 | |
| Micro TUBE AFA Fiber Pre-slit snap cap 6x16mm vials | Covaris | 520045 | For sonication |
| SPRI beads (Agencourt AMPure XP) | Beckman Coulter | A63881 | |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| Tween 20 | Sigma | P9416 | |
| 10 mM dATP | Life Technologies | 18252-015 | |
| T4 DNA polymerase 3000 units/mL | New England Biolabs | M0203 | |
| T4 PNK 10000 units/mL | New England Biolabs | M0201 | |
| Klenow exo minus 5000 units/mL | New England Biolabs | M0212 | |
| Quick ligation reaction buffer | New England Biolabs | B6058 | |
| NEB DNA Quick ligase | New England Biolabs | M2200 | |
| PE adapter 1.0 (5'-P-GATCGGAAGAGCGGTTCAGC AGGAATGCCGAG-3') | Illumina | ||
| PE adapter 2.0 (5'-ACACTCTTTCCCTACACGACGCT CTTCCGATCT-3') | Illumina | ||
| NEB Phusion PCR kit | New England Biolabs | M0530 | |
| PE PCR primer 1.0 (5'-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTAC ACGACGCTCTTCCGATCT-3') | Illumina | ||
| PE PCR primer 2.0 (5'-CAAGCAGAAGACGGCATACGA GATCGGTCTCGGCATTCCT GCTGAACCGCTCTTCCGATCT-3') | Illumina | ||
| PCR strips | Agilent Technologies | 410022 and 401425 | |
| SureSelect SSEL TE Reagent ILM PE full adaptor kit | Agilent Technologies | 931108 | |
| SureSelect custom 3-5.9 Mb library | Agilent Technologies | 5190-4831 | custom design mouse or human PCHi-C system |
| Dynabeads MyOne Streptavidin T1 beads | Invitrogen | 65601 | |
| E220 high-performance focused ultra-sonicator | Corvaris | E220 |
Referencias
- Osborne, C. S., et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nature Genetics. 36, 1065-1071 (2004).
- Schoenfelder, S., et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature Genetics. 42, 53-61 (2010).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501, 227-231 (2013).
- Bantignies, F., et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 144, 214-226 (2011).
- Engreitz, J. M., et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 341, 1237973(2013).
- Denholtz, M., et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 13, 602-616 (2013).
- Schoenfelder, S., et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics. 47, 1179-1186 (2015).
- Kundu, S., et al. Polycomb Repressive Complex 1 generates discrete compacted domains that change during differentiation. Molecular Cell. 65, 432-446 (2017).
- Skok, J. A., Gisler, R., Novatchkova, M., Farmer, D., de Laat, W., Busslinger, M. Reversible contraction by looping of the Tcra and Tcrb loci in rearranging thymocytes. Nature Immunology. 8, 378-387 (2007).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148, 908-921 (2012).
- Aymard, F., et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology. 24, 353-361 (2017).
- Ryba, T., et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research. 20, 761-770 (2010).
- Pope, B. D., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 515, 402-405 (2014).
- Chandra, T., et al. Global reorganization of the nuclear landscape in senescent cells. Cell Reports. 10, 471-483 (2015).
- Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., Fraser, P. Long-range chromatin regulatory interactions in vivo. Nature Genetics. 32, 623-626 (2002).
- Tolhuis, B., Palstra, R. J., Splinter, E., Grosveld, F., de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Molecular Cell. 10, 1453-1465 (2002).
- Amano, T., Sagai, T., Tanabe, H., Mizushina, Y., Nakazawa, H., Shiroishi, T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Developmental Cell. 16, 47-57 (2009).
- Zuniga, A., et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes & Development. 18, 1553-1564 (2004).
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132, 797-803 (2005).
- D'Haene, B., et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 5, e1000522(2009).
- Sur, I. K., et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 338, 1360-1363 (2012).
- Herranz, D., et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature Medicine. 20, 1130-1137 (2014).
- Deng, W., et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 149, 1233-1244 (2012).
- Groschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157, 369-381 (2014).
- Lupianez, D. G., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 161, 1012-1025 (2015).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538, 265-269 (2016).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295, 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38, 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genetics. 38, 1341-1347 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research. 16, 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289-293 (2009).
- Belton, J. M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., Dekker, J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 58, 268-276 (2012).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47, 598-606 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582-597 (2015).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167, 1369-1384 (2016).
- Wilson, N. K., et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 127, e12-e23 (2016).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife. 6, (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49, 1522-1528 (2017).
- Siersbaek, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66, 420-435 (2017).
- Sahlen, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Nagano, T., et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64 (2013).
- Nagano, T., Varnai, C., Schoenfelder, S., Javierre, B. M., Wingett, S. W., Fraser, P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology. 16, 175(2015).
- Wingett, S., et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Res. 4, 1310(2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24, 1854-1868 (2014).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- McGovern, A., et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol.ogy. 17, 212(2016).
- Hodge, D., et al. A global role for EKLF in definitive and primitive erythropoiesis. Blood. 107, 3359-3370 (2006).
- Jager, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter Interactions between two states of pluripotency. Cell Stem Cell. 17, 748-757 (2015).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46, 205-212 (2014).
- Kolovos, P., et al. Targeted Chromatin Capture (T2C): A novel high-resolution high-throughput method to detect genomic interactions and regulatory elements. Epigenetics Chromatin. 7, 10(2014).
- Davies, J. O., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13, 74-80 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13, 919-922 (2016).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462, 58-64 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337, 1190-1195 (2012).
- Petersen, R., et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat. Commun. 8, 16058(2017).
- Akhtar, W., et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 154, 914-927 (2013).
- Cairns, J., et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biology. 17, 127(2016).
- Rajagopal, N., et al. High-throughput mapping of regulatory DNA. Nature Biotechnology. 34, 167-174 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados