
Method Article
Promotore cattura Ciao-c: Ad alta risoluzione, Genome-wide profilatura delle interazioni del promotore
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Elementi regolatori del DNA, come rinforzatori, controllano l'espressione genica contattando fisicamente i promotori di geni bersaglio, spesso attraverso interazioni a lungo raggio cromosomiche, che coprono grandi distanze genomiche. Promotore Capture Hi-C (PCHi-C) identifica interazioni significative tra promotori e regioni distali, che consente l'assegnazione di potenziali sequenze regolatrici a loro geni bersaglio.
Abstract
L'organizzazione tridimensionale del genoma è legata alla sua funzione. Ad esempio, elementi regolatori come rinforzatori trascrizionali controllano l'espressione spazio-temporale dei loro geni bersaglio attraverso il contatto fisico, spesso colmare distanze genomiche considerevole (in alcuni casi centinaia di kilobasi) e bypassando geni vicini. Il genoma umano porti un stimato rinforzatori 1 milione, la maggior parte dei quali hanno sconosciuto geni bersaglio. Assegnazione regioni regolative distale ai loro geni bersaglio è dunque fondamentale per comprendere il controllo di espressione genica. Abbiamo sviluppato promotore Capture Hi-C (PCHi-C) per abilitare il rilevamento del genoma di regioni promotore-interacting distali (PIRs), per tutti i promotori in un singolo esperimento. In PCHi-C, altamente complesso Hi-C biblioteche specificamente sono arricchiti per sequenze promotrici attraverso selezione in soluzione ibrida con migliaia di esche biotinilati RNA complementari alle estremità dei frammenti di restrizione promotore-contenenti tutti. L'obiettivo è quello di sequenze promotrici poi discesa e i loro partner di frequente interazione come esaltatori e altri potenziali elementi regolatori. Dopo la sequenziazione di alto-rendimento accoppiato-fine, viene applicato un test statistico per ogni frammento di restrizione promotore-legati per identificare PIRs significativi a livello di frammento di restrizione. Abbiamo usato PCHi-C per generare un Atlante delle interazioni a lungo raggio promotore in decine di umani e tipi di cellule del mouse. Queste mappe Interactoma promotore hanno contribuito ad una maggiore comprensione del controllo di espressione genica dei mammiferi assegnando putative regioni regolative ai loro geni bersaglio e rivelando reti di interazione spaziale preferenziale promotore-promotore. Questa informazione ha anche alta rilevanza per la comprensione di malattie genetiche umane e l'identificazione di potenziali geni-malattia, collegando malattia-collegati non codificanti varianti a o vicino a sequenze di controllo a loro geni bersaglio di sequenza.
Introduzione
Raccogliendo la prova suggerisce che l'organizzazione tridimensionale del genoma svolge un importante ruolo funzionale in una gamma di processi nucleari, tra cui gene attivazione1,2,3, repressione4 ,5,6,7,8, ricombinazione9,10, DNA riparazione11, DNA replica12,13, e senescenza cellulare14. Esaltatori di distanti si trovano in prossimità spaziale ai promotori che regolano15,16,17, che è essenziale per il controllo di espressione genica spazio-temporale adeguato. Delezioni Enhancer mostrano che rinforzatori distali sono essenziali per destinazione gene trascrizione18,19,20,21,22e 'costretto cromatina loop' dimostra che derivati dal tethering tra un enhancer e relativo promotore di destinazione il locus Hbb è sufficiente per guidare l'attivazione trascrizionale23. Ulteriormente, le riorganizzazioni di genoma che portano geni sotto il controllo di esaltatori di ectopici possono causare l'attivazione del gene inappropriati e malattia24,25,26. Insieme, questi esempi illustrano che interazioni promotore-enhancer sono essenziali per il controllo del gene e richiedono regolazione stretta affinché l'espressione genica appropriato. L'essere umano e genomi del mouse sono stimati ciascuno per harbor esaltatori di circa 1 milione. Per la stragrande maggioranza di questi rinforzatori, geni bersaglio sono sconosciuti, e le regole di ingaggio tra promotori ed esaltatori di sapidità sono capite male. Assegnazione di esaltatori di transcriptional ai loro geni bersaglio così rimane una grande sfida nel decifrare il controllo di espressione genica dei mammiferi.
La nostra comprensione dell'architettura tridimensionale del genoma è stato rivoluzionato con l'introduzione di 3C27 (cattura di conformazione del cromosoma) e sue varianti28,29,30,31 . Il più potente di queste tecniche, Hi-C (cattura di conformazione di throughput elevato del cromosoma) è progettato per identificare l'intero ensemble di cromosomiche interazioni all'interno di una popolazione delle cellule. Hi-C biblioteche, tipicamente generati da milioni di cellule, sono molto complessi con un prodotti di legatura indipendente11 10 stimato tra frammenti di ~ 4 kb nel genoma umano32. Come una conseguenza, affidabile e riproducibile identificazione delle interazioni tra restrizione singoli frammenti (come quelle che contengono un promotore o enhancer) da Hi-C dati non sono fattibili, a meno che le librerie Hi-C sono sottoposti a sequenziamento ultra-profondo, che non è una soluzione economicamente conveniente per i laboratori di preparazione librerie Hi-C ordinariamente. Per ovviare a questo inconveniente, abbiamo sviluppato il promotore Capture Hi-C per arricchire in modo specifico promotore-contenente i prodotti di legatura da librerie Hi-C. Ci siamo concentrati sui promotori per due motivi. In primo luogo, contatti promotore-enhancer sono stati indicati per essere cruciale per i livelli di espressione di gene corretto nei numerosi studi (vedi riferimenti sopra), e in secondo luogo, come i promotori sono in gran parte invarianti tra tipi di cellule, lo stesso sistema di cattura dell'esca può essere utilizzato per interrogare il circuito di regolamentazione nei diversi tipi cellulari e condizioni. Il nostro approccio si basa sull'ibridazione nella soluzione delle librerie Hi-C con decine di migliaia di 120mers di biotinylated RNA complementare al promotore-contenente i prodotti di legatura Hi-C e successiva acquisizione su biglie magnetiche rivestite con streptavidina. Ciò si traduce in librerie PCHi-C con molto ridotta complessità rispetto alla libreria originale Hi-C, concentrandosi solo sull'identificazione di frammenti che sono legati ai promotori alle significativamente alte frequenze.
Abbiamo usato PCHi-C in un numero di umani e tipi di cellule del mouse di contribuire ad una migliore comprensione del controllo di espressione genica da scoprire regioni interagenti promotore distale a lungo raggio con funzione regolatrice putativo, come pure non casuale contatti di promotore-promotore nello spazio tridimensionale del nucleo. Gli studi hanno mappato centinaia di migliaia di contatti promotore-enhancer in tutto albergo numerose cellule tipi33,34,35,36,37,38, 39, identificato organizzazione genoma spaziale Polycomb Repressive Complex-mediata in cellule staminali embrionali di topo7, dimostrato ricablaggio su larga scala del promotore Interattomi durante la differenziazione cellulare37, 38 , 39e collegato non codificanti malattia-collegati di sequenza varianti a gene promotori35.
PCHi-C è un metodo ideale per mappare il genoma ensemble di sequenze di DNA che interagiscono con i promotori. Approcci correlati, ad esempio catturare Hi-C di regioni genomiche continue (Vedi discussione) sono il metodo di scelta per ottenere profili di interazione ad alta risoluzione per regioni genomiche selezionate. PCHi-C e catturare Hi-C sono estremamente simili da un punto di vista sperimentale (l'unica differenza è la scelta del sistema di cattura), così che i consigli e le linee guida che forniamo sono applicabili a entrambi gli approcci. Qui, presentiamo una descrizione dettagliata di PCHi-C. Delineare la spiegazione razionale e la progettazione di un esperimento di PCHi-C, forniamo un dettagliata PCHi-C protocollo di generazione in biblioteca e illustrare come la qualità delle librerie PCHi-C può essere monitorata a varie fasi nel protocollo per produrre dati di alta qualità.
Protocollo
1. formaldeide fissazione
-
Preparazione di cella: iniziare con un minimo di 2 x 107 cellule ogni esperimento.
- Per le cellule in coltura, risospendere le cellule in coltura. Per ex vivo di cellule, risospendere in 1 x di Dulbecco per volta Eagle Medium (DMEM), completati con 10% (vol/vol) siero bovino fetale (FBS).
- Per le celle aderenti, rimuovere il terreno di coltura e aggiungere 30,625 mL di terreno nuovo con 10% (vol/vol) FBS a temperatura ambiente (RT; 20 – 25 ° C).
- Per cellule in sospensione, raccogliere e centrifugare le cellule a 400 x g e a 20 ° C per 3 min. Remove supernatante e risospendere il pellet cellulare in 30,625 mL di terreno con 10% (vol/vol) FBS a TA.
- Per tessuti solidi, utilizzare tripsina (concentrazione finale 0.05% al 2,5%, a seconda del tipo di cellula) o lisata omogeneizzazione per ottenere una sospensione di singola cellula. Dopo questo ulteriore passo, trattare le cellule come cellule in sospensione.
-
Aggiungere mL 4,375 di 16% privo di metanolo paraformaldeide (fiala aperta appena prima dell'uso) ad una concentrazione finale di 2% (vol/vol). Difficoltà per 10 min a RT con miscelazione delicata su un agitatore meccanico.
Attenzione: Paraformaldeide è un prodotto chimico pericoloso. Seguire le norme di salute e sicurezza adeguate. - Placare la reazione aggiungendo 5 mL di preparata 1m ghiacciata glicina. Mescolare per 5 min con dolce dondolo a RT e poi incubare in ghiaccio per 15 minuti con inversione occasionale.
-
Lavare e raccogliere le cellule fisse.
- Per le celle aderenti, rimuovere il surnatante, aggiungere 10 mL di gelida 1X PBS pH 7.4 sulla parete piastra e rimuoverlo. Aggiungere 1 mL di gelida 1X PBS pH 7.4, raccogliere le cellule utilizzando un raschietto di cella e il trasferimento in una provetta da 50 mL. Ripetere l'operazione due volte per raccogliere il numero di celle possibili. Aggiungere PBS ghiacciata fino a 50 mL di volume finale.
- Per cellule in sospensione, centrifuga cellule a 760 x g e a 4 ° C per 5 min, rimuovere il surnatante e risospendere il pellet cellulare in 50 mL di PBS ghiacciata a pH 7,4.
- Centrifugare le cellule a 400 x g e a 4 ° C per 10 minuti e rimuovere con cautela il surnatante. Il pellet cellulare può essere snap congelati in azoto liquido e successivamente conservati a-80 ° C per diversi mesi.
2. lisi cellulare
- Pellet e risospendere in 50 mL di tampone di lisi ghiacciata preparata (10 mM Tris-HCl a pH 8, 0,2% (vol/vol) Igepal CA-630, 10 millimetri di NaCl e inibitore della proteasi una compressa cocktail) e mescolare. Incubare in ghiaccio per 30 minuti, ogni tanto mescolare capovolgendo. Centrifugare i nuclei a 760 x g e a 4 ° C per 5 minuti e rimuovere il surnatante.
3. hindIII digestione
- Lavare i nuclei delle cellule con buffer di restrizione 1,25 x 2. Risospendere il pellet cellulare in 1 mL di tampone di restrizione ghiacciata 1,25 x 2 e trasferimento in una provetta da 1,5 mL. Gira i nuclei a 760 x g e a 4 ° C per 5 minuti e rimuovere il surnatante.
- Risospendere il pellet cellulare in 1790 µ l di tampone di restrizione 1,25 x 2. Fare 5 aliquote, ciascuna contenente 5 milioni di cellule in µ l 358 di 1.25 x buffer di restrizione 2.
- Aggiungere 11 µ l di 10% (wt/vol) SDS all'aliquota e agitare a 950 giri / min (rpm) per 30 min a 37 ° C in un thermomixer. Se viene visualizzata la finestra di cella ciuffi, dissociare pipettando, evitando bolle.
- Aggiungere 75 µ l di 10% Triton X-100 (vol/vol) per aliquota e scossa a 950 giri/min e 37 ° C per 15 min in un thermomixer. Se viene visualizzata la finestra di cella ciuffi, dissociare pipettando, evitando bolle.
-
Aggiungere 12 µ l di 100 U / µ l HindIII 100 (1.200 unità in totale) per aliquota e incubare a 37 ° C durante la notte (O/N) mentre si stringono a 950 giri/min in un thermomixer.
- Per il controllo di digestione, 25 µ l di campione (5 µ l di ciascuna aliquota) in un nuovo tubo di trasferimento prima di aggiungere l'enzima (controllo non digerito) e ripetere la stessa procedura dopo aver aggiunto l'enzima (digerito control). Incubare entrambi i tubi nello stesso modo come la libreria Hi-C.
- La mattina seguente, aggiungere 5 µ l di 100 U / µ l HindIII (500 unità in totale) per aliquota e incubare a 37 ° C per 2 h mentre si stringono a 950 giri/min in un thermomixer.
-
Controllo di digestione: per i controlli non digeriti e digeriti (Vedi 3.5.1), eseguire crosslink inversione (passaggio 6), l'estrazione del fenolo: cloroformio e precipitazione del DNA (passaggio 7).
- Progettare un paio degli iniettori che si estendono su un sito di dIII Hin. Nella stessa regione, progettare un'altra coppia di primer che non includono un sito dIII Hin. Disegnare primers per PCR quantitativa (Q-PCR) utilizzando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) e i seguenti parametri:
Dimensione del primer: ottimale 20 (min.: 18, max.: 27); Primer Tm: Ottimale 60 (min: 57, max.: 63); % Di contenuto di primer CG: min.: 20, max.: 80; Dimensione Amplicone: RT-PCR ~ 100 bp (per PCR convenzionale ~ 300 bp); Mispriming biblioteca: umani (human primer) o semplice (primer del mouse) e del roditore. - Eseguire Q-PCR per ottenere 4 media Cts (ciclo soglia): Ct [D; H], ottenuti dal campione digerito [D] con la coppia di primer che si estendono su un sito di dIII Hin[H]; CT [D;-], ottenuta dal campione digerito [D] con la coppia di primer che non abbracciano un HindIII sito [--]; CT [U; H], ottenuti dal campione non digerito [U] con la coppia di primer che si estendono su un sito di dIII Hin; CT [U;-], ottenuta dal campione non digerito [U] con la coppia di primer che non abbracciano un HindIII sito [--]. Calcolare la percentuale di digestione come: digestione % = 100-100/2(Ct[D,H]-Ct[D,-]) - (Ct[U,H]-Ct[U,-]).
- Progettare un paio degli iniettori che si estendono su un sito di dIII Hin. Nella stessa regione, progettare un'altra coppia di primer che non includono un sito dIII Hin. Disegnare primers per PCR quantitativa (Q-PCR) utilizzando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) e i seguenti parametri:
4. Biotinylation delle sporgenze di frammento di restrizione
- Preparare mix master biotinylation: 30,6 µ l di tampone di restrizione 2, 10,2 µ l di H2O 10x (grado di biologia molecolare), 7.65 µ l di 10 millimetri di dCTP, 7.65 µ l di 10 millimetri di dGTP, 7.65 µ l di 10 millimetri di dTTP, 191.25 µ l di 0,4 mM biotina-14-dATP e 51 µ l di 5.000 U/mL DNA polimerasi I (grande Frammento di Klenow).
- Aggiungere 60 µ l di mix master biotinylation per aliquota, mix e incubare a 37 ° C per 1 h agitazione a 700 giri/min (thermomixer) per 5 s, ogni 30 s. Dopo 1 h, mettere le aliquote sul ghiaccio.
5. in-nucleo legatura
- Preparare il mix master legatura: 510 µ l di tampone di T4 DNA ligasi, 51 µ l di 10 mg/mL albumina di siero bovino 10x (100 x BSA), 1754.4 µ l di acqua (grado di biologia molecolare) e 127,5 µ l di 1 U / µ l T4 DNA ligasi (Vedi Tabella materiali).
- 479 µ l di mix master legatura al mix aliquotare e incubare a 16 ° C per 4 h agitazione a 700 rpm per 5 s ogni 2 min in un thermomixer.
- Incubare 30 minuti a TA.
6. Crosslink inversione
- Combinare tutte le aliquote in una provetta da centrifuga 50ml (adatta per centrifugazione ad alta velocità).
- 62,5 µ l di 10 mg/mL RNasi A, mix e incubare per 30 min a 37 ° C.
- Aggiungere 300 µ l di proteinasi K, mix, 10 mg/mL ed incubare per 30 min a 37 ° C.
- Incubare la reazione O/N (o almeno 4 ore) a 65 ° C. La mattina seguente, aggiungere 300 µ l di proteinasi K, mix, 10 mg/mL e incubare per 1 h a 65 ° C.
7. purificazione di DNA
- 4337.5 µ l di tampone TLE (10 mM Tris-HCl a pH 8.0; pH di 0,1 mM EDTA 8.0) e mescolare.
- Aggiungere 1 pH in volume (10ml) fenolo 8.0, vortexare per 10 s e centrifugare a RT e 20.000 x g per 3 min. trasferire 9 mL di superiore (acquosa) ad un nuovo tubo 50 mL.
Attenzione: Il fenolo è un prodotto chimico pericoloso. Seguire le norme di salute e sicurezza adeguate. - Aggiungere 2 mL di tampone TLE alla fase acquosa rimanente, vortice per 10 s e centrifugare a RT e 20.000 x g per 3 min. 2,5 mL della fase acquosa nel tubo di nuovo il trasferimento da passo 7.2, rendendo il volume finale 11,5 mL. Gettare la provetta contenente la fase inferiore (biologica).
- Aggiungere 1 volume (11,5 mL) di fenolo: cloroformio: alcool isoamilico (25:24:1), vortex per 10 s e centrifugare a RT e 20.000 x g per 3 min trasferimento 11 mL della fase superiore (acquosa) ad un nuovo tubo 50 mL. Ripetere il punto 7.3. Il volume totale del campione sarà ora 13,5 mL.
- Aggiungere 1,35 mL di 3M sodio acetato pH 5.2 e 33.75 mL di etanolo al 100% ghiacciata, mix e incubare a-80 ° C per 45 minuti o in alternativa una notte a-20 ° C.
- Centrifugare a 4 ° C e 20.000 x g per 10 min, rimuovere il surnatante, risospendere il pellet in 1 mL di preparata (vol/vol) etanolo al 70% e trasferire in una nuova provetta.
- Centrifugare a 4 ° C e a tutta velocità per 3 minuti in una centrifuga da banco, quindi rimuovere il surnatante.
- Risospendere il pellet in 1 mL di etanolo (vol/vol) 70% freddo di ghiaccio e ripetere il punto 7.7. Asciugare il pellet a 37 ° C per 10 min e risospendere in 650 µ l di tampone TLE. Determinare il rendimento del DNA utilizzando un'analisi di fluorescenza-basata per quantificare il DNA double-stranded.
Nota: Il protocollo può essere sospesa qui da snap congelare e conservare il campione a-80 ° C per diversi mesi, o a-20 ° C per un breve periodo di tempo.
8. controlli di qualità
- Monitorare l'integrità biblioteca e legatura tramite l'elettroforesi del DNA. Eseguire 200 ng di libreria su un gel di x TBE di agarosio/1 0,8%. Il DNA deve eseguire come una band oltre 10 kb.
- Rilevare il cellula-tipo noto invarianti a breve e lungo raggio le interazioni tramite PCR convenzionale. Uso 100 ng di DNA campione per reazione di PCR. Progettare gli iniettori PCR nelle vicinanze e verso i siti di restrizione seguendo le istruzioni sopra (Vedi 3.7.1). Sequenze dell'iniettore per il controllo di qualità di mouse e umano Hi-C biblioteche sono elencate nella tabella 1.
-
Controllo di Fill-in e la legatura: taglia fuori le fasce di gel contenente gli ampliconi di controllo 8.2, Estratto del gel del DNA e utilizzare il DNA come modello per 4 singole reazioni di PCR con combinazioni di primer identici.
- Ampliconi utilizzando un kit di purificazione di PCR di purificare e quantificare la concentrazione di DNA.
- Preparare quattro digestione reazioni (HindIII [a], NheI [b], HindIII + NheI [c] e nessun enzima [d]) per ogni amplicone in un volume finale di 15 µ l: 500 ng di amplicon, 1,5 µ l di tampone di restrizione 2.1, 0.15 µ l di 10 mg/mL albumina di siero bovino 10x (100 x BSA) e 0,1 µ l (10 unità) dell'enzima (HindIII [a], NheI [b], HindIII + NheI [c] o acqua [d]).
- Digerire per 1h a 37 ° C, quindi eseguire reazioni di digestione su un gel di x TBE di agarosio/1 1,5% (wt/vol).
9. DNA frammentazione
- Trasferimento 50,5 µ g di campione in una provetta di nuovo e aggiungere buffer di TLE ad un volume finale di campione 655 µ l. Split in 5 flaconcini di sonicazione (Vedi Tabella materiali) aggiungendo 130 µ l di biblioteca (10 µ g) per ogni flaconcino. Cesoia per una dimensione di ~ 400 bp in un ultra-sonicatore (Vedi Tabella materiali) utilizzando i seguenti parametri: fattore di utilizzo: 10%; potenza incidente di picco (w): 140; cicli di burst: 200; tempo: 55 s.
- Raccogliere lisati mediante campione in una provetta di fresco da 2 mL.
10. selezione dimensione bifacciale SPRI-perlina
- Soluzione perlina mix SPRI (solido fase reversibile immobilizzazione) ben invertendo, 1,85 mL di soluzione di perlina di trasferimento ad un nuovo tubo e portare a RT per 15 min.
- Aggiungere 350 µ l di acqua (grado di biologia molecolare) al campione (volume finale 1 mL).
- Aggiungere 600 µ l di soluzione perlina SPRI nell'esempio (volume totale 1,6 mL; rapporto di soluzione SPRI al DNA: 0,6 a 1), incubare per 5 min a RT e spin campione in una centrifuga da banco per 2 – 3 e raccogliere il campione.
- Aprire il coperchio, posizionare il campione sul cavalletto separazione magnetica per 5 min, trasferimento chiaro surnatante in una nuova provetta e scartare perline.
- Concentrarsi perline SPRI per la seconda fase di selezione dimensione: trasferimento 930 µ l di SPRI perline in un nuovo tubo, posto nello stand di separazione magnetica per 5 min e scartare chiaro surnatante. Risospendere le sfere in 310 µ l di soluzione perlina SPRI.
- Aggiungere 300 µ l di concentrato perline SPRI (passo 10.5) al campione (volume totale 1,9 mL; rapporto SPRI soluzione al DNA è ora 0,9 a 1), incubare a temperatura ambiente per 5 min e rotazione del campione in una centrifuga da banco per 2 – 3 s. attentamente aperto il coperchio , posizionare il tubo nello stand di separazione magnetica per 5 min ed eliminare il surnatante.
- Aggiungere 1 mL di etanolo al 70% preparata al momento (vol/vol) alla provetta del campione allo stand di separazione magnetica, incubare per 30 s e scartare surnatante. Ripetere due volte.
- Perline a secco a 37 ° C in un thermomixer (coperchio del tubo aperto) per non più di 5 min, aggiungere 300 µ l di tampone TLE al campione, mescolare e incubare per 10 minuti a temperatura ambiente.
- Rotazione del campione in una centrifuga da banco per 2 – 3 s, aprire il coperchio e posto il tubo sul magnetico stand per 5 min, trasferimento deselezionare surnatante in una nuova provetta e scartare perline.
11. biotina/streptavidina Pull-down di legatura prodotti
- Preparare buffer: 1x buffer di TB (0,5 mM EDTA; 1 M NaCl, 5mM Tris-HCl a pH 8.0; 0.05% Tween 20); 2 x buffer di NTB (10 mM Tris-HCl pH 8.0; 1 mM EDTA; 2 M NaCl); 1 tampone di x NTB (5 mM Tris-HCl a pH 8.0; 0,5 mM EDTA; NaCl di 1m).
- Aggiungere 200 µ l di biglie magnetiche streptavidina-accoppiati (Vedi Tabella materiali) in una nuova provetta, inserirlo nello stand di separazione magnetica per 1 min e rimuovere il surnatante.
-
Lavare le perle due volte con 500 µ l di tampone di TB di 1x.
- Per ogni passaggio di lavaggio durante la discesa la biotina, la riparazione di fine e la rimozione di biotina alle estremità del DNA di non-legati, dATP tailing e adattatore legatura passi, risospendere le perline nel buffer corrispondente, ruotare a RT e 15 giri/min per 3 minuti, girare il tubo in una centrifuga da banco per 2 – 3 s, posizionare il tubo nello stand di separazione magnetica per 3 minuti e rimuovere il surnatante.
- Risospendere perline in 300 µ l di tampone di x NTB 2. Mix perline e campione (600 µ l di volume totale) e incubare a + RT 15 min su una ruota rotante a 3 giri/min.
- Reclamare perline allo stand di separazione magnetica per 3 minuti e rimuovere il sovranatante chiaro. Lavare perline due volte in 500 µ l di 1 x NTB tampone prima e poi a 200 µ l di tampone legatura 1x. Risospendere le sfere in 50 µ l di tampone di legatura x 10.
12. fine riparazione e rimozione di biotina alle estremità del DNA di Non-legati
- Il campione (50 µ l in totale) si combinano con 50 µ l di 2,5 mM dNTP mix (12,5 µ l di 10 mM di ogni dNTP), 18,1 µ l di 3.000 U/mL T4 DNA polimerasi, 18,1 µ l di 10.000 U/mL T4 PNK, 3,7 µ l di 5.000 U/mL DNA polimerasi sono grandi (Klenow) frammento e 360.1 µ l di H2O.
- Mescolare e incubare a 20 ° C per 1 h, agitando 5 s a 700 giri/min ogni 2 min in un thermomixer.
- Recuperare perle nello stand di separazione magnetica, rimuovere il supernatante chiaro e lavare perline due volte in 500 µ l di tampone di TB 1x.
- Lavare perline in 500 µ l di tampone di x NTB 1, seguita da un lavaggio in 500 µ l di 1 x TLE.
- Recuperare perle nello stand di separazione magnetica, rimuovere il supernatante chiaro e risospendere perline in 415 µ l di tampone x TLE 1.
13. dATP Tailing
- Campione (415 µ l) si combinano con 50 µ l di tampone di restrizione 2, 5 µ l di 10 millimetri di dATP e 30 µ l di 5 U / µ l Klenow exo-minus 10x.
- Mescolare e incubare a 37 ° C per 30 minuti, agitando 5 s a 700 giri/min ogni 2 min in un thermomixer.
- Recuperare perle nello stand di separazione magnetica, rimuovere il supernatante chiaro e lavare perline due volte in 500 µ l di tampone di TB 1x.
- Lavare perline in 500 µ l di tampone di x NTB 1.
14. adattatore legatura
- Lavare perline in 200 µ l di tampone di reazione di legatura: 1x (Vedi Tabella materiali).
- Risospendere perline in 200 µ l di tampone di reazione 1 x legatura. Aggiungere 4 µ l di DNA ligasi (Vedi Tabella materiali) e 16 µ l di 15 µM pre-temprato adattatori PE (pre-tempri gli adattatori PE mescolando volumi uguali di PE scheda 1 e scheda di PE 2 (entrambi a 30 µM) e incubando per pochi minuti alla RT). Incubare a temperatura ambiente per 15 min.
- Recuperare perle nello stand di separazione magnetica, rimuovere il supernatante chiaro e lavare perline due volte in 500 µ l di tampone di TB 1x.
- Lavare perline in 500 µ l di tampone di x NTB 1. Poi, lavare perline in 100 µ l di tampone di restrizione 2, 1x risospendere perline in 50 µ l di tampone di restrizione 2 1x e trasferire in una nuova provetta.
15. Ciao-C Library amplificazione
- Preparare il mix master PCR: 100 µ l di buffer di x Phusion 5; 6 µ l di primer di PCR PE 25 µM 1.0; 6 µ l di primer di 25 µM PCR PE 2.0; 14 µ l di miscela del dNTP (10 mM); 6 µ l della polimerasi Phusion; 318 µ l di H2O.
- Il mix master mix PCR con le perline (500 µ l in totale), dividere in 10 aliquote di 50 µ l e amplificare mediante PCR utilizzando le seguenti condizioni:
30s a 98 ° C
7 cicli di: 10 s a 98 ° C; 30 s a 65 ° C; 30 s a 72 ° C
7 min a 72 ° C - Raccogliere le reazioni di PCR in un nuovo tubo, recupero di perline sul basamento di separazione magnetica e il trasferimento surnatante (500 µ l) in una nuova provetta.
-
Purificare il DNA di libreria usando le perle di SPRI.
- Perline mix SPRI, trasferire 460 µ l di perle in un nuovo tubo e portare a RT per 15 min aggiungere 450 µ l di perline SPRI per le reazioni di PCR (volume finale 950 µ l), incubare per 5 min a RT e spin campione in una centrifuga da banco per 2 – 3 e raccogliere il campione.
- Aprire il coperchio, posizionare il campione sul cavalletto separazione magnetica per 5 min e rimuovere il surnatante.
- Mantenendo le perle nello stand di separazione magnetica, aggiungere 1 mL di etanolo al 70% (vol/vol) filmato di esempio su una superficie chiara di perline, lasciare per 30 s e scartare surnatante.
- Ripetere il passaggio 15.4.3 altre due volte.
- A secco di perline a 37 ° C in un thermomixer (coperchio del tubo aperto) per non più di 5 min.
- Aggiungere 51 µ l di tampone TLE al campione, mix e incubare per 10 min a 37 ° C, agitando a 950 giri/min in un thermomixer.
- Rotazione del campione in una centrifuga da banco per 2 – 3 s, aprire il coperchio e posto il tubo sul magnetico stand per 5 min, trasferimento deselezionare surnatante in una nuova provetta e scartare perline.
- Quantificare la concentrazione della libreria Hi-C. Dopo 7 turni di amplificazione di PCR, otteniamo ordinariamente 500 – 1.500 ng di libreria Hi-C.
16. hybrid Capture In soluzione
Nota: Blocker e buffer (SHS1-4) soluzioni riportate di seguito sono dal SureSelect kit (Vedi Tabella materiali).
- Trasferimento 500 ng di 1 µ g di Hi-C libreria in una nuova provetta e far evaporare il campione su un concentratore di vuoto (vedere Tabella materiali; 45 ° C; pressione di vuoto: livello 30.0, rampa 5) fino a secco.
- Risospendere evaporato Hi-C biblioteca aggiungendo 3,6 µ l di H2O (grado di biologia molecolare), 2,5 µ l di blocker 1, 2,5 µ l di blocker 2 e 0,6 µ l di blocco personalizzato.
- Trasferire il campione in un pozzo di una nuova striscia di tubo PCR, chiudere con una striscia di tappo PCR e metterli su ghiaccio. Etichetta come "D" (per DNA Hi-C).
- Preparare il tampone di ibridazione: 12,5 µ l di tampone di SHS1; 0,5 µ l di tampone SHS2; 5 µ l di tampone di SHS3; 6,5 µ l di tampone di SHS4.
- Incubare a 65 ° C per 5 min in un thermomixer. Trasferire in un pozzo di una nuova striscia di tubo PCR, chiudere con una striscia di tappo PCR e mantenere a RT. etichetta come "H" (per tampone di ibridazione).
- In un pozzo di una nuova striscia di tubo PCR, mescolare 5 µ l di 100 ng / µ l biotinilati le sonde del RNA (conservare a-80 ° C e scongelare su ghiaccio appena prima dell'uso); 0,5 µ l di sRNasi B (inibitore di RNasi) e 1,5 µ l di H2O (grado di biologia molecolare).
- Chiudere la striscia di tubo PCR con una striscia di tappo PCR e posto sul ghiaccio. Etichetta come "R" (per RNA).
- Impostare la macchina PCR utilizzando i seguenti parametri:
5 min a 95 ° C; 25 h a 65 ° C; coperchio riscaldato; Volume di reazione di PCR 29 µ l.
Nota: Procedere più rapidamente possibile durante tutte le procedure, mentre la macchina PCR viene eseguita al fine di evitare l'evaporazione del campione. - Posizionare la striscia di tubo "D" PCR nella macchina PCR, il coperchio della macchina PCR e avviare la reazione di PCR. Quando il programma PCR raggiunge i 65 ° C, aprire il coperchio della macchina PCR e posizionare la striscia di tubo "H" PCR nella macchina PCR. Il coperchio della macchina PCR e incubare per 3 min. Apri il coperchio della macchina PCR, posto il tubo "R" PCR striscia sulla macchina PCR e chiudere la macchina PCR.
- Dopo 2 minuti, aprire il coperchio della macchina PCR e tutte le strisce di tubo PCR. Trasferire 13 µ l di ben "H" in ben "R", quindi tutto il volume di ben "D" in ben "R". Dispensare su e giù per 3 volte per mescolare la reazione, chiudere la striscia di tubo PCR, Rimuovi la "H" e "D" PCR tube strisce e chiudere il coperchio della macchina PCR. Incubare la reazione a 65 ° C per 24 h.
17. isolamento del promotore frammento-contenente prodotti di legatura
Nota: I passaggi seguenti sono raccomandati per essere fatto con kit adattatore SureSelect e biblioteca (Vedi Tabella di Materals).
- Pre-riscaldare 1,5 mL di tampone di lavaggio 2 per campione a 65 ° C in anticipo.
- Aggiungere 60 µ l di biglie magnetiche streptavidina-accoppiati (Vedi Tabella materiali) in una nuova provetta, posizionare lo stand di separazione magnetica per 1 min e rimuovere il surnatante.
- Lavare perle tre volte con 200 µ l di tampone di legame 1x.
Nota: Per ogni passaggio di lavaggio durante l'isolamento post-acquisizione di prodotti contenenti promotore di legatura, risospendere perline nel buffer corrispondente, ruotare per 3 min a RT e 15 giri/min su una ruota in movimento, spin dolcemente il tubo in una centrifuga da banco per 2 – 3 e di raccogliere campione, posto il tubo sul magnetico stand per 3 min e Rimuovi surnatante. - Risospendere perline in 200 µ l di tampone di legame 1x. Aprire la macchina PCR e la striscia di tubo PCR (mentre è ancora in esecuzione il programma PCR) e trasferire la reazione di ibridazione nella provetta con i branelli magnetici. Incubare per 30 minuti su un disco rotante a 3 giri/min a RT.
- Reclamare perline allo stand di separazione magnetica e rimuovere il sovranatante chiaro. Risospendere perline in 500 µ l di tampone di lavaggio 1, mix e incubare per 15 minuti a 20 ° C mentre si stringono a 950 giri/min in un thermomixer.
- Reclamare perline allo stand di separazione magnetica per 3 minuti e rimuovere il sovranatante chiaro. Risospendere perline in 500 µ l di tampone di lavaggio 2, miscelare e incubare 10 min a 65 ° C mentre si stringono a 950 giri/min in un thermomixer. Ripetere il passaggio due volte più di 17,5.
- Recuperare perle nello stand di separazione magnetica, rimuovere il supernatante chiaro e risospendere perline in 200 µ l di tampone di restrizione 2 1x. Recuperare perle nello stand di separazione magnetica, rimuovere il surnatante e risospendere perline in 30 µ l di tampone di restrizione 2 1x.
18. PCHi-C Library amplificazione
- Preparare il mix master PCR: 60 µ l di buffer di 5 x PCR (Phusion buffer), 3,6 µ l di primer 25 di PE PCR di µM 1.0, 3,6 µ l di primer 25 di PE PCR di µM 2.0, 8,4 µ l di miscela del dNTP (10 mM) e 3,6 µ l della polimerasi Phusion 190,8 µ l di H2O.
- Il mix master mix PCR con le perline (300 µ l in totale), si dividono in 6 aliquote di 50 µ l e PCR-amplificare usando le seguenti condizioni:
30 s a 98 ° C
4 cicli di: 10 s a 98 ° C, 30 s a 65 ° C, 30 s a 72 ° C
7 min a 72 ° C - Raccogliere tutte le reazioni di PCR in un nuovo tubo, recuperare le perline sul magnete e trasferire il surnatante (300 µ l; contiene libreria PCHi-C) in ad un nuovo tubo.
- Purificare la libreria PCHi-C usando i branelli SPRI, seguendo la procedura descritta sopra sotto 15,4.
- Quantificare la concentrazione della libreria PCHi-C.
Risultati
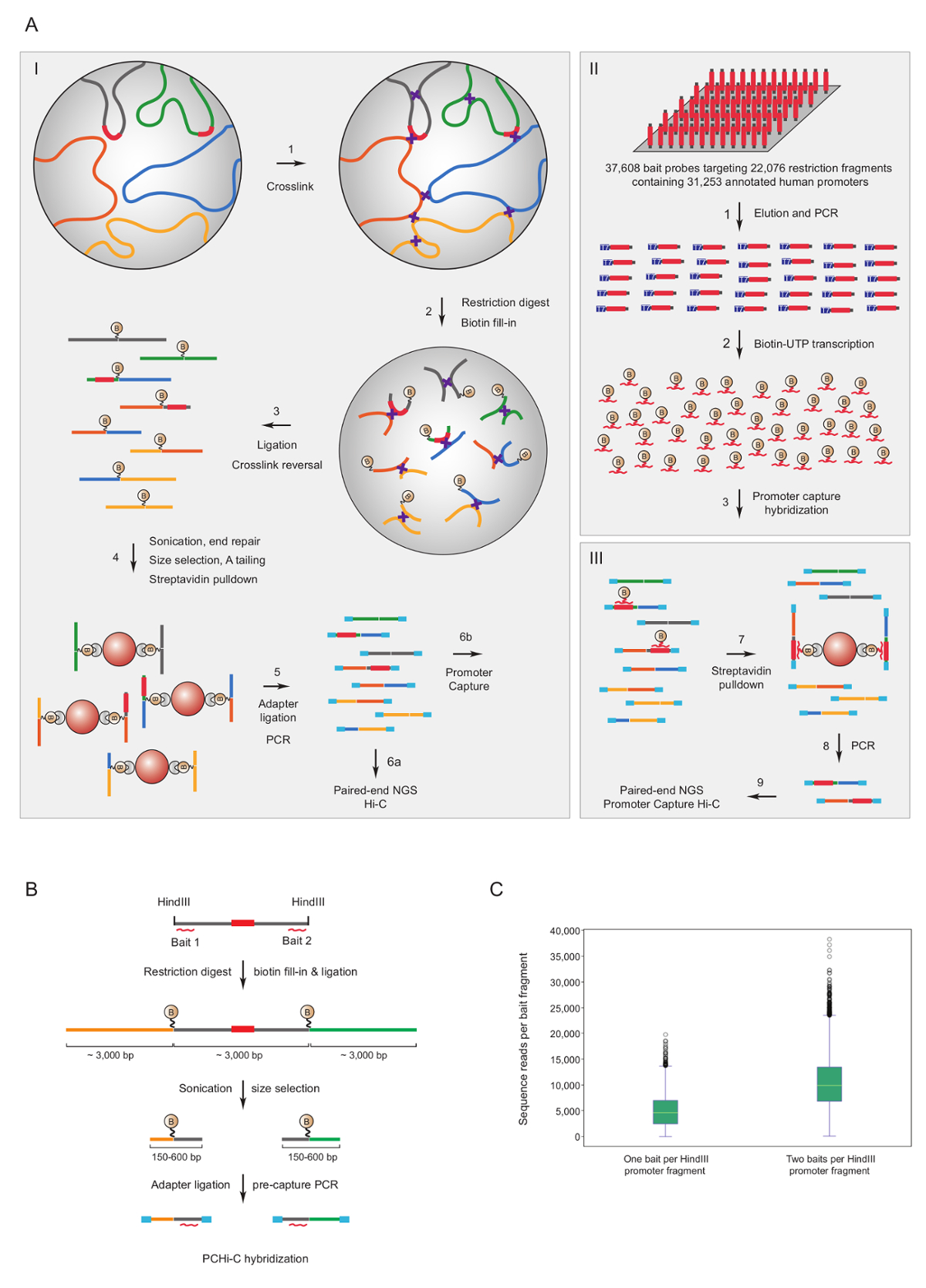
Promotore Capture Hi-C è stato utilizzato per arricchire del mouse7,34,36,39 e umano33,35,37,38 Hi-C biblioteche per interazioni di promotore. Un simile protocollo (denominato HiCap) è stata descritta dal gruppo Sandberg40. Figura 1A Mostra il flusso di lavoro schematico per promotore Capture Hi-C. Nel protocollo descritto qui, librerie Hi-C sono generate tramite la legatura nel nucleo41, che si traduce in un numero significativamente ridotto di legatura spurie prodotti42. Per PCHi-C, mouse altamente complesso o librerie Hi-C umane sono sottoposti a nella soluzione di ibridazione e catturano utilizzando 39.021 biotinilati RNA complementare a frammenti di restrizione HindIII promotore-contenenti del mouse 22.225, o 37.608 biotinilati RNAs targeting 22.076 umano contenenti promotore HindIII frammenti di restrizione, rispettivamente. Promotore contenenti frammenti di restrizione possa essere mirati ad una o entrambe le estremità di singoli biotinilati RNAs (Figura 1B). Abbiamo trovato che la cattura di entrambi finisce copertura migliorata di singoli promotori (Figura 1; prime sequenze letture) quasi duplice, come previsto. Così, in ogni momento possibile (cioè, nelle regioni non-ripetitive), si consiglia di utilizzare biotinilati RNA complementare ad entrambe le estremità di un frammento di restrizione per essere catturato.
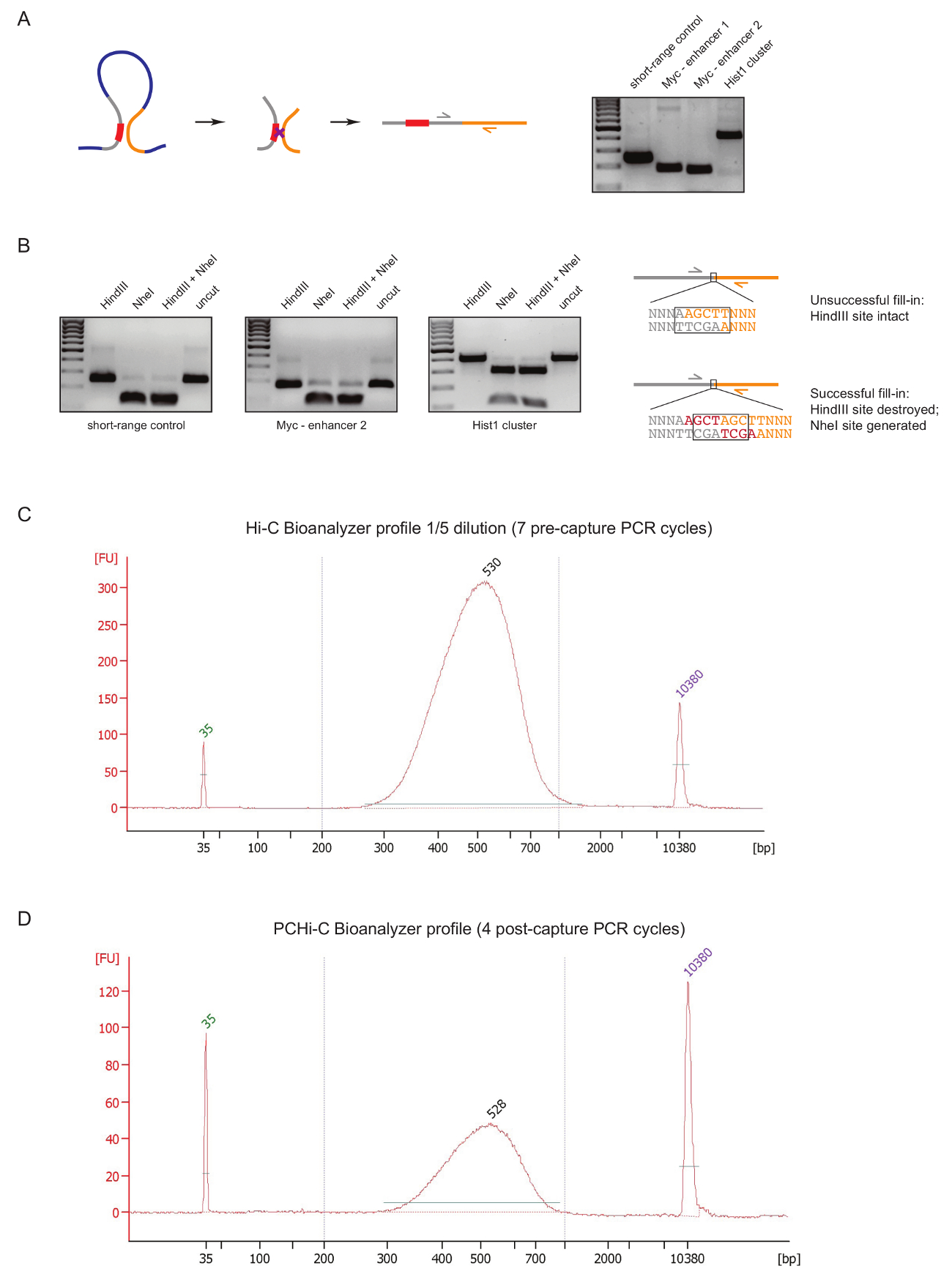
Per valutare la qualità biblioteca PCHi-C in una fase iniziale durante la preparazione della biblioteca, eseguiamo due controlli dopo legatura del DNA e purificazione, come descritto in precedenza31. Il primo consiste nell'utilizzare coppie di primer specifici per amplificare i prodotti di legatura come in 3 C27. Usiamo le coppie di primer (tabella 1) per amplificare i prodotti di cellula-tipo invariante legatura a lungo raggio, ad esempio tra il gene Myc e suoi esaltatori di noti si trova circa 2 Mb (Figura 2A) o tra geni del locus ( Hist1 separati da 1,5 Mb) e tra due regioni situate nelle immediate vicinanze lineare ('a corto raggio control').
Il secondo controllo di qualità è effettuato per determinare l'efficienza dell'incorporazione di biotina durante Fill-in di Klenow-mediata della restrizione sito strapiombi con biotina-dATP. Successo Klenow Fill-in e la legatura successiva smussato-fine risultati nella scomparsa del sito di restrizione originale tra le molecole di DNA di un prodotto di legatura e nel caso di HindIII nella formazione di un nuovo sito di riconoscimento NheI (Figura 2B ). Il rapporto tra il HindIII al prodotto digerito la legatura NheI è una lettura diretta dell'efficienza di incorporazione di biotina. Una libreria di Hi-C di scarsa qualità mostrerà un elevato livello di digestione HindIII, considerando che sono librerie di alta qualità vicino-completa NheI digestione dei prodotti di legatura (Figura 2B).
Dopo la preparazione di biblioteca Hi-C (vale a dire, dopo che la biotina-streptavidina tirare verso il basso della dimensione selezionata di prodotti di legatura Hi-C, adattatore legatura e pre-cattura PCR), la distribuzione l'integrità e la dimensione della libreria Hi-C è valutata da Bioanalyzer (Figura 2 C). lo stesso controllo viene effettuato alla fine della preparazione di libreria PCHi-C (vale a dire, dopo la cattura di ibridazione dei prodotti contenenti promotore di legatura e post-cattura PCR). Confronto dei profili Hi-C e C PCHi Bioanalyzer dimostra che come previsto, librerie Hi-C sono molto più concentrate rispetto le librerie corrispondenti PCHi-C, ma la distribuzione di dimensione delle librerie è molto simile, che indica che la cattura un passo in PCHi-C non introduce un bias di dimensione (Figura 2, D).
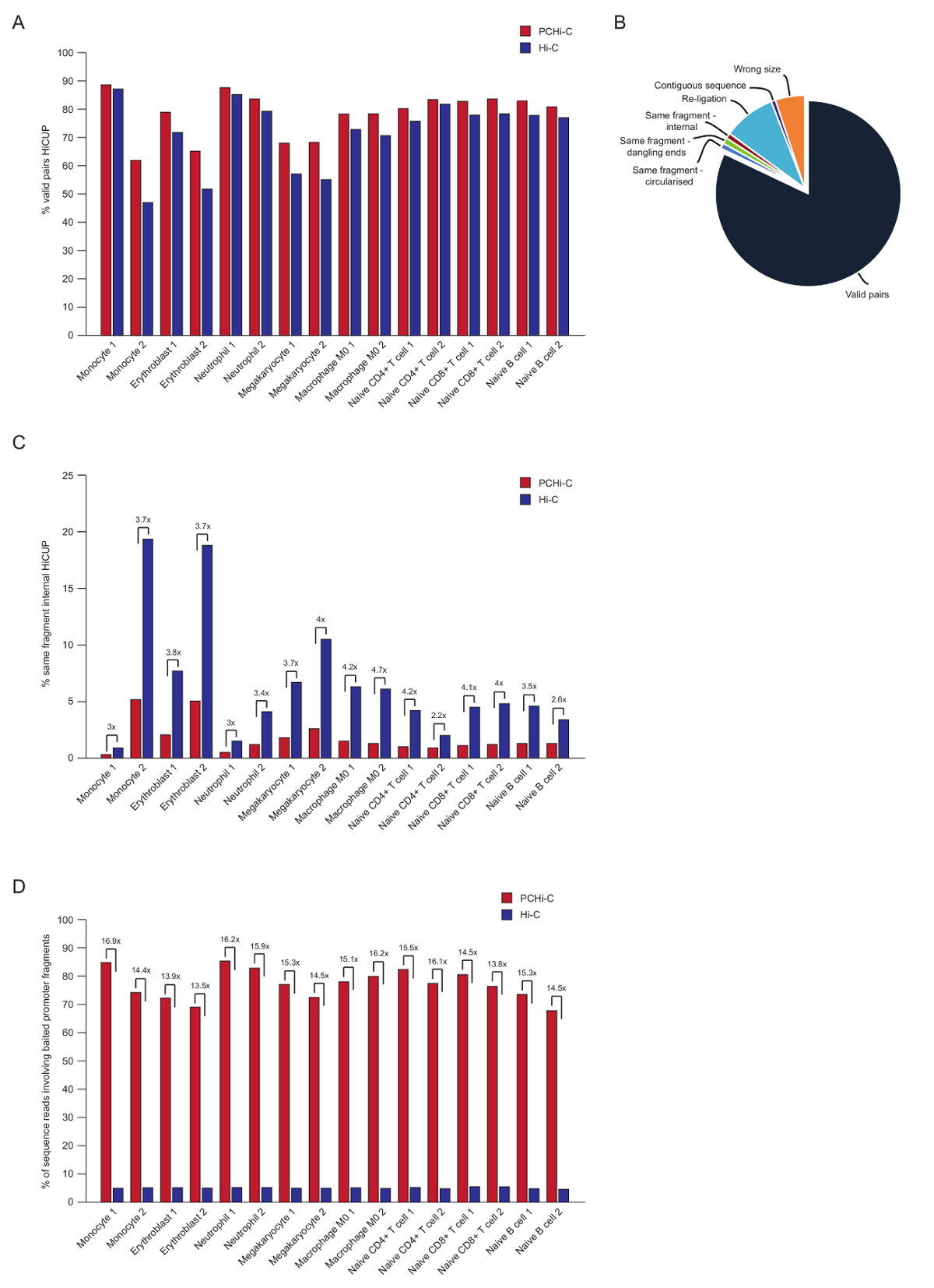
Dopo la sequenziazione accoppiato-fine, la legge di PCHi-C vengono mappata, qualità controllata e filtrata utilizzando il HiCUP della pipeline43. Alta qualità PCHi-C librerie contengono tra 70-90% 'coppie valide' (cioè, accoppiato-fine sequenza legga tra due frammenti di restrizione che non sono vicine sulla mappa genomica lineare; Figura 3A, B). Utilizzando la legatura nel nucleo protocollo41,42, la percentuale di trans legge coppie (cioè, accoppiato-fine sequenza legga tra due frammenti di restrizione che si trovano su cromosomi diversi) sono di solito basse, tra 5 e 25%, che riflette l'esistenza di territori cromosomici e che indica la qualità di buona biblioteca. Confronto diretto della percentuale di 'coppie valide' tra librerie Hi-C e i loro corrispondenti PCHi-C biblioteche35, spettacoli che in tutti i casi la percentuale di coppie valide è più alto nelle librerie PCHi-C (Figura 3B). Questo è accompagnato da una riduzione della percentuale di letture non valido 'stesso frammento interno' in PCHi-C (Figura 3). Questo è previsto, come il passo di cattura non solo arricchisce per prodotti contenenti promotore di legatura, ma anche per fini di frammento di restrizione, a causa della posizione della cattura i oligos sulla restrizione frammenti (Vedi Figura 1B).
Dopo HiCUP filtraggio, determiniamo l'efficienza di cattura. PCHi-C librerie contengono tre tipi di sequenza valida letture dopo il filtraggio di HiCUP:
1.) promotore: genoma legge (cioè, letture tra un frammento catturato promotore e un frammento di restrizione HindIII non-promotore ovunque nel genoma)
2.) promotore: promotore legge (letture tra due frammenti catturati promotore)
3.) genoma: genoma legge (prodotti di legatura di sfondo Hi-C dove nessuno dei partner di prodotto di legatura esegue il mapping a un promotore catturato). Queste vengono eliminate prima dell'analisi a valle.
Librerie di alta qualità PCHi-C hanno efficienze di cattura (somma delle categorie 1 e 2 sopra) tra 65-90% (Figura 3D). Un confronto diretto a librerie Hi-C Mostra che PCHi-C si traduce in una ~ 15 volte arricchimento per promotore-legatura prodotti contenenti (Figura 3D), in alcuni casi 17-fold. Questo è vicino al massimo ipotetico (19.6-fold) arricchimento per PCHi-C, che è dipendente la percentuale dei frammenti di restrizione di genoma coperte dal sistema di acquisizione. Maggiore arricchimento può essere ottenuta progettando sistemi di cattura mira meno restrizione frammenti44,45,46.
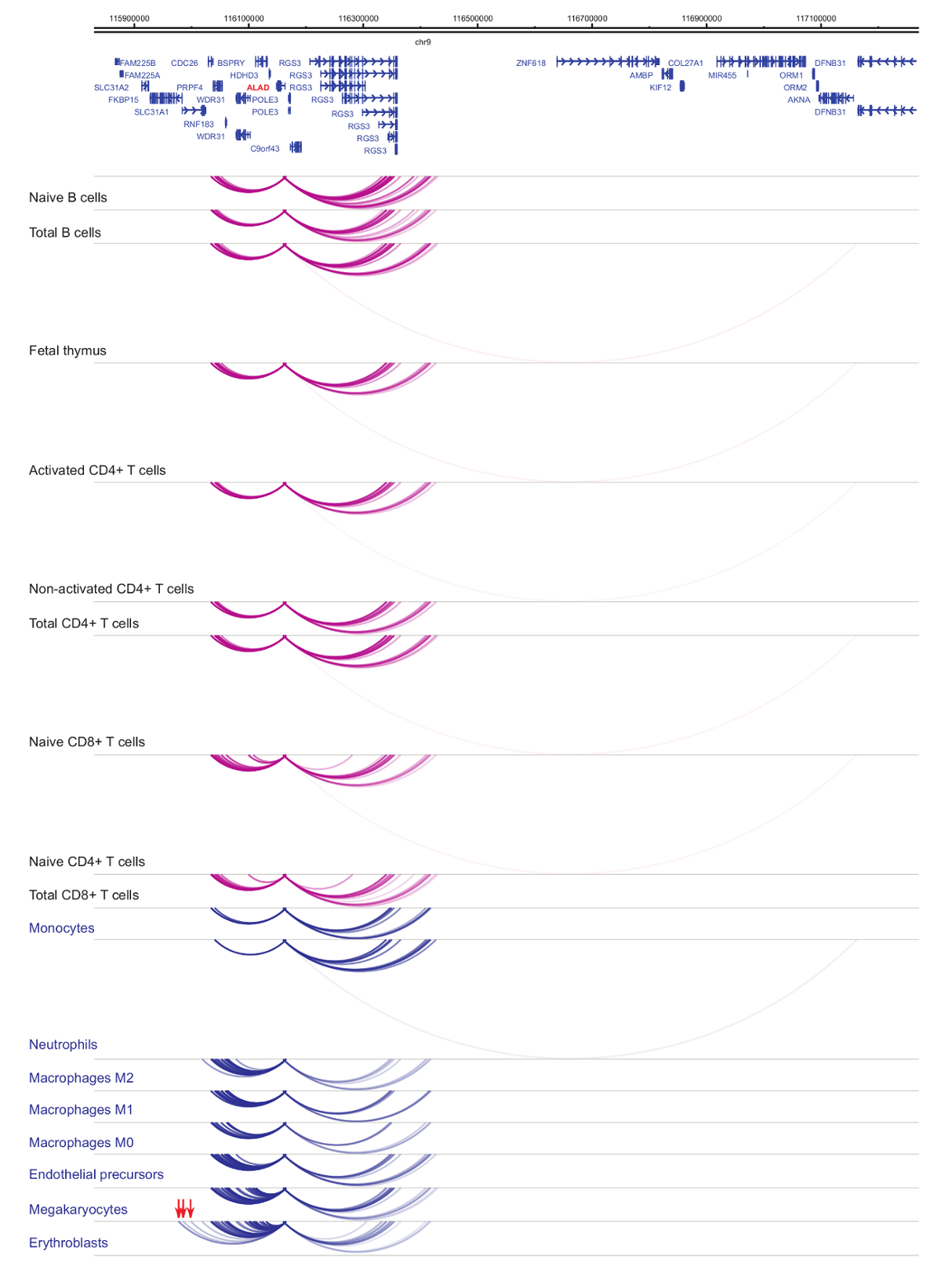
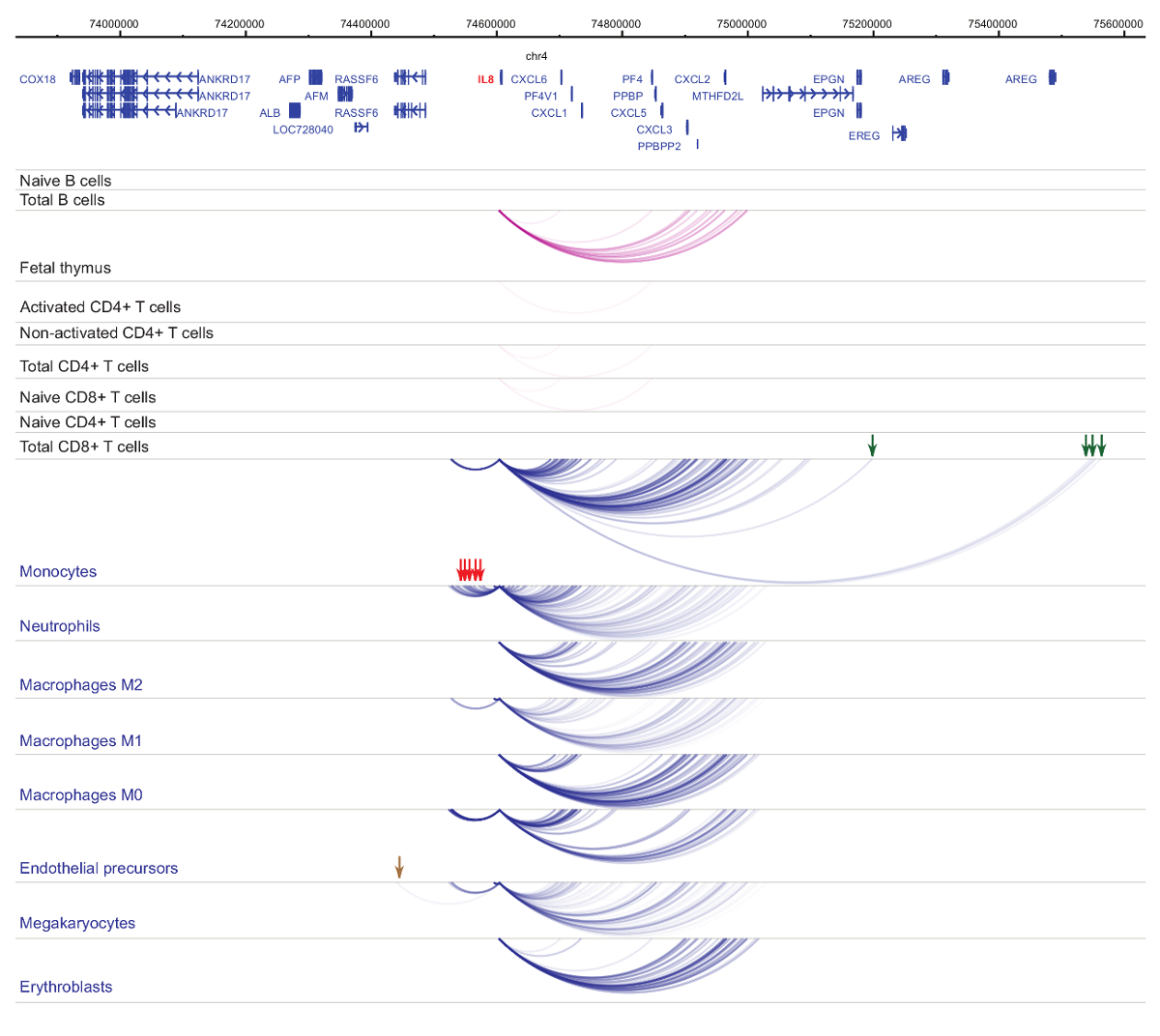
Analisi del promotore Interattomi dimostra cella tipo e specificità di lineage33,34,35, con cambiamenti pronunciati durante la differenziazione cellulare37,38,39 . Figure 4 e 5 mostrano esempi di dinamiche di specificità e differenziazione di lignaggio a promotori specifici. Ad esempio, ALAD è costitutivamente espressa in tutte le cellule, ma la sua espressione è upregulated in eritroblasti47. Il promotore ALAD contatti diversi frammenti distali in tutte le cellule ematopoietiche e si impegna in ulteriori interazioni specificamente negli eritroblasti (Figura 4). IL-8 non mostra nessuna interazione statisticamente significativa in cellule di B, molto poche interazioni in cellule di T, ma decine di interazioni in cellule della linea mieloide, comprese specifiche interazioni cellula-tipo in monociti, neutrofili e megacariociti ( Figura 5). Questi esempi dimostrano come PCHi-C può essere utilizzato per svelare Interattomi specifico tipo di cella e identificare regioni promotore-interagendo con potenziale normativo.

Figura 1 : Spiegazione razionale promotore Capture Hi-C e la cattura dell'esca design. Flusso di lavoro schematico (A) di PCHi-C. Nel nucleo la legatura Hi-C41,42 (I) è seguita dall'ibridazione nella soluzione con esche biotinilati RNA (II) targeting i frammenti di restrizione di tutto l'essere umano (qui raffigurato) o promotori di geni (III) il mouse. (B) esca design per PCHi-C. Biotinilati RNA cattura esche (linee curve rosse) sono destinate contro le estremità dei frammenti di restrizione promotore-contenenti (grigio; nota che le sequenze del promotore stessi (rosso) sono solo mirate dalle esche di cattura di RNA se essi si trovano a restrizione estremità del frammento). Prodotti di legatura costituito promotore contenenti frammenti di restrizione (grigi) e loro interagenti frammenti di restrizione (giallo e verde) sono isolati mediante ibridazione di sequenza-complementarità tra esca di RNA e DNA bersaglio e successive biotina-streptavidina pulldown, come mostrato in a. (C) confronto di PCHi-C l'efficienza di cattura per promotore contenenti frammenti di restrizione mirati da un RNA esca cattura sonda vs due sonde di RNA esca cattura (Vedi schema b). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2 : Controlli di qualità di pre-sequenza PCHi-C. (A) a sinistra, schema di contrapposizione spaziale tra promotore e PIR con conseguente Hi-C legatura prodotto costituito da un frammento di restrizione promotore-contenenti (grigio; sequenza del promotore in rosso) e un frammento di restrizione del PIR (giallo). Diritto, DNA elettroforesi mostrando esempi di Hi-C legatura prodotti amplificati utilizzando coppie di primer specifici (come raffigurato nello schema a sinistra). (B) a sinistra, esempi rappresentativi di restrizione HindIII, NheI e HindIII/NheI digerisce di Hi-C legatura (prodotti PCR mostrato nella A). Bene, disegno schematico del DNA sequenza dopo legatura Hi-C seguito infruttuoso (in alto) o Fill-in Klenow di dNTP successo (in basso) di giunzioni di restrizione e la legatura successiva. (C) rappresentante Hi-biblioteca bioanalyzer il profilo C (diluizione 1/5). Profilo bioanalyzer rappresentante PCHi-C biblioteca di (D) (Nessuna diluizione). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3 : Controlli di qualità post-sequenziamento PCHi-C. (A) confronto di sequenza valida percentuale legga le coppie dopo HiCUP43 elaborazione a PCHi-C vs corrispondenti librerie Hi-C (dati da Javierre et al., 201635). (B) risultato rappresentante HiCUP PCHi-C risultati validi legge coppie e altre categorie di sequenza che vengono eliminate prima dell'analisi a valle (dati da Javierre et al., 201635). (C) confronto della percentuale 'stesso frammento interno' legge dopo HiCUP elaborazione a PCHi-C vs corrispondenti librerie Hi-C (dati da Javierre et al., 201635). (D) confronto di sequenza percentuale legge frammenti innescati promotore che coinvolgono (efficienza di cattura) a PCHi-C vs corrispondenti librerie Hi-C (dati da Javierre et al., 201635). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: ALAD profilo PCHi-C in cellule ematopoietiche umane. Interazioni di promotore di tipi delle cellule mieloidi sono mostrati come archi blu, e interazioni di promotore di tipi delle cellule linfoidi come archi viola. Interazioni dell'eritroblasto specifiche sono indicati dalle frecce rosse (dati da Javierre et al., 201635). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5 : IL8 Profilo di PCHi-C in cellule ematopoietiche umane. Interazioni di promotore di tipi delle cellule mieloidi sono mostrati come archi blu, e interazioni di promotore di tipi delle cellule linfoidi come archi viola. Interazioni del monocito-specifiche sono indicati dalle frecce verdi, interazioni del neutrofilo-specifiche sono indicati dalle frecce rosse e un'interazione megacariocita specifico è indicata da una freccia marrone (dati da Javierre et al., 201635). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Umano | ||||||||
| Nome | Sequenza | Cromosoma | Strand | Inizio GRCh38/hg38 | Fine GRCh38/hg38 | Combinazioni di primer per testare 3C interazioni e incorporazione di biotina | ||
| HS AHF64 Dekker | GCATGCATTAGCCTCTGCTGTTCTCTGAAATC | 11 | + | 116803960 | 116803991 | utilizzare in combinazione con hs AHF66 Dekker | ||
| HS AHF66 Dekker | CTGTCCAAGTACATTCCTGTTCACAAACCC | 11 | + | 116810219 | 116810248 | utilizzare in combinazione con hs AHF64 Dekker | ||
| locus MYC HS | GGAGAACCGGTAATGGCAAA | 8 | - | 127733814 | 127733833 | utilizzare in combinazione con hs MYC + 1820 o hs MYC-538 | ||
| HS MYC + 1820 | AAAATGCCCATTTCCTTCTCC | 8 | + | 129554527 | 129554547 | utilizzare in combinazione con locus MYC hs | ||
| HS MYC-538 | TGCCTGATGGATAGTGCTTTC | 8 | - | 127195696 | 127195716 | utilizzare in combinazione con locus MYC hs | ||
| HS HIST1 F | AAGCAGGAAAAGGCATAGCA | 6 | + | 26207174 | 26207193 | utilizzare in combinazione con hs HIST1 R | ||
| HS HIST1 R | TCTTGGGTTGTGGGACTTTC | 6 | + | 27771575 | 27771594 | utilizzare in combinazione con hs HIST1 F | ||
| Mouse | ||||||||
| Sequenza | Cromosoma | Strand | Inizio GRCm38/mm10 | Fine GRCm38/mm10 | Combinazioni di primer per testare 3C interazioni e incorporazione di biotina | |||
| TCATGAGTTCCCCACATCTTTG | 8 | + | 84841090 | 84841111 | utilizzare in combinazione con mm Calr2 | |||
| CTGTGGGCACCAGATGTGTAAAT | 8 | + | 84848519 | 84848541 | utilizzare in combinazione con mm Calr1 | |||
| TATCAAGGGTGCCCGTCACCTTCAGC | 6 | + | 125163098 | 125163123 | utilizzare in combinazione con Gapdh4 Dekker | |||
| GGGCTTTTATAGCACGGTTATAAAGT | 6 | + | 125163774 | 125163799 | utilizzare in combinazione con Gapdh3 Dekker | |||
| GGAGGAGGGAAAAGGAGTGATT | 6 | + | 52212829 | 52212850 | utilizzare in combinazione con mm Hoxa13 | |||
| CAGGCATTATTTGCTGAGAACG | 6 | - | 52253490 | 52253511 | utilizzare in combinazione con mm Hoxa7 | |||
| GGGTAATGGTGTCACTAACTGG | 13 | + | 23571284 | 23571305 | utilizzare in combinazione con Hist1h3e o mm Hist1h4i | |||
| GGGTTTGATGAGTTGGTGAAG | 13 | + | 23566541 | 23566561 | utilizzare in combinazione con mm Hist1h2ae | |||
| TTGGGCCAAAGCCTATATGA | 13 | + | 22043085 | 22043104 | utilizzare in combinazione con mm Hist1h2ae | |||
Tabella 1: Sequenze Primer per il controllo di qualità di essere umano e le librerie del mouse Hi-C.
Discussione
Design modulare del promotore Capture Hi-C
Promotore Capture Hi-C è progettato per arricchire in particolare librerie Hi-C per le interazioni che coinvolgono i promotori. Queste interazioni comprendono solo un sottoinsieme di legatura prodotti presenti in una libreria di Hi-C.
Cattura Hi-C può essere facilmente modificato per arricchire le librerie Hi-C per qualsiasi regione genomica o regioni di interesse modificando il sistema di acquisizione. Cattura regioni possono essere continuo segmenti genomici44,45,46,48, esaltatori di che sono stati identificati in PCHi-C ('Reverse Capture Hi-C'35), o dnasi I siti ipersensibili49 . La dimensione del sistema di acquisizione può essere regolata a seconda dell'ambito sperimentale. Ad esempio, Dryden et al. destinazione 519 esca frammenti nei tre deserti di gene associati al seno cancro44. Il sistema di acquisizione da Martin et al. gli obiettivi di entrambi i segmenti genomici continui ('Regione Capture': 211 regioni genomiche in totale; 2.131 frammenti di restrizione) e selezionato promotori (3.857 promotori di geni)45.
SureSelect librerie sono disponibili in dimensioni diverse gamme: 1 kb per 499 kb (5.190 – 4.806), 500 kb a 2,9 Mb (5.190 – 4.816) e 3 Mb a 5,9 Mb (5.190 – 4.831). Come ogni acquisizione singola biotina-RNA è 120 nucleotidi lunghi, queste acquisizione sistemi di ospitare un massimo di 4.158, 24.166 e 49.166 individuali cattura sonde, rispettivamente. Ciò corrisponde a 2.079, 12.083 e 24.583 frammenti di restrizione mirata, rispettivamente (si noti che i numeri per i frammenti di restrizione sono limiti inferiori basati sul presupposto che le due sonde di acquisizione singola possono essere progettati per ogni restrizione frammento — in realtà a causa di sequenze ripetitive questo non sarà il caso per ogni restrizione frammento (Vedi anche Figura 1B, C), determinando un maggior numero di frammenti di restrizione indirizzabile per un numero costante di acquisizione disponibili sonde ).
Il protocollo qui descritto si basa sull'uso di un enzima di restrizione con un sito di riconoscimento bp 6 per scoprire interazioni a lungo raggio. Usando un enzima di restrizione con un sito di riconoscimento bp 4 per una maggiore risoluzione delle interazioni più prossimale è anche possibile40,49.
Limitazioni di PCHi-C
Un limite intrinseco di ogni cromosoma conformazione cattura saggi è che la loro risoluzione è determinata con l'enzima di restrizione utilizzato per la generazione della libreria. Interazioni che avvengono tra elementi di DNA situati sullo stesso frammento di restrizione sono invisibili per analisi di 'tipo C'. Ulteriormente, in PCHi-C, in alcuni casi più di un sito di inizio della trascrizione possa trovarsi sullo stesso frammento di restrizione contenenti promotore, e PIRs in alcuni casi porto entrambi i contrassegni istone attivo e repressiva, che lo rende difficile individuare quale regolamentazione elementi di mediano le interazioni e di prevedere l'output regolamentazione delle interazioni del promotore. Usando gli enzimi di restrizione con 4 siti di riconoscimento di bp attenua questo problema ma a scapito di molto maggiore complessità di biblioteca Hi-C (Hi-C biblioteche generate con 4 degli enzimi di restrizione del sito di riconoscimento di bp sono almeno 100 volte più complessi di Hi-C le librerie generate con 6 degli enzimi di restrizione del sito di riconoscimento di bp) e i costi associati per il sequenziamento di nuova generazione.
Un'altra limitazione è che l'attuale protocollo di PCHi-C richiede milioni di cellule come materie prime, precludendo l'analisi delle interazioni di promotore in tipi cellulari rara. Una versione modificata di PCHi-C per attivare l'interrogatorio di contatti promotore in popolazioni di cellule con 10.000 a 100.000 cellule (ad esempio cellule durante lo sviluppo embrionale precoce o di cellule staminali ematopoietiche) sarebbe pertanto una preziosa aggiunta alla cattura Hi-C della casella degli strumenti.
Infine, come tutti i metodi che si basano sulla fissazione di formaldeide, PCHi-C registra solo le interazioni che sono 'congelate' presso il punto di tempo di fissazione. Così, per studiare la cinetica e la dinamica delle interazioni di promotore, metodi come microscopia di Super-risoluzione cellule vive sono tenuti insieme a PCHi-C.
Metodi di sezionare l'organizzazione spaziale del cromosoma ad alta risoluzione
La complessità vasta delle librerie di interazione cromosomiche vieta l'identificazione affidabile di prodotti di interazione tra due frammenti di restrizione specifici con significatività statistica. Per aggirare questo problema, acquisizione di sequenza è stato utilizzato per arricchire o Hi-C33,34,40,44 o 3 C50,51 librerie per interazioni specifiche. Il principale vantaggio dell'utilizzo di librerie librerie oltre 3C Hi-C per la fase di arricchimento è che Hi-C, a differenza di C 3, include una fase di arricchimento per i prodotti genuini di legatura. Di conseguenza, la percentuale di legge valide in librerie PCHi-C è di circa 10 volte superiore a quella in Capture-C biblioteche50, che conteneva circa 5 – 8% valido legge dopo il filtraggio di HiCUP. Serifoski et al hanno confrontato direttamente cattura-C a HiCap, che come PCHi-C utilizza librerie Hi-C per l'arricchimento di cattura, in contrasto con Capture-C che utilizza librerie C 3. Coerente con i nostri risultati, hanno trovato che cattura-C biblioteche sono costituiti principalmente da frammenti ONU-dei40. Inoltre, le librerie HiCap avevano una complessità maggiore di cattura-C biblioteche40.
Una variante di cattura-C, chiamato generazione cattura-C52 (NG cattura-C) utilizza un oligo al fine di frammento di restrizione, come precedentemente stabilito in PCHi-C33,34, invece che sovrapposte sonde utilizzate nell'originale Protocollo di acquisizione-C50. Questo aumenta la percentuale di letture valide rispetto all'acquisizione-C modestamente, ma NG cattura-C impiega due turni sequenziale di arricchimento di cattura, e un numero relativamente elevato di PCR cicli (cicli di 20-24 in totale, rispetto a 11 cicli in genere per PCHi-C), che inevitabilmente si traduce in più alti numeri di sequenza duplicati e minore complessità di biblioteca. In esperimenti di prova durante l'ottimizzazione di PCHi-C, abbiamo trovato che la percentuale di unico (cioè, non duplicati) legge coppie era solo circa 15% quando abbiamo usato 19 cicli di PCR (13 cicli pre-capture + 6 cicli post-acquisizione; dati non indicati), tuttavia ottimizzazione di un minor numero di cicli di PCR, in genere produce 75 – 90% coppie di lettura uniche. Quindi, riducendo il numero di cicli di PCR sostanzialmente aumenta la quantità di dati di sequenza informativa.
Un metodo recente combina ChIP con Hi-C di concentrarsi sulle interazioni cromosomiche mediati da una proteina specifica di interesse (HiChIP53). Rispetto alla ChIA-PET54, che si basa su una logica simile, HiChIP dati contengano un numero maggiore di letture di sequenza informativo, consentendo l'interazione maggiore fiducia chiamando53. Sarà molto interessante confrontare direttamente il corrispondente HiChIP e insiemi di dati di cattura Hi-C una volta che diventano disponibili (ad esempio HiChIP usando un anticorpo contro la coesina unità Smc1a53 con Capture Hi-C per tutti i Smc1a associato restrizione frammenti) fianco a fianco. Una differenza intrinseca tra questi due approcci è che catturare Hi-C non si basa su immunoprecipitazione della cromatina e pertanto è in grado di interrogare cromosomiche interazioni indipendentemente dall'occupazione di proteina. In questo modo il confronto dell'organizzazione del genoma 3D in presenza o in assenza di specifico fattore legante, come è stato utilizzato per identificare PRC1 come regolatore chiave del mouse ESC genoma spaziale architettura7.
PCHi-C e GWAS
Studi di associazione genome-wide (GWAS) hanno rivelato che superiore al 95% della malattia-collegati varianti di sequenza si trovano in regioni non codificanti del genoma, spesso a grandi distanze di proteina-codificazione geni55. Varianti di GWAS sono spesso trovati in prossimità di dnasi I siti ipersensibili, che è un segno distintivo delle sequenze con potenziale attività regolatoria. PCHi-C e catturare Hi-C sono stati ampiamente utilizzati per collegare i promotori ai loci GWAS rischio implicati nel cancro di seno44, cancro colorettale48e malattia autoimmune35,45,46. Un PCHi-C studio su 17 cellule ematopoietiche umane differenti tipi trovano SNPs associati con la malattia autoimmune sono stati arricchiti in PIRs in cellule linfoidi, mentre varianti di sequenza associate tratti specifici delle piastrine e globuli rossi sono stati trovati principalmente in i macrofagi ed eritroblasti, rispettivamente35,56. Così, promotore specifico tessuto tipo Interattomi scoperto da PCHi-C possono aiutare a capire la funzione della malattia-collegati non codificanti varianti di sequenza e identificare nuovi geni malattia potenziale per intervento terapeutico.
Caratteristiche delle regioni promotore-interagire
Parecchie linee di prova link promotore Interattomi a controllo di espressione genica. In primo luogo, parecchi PCHi-C studi hanno dimostrato che le regioni genomiche che interagiscono con i promotori di geni espressi (altamente) sono arricchite in segni associati con attività di enhancer, come H3K27 acetilazione e p300 associazione33,34 , 37. abbiamo trovato una correlazione positiva tra livello di espressione genica e il numero di rinforzatori interagenti, suggerendo che gli effetti additivi di risultato rinforzatori in aumentata espressione genica livelli34,35. In secondo luogo, di origine naturale espressione loci quantitative trait (QTLs) sono arricchiti in PIRs connessi agli stessi geni cui espressione è influenzata dal QTLs35. In terzo luogo, integrando dati PCHi-C e viaggio57 , Cairns et al trovato che geni reporter di viaggio mapping a PIRs in mouse CES mostrano reporter più forte espressione genica di geni reporter presso siti di integrazione nelle regioni non-interagenti promotore 58, che indica che il PIRs possiedono attività trascrizionale di regolamentazione. Insieme, questi risultati suggeriscono che il promotore Interattomi scoperti da PCHi-C in vari tipi di cellule umane e del mouse includono moduli regolatori chiave per controllo di espressione genica.
Vale la pena notare che i rinforzatori rappresentano solo una piccola frazione (~ 20%) di tutte le PIRs scoperto da PCHi-C33,34. Altri PIRs potrebbe avere ruoli strutturali o topologici anziché funzioni di regolamentazione trascrizionale dirette. Tuttavia, c'è anche prova che PCHi-C può svelare elementi di DNA con funzione regolatrice che non porto classica enhancer marchi. In una linea di cellule linfoidi umane, il promotore di BRD7 è stato trovato per interagire con una regione priva di segni di enhancer che è stata indicata per possedere l'attività enhancer in reporter gene saggi33. Elementi regolatori con caratteristiche simili possono essere più abbondanti che attualmente apprezzato. Ad esempio, una schermata basata su CRISPR per DNA elementi identificati contrassegno normativi elementi regolatori (UREs) che controllano l'espressione genica, ma sono privi del rinforzatore segna59.
In altri casi, PIRs hanno dimostrato di marchi di cromatina associate di repressione trascrizionale del porto. PIRs e interagenti promotori vincolati PRC1 in mouse CES erano impegnati in una vasta rete spaziale dei geni repressi recanti che il repressivo Marco H3K27me37. In cellule linfoblastoidi umane, un elemento distante interagendo con il promotore di BCL6 represso transgene reporter gene espressione33, suggerendo che potrebbe funzionare per reprimere la trascrizione di BCL6 nel suo contesto nativo.
PIRs arricchiti per occupazione della proteina isolante della cromatina CTCF umano CES e NECs37 può rappresentare un'altra classe di PIRs. Collettivamente, questi risultati suggeriscono che il PIRs harbor una raccolta di attività di regolamentazione gene ancora ad essere funzionalmente caratterizzato.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Ringraziamo Valeriya Malysheva per lettura critica del manoscritto e aiuto di esperti con la figura 1. Questo lavoro è stato supportato dal Medical Research Council, UK (MR/L007150/1) e UK Biotechnology and Biological Sciences Research Council, UK (BB/J004480/1).
Materiali
| Name | Company | Catalog Number | Comments |
| 16% (vol/vol) paraformaldehyde solution | Agar Scientific | R1026 | |
| Dulbecco's Modified Eagle Medium (DMEM) 1x | Life Technologies | 41965-039 | |
| Fetal bovine serum (FBS) sterile filtered | Sigma | F9665 | |
| Low-retention filter tips | Starlab | S1180-3810, S1180-1810, S1180-8810 and S1182-1830 | |
| 10x PBS pH 7.4 | Life Technologies | 70011-036 | |
| Molecular biology grade water | Sigma-Aldrich | W4502 | |
| 1 M Tris-HCl pH 8.0 | Life Technologies | 15568-025 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 5 M NaCl | Life Technologies | 24740-011 | |
| Protease inhibitor cocktail (EDTA-free) | Roche Diagnostics | 11873580001 | |
| Restriction buffer 2 (10x NEBuffer 2) | New England Biolabs | B7002 | |
| DNA LoBind tube, 1.5 mL | Eppendorf | 0030 108.051 | |
| DNA LoBind tube, 2 mL | Eppendorf | 30108078 | |
| 20% (wt/vol) SDS | Bio-Rad Laboratories | 161-0418 | |
| 20% (vol/vol) Triton X-100 | Sigma-Aldrich | T8787 | |
| HindIII, 100 U/uL | New England Biolabs | R0104 | |
| 10 mM dCTP | Life Technologies | 18253-013 | |
| 10 mM dGTP | Life Technologies | 18254-011 | |
| 10 mM dTTP | Life Technologies | 18255-018 | |
| 0.4 mM Biotin-14-dATP | Life Technologies | 19524-016 | |
| DNA polymerase I large (Klenow) fragment 5000 units/mL | New England Biolabs | M0210 | |
| 10x T4 DNA ligase reaction buffer | New England Biolabs | B0202 | |
| 100x 10mg/ml Bovine Serum Albumin | New England Biolabs | B9001 | |
| T4 DNA ligase, 1 U/μL | Invitrogen | 15224-025 | |
| RNase A | Roche | 10109142001 | |
| Proteinase K, recombinant, PCR grade | Roche | 3115836001 | |
| 20 000×g 50 ml centrifuge tube | VWR | 525-0156 | |
| 0.5 M EDTA pH 8.0 | Life Technologies | 15575-020 | |
| Phenol pH 8.0 | Sigma | P4557 | |
| Phenol: Chloroform: Isoamyl Alcohol 25:24:1 | Sigma | P3803 | |
| Sodium acetate pH 5.2 | Sigma | S7899 | |
| Quant-iT PicoGreen | Invitrogen | P7589 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| Restriction buffer 2.1 (10x NEBuffer 2.1) | New England Biolabs | B7202 | |
| NheI, 100U/uL | New England Biolabs | R0131 | |
| Micro TUBE AFA Fiber Pre-slit snap cap 6x16mm vials | Covaris | 520045 | For sonication |
| SPRI beads (Agencourt AMPure XP) | Beckman Coulter | A63881 | |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| Tween 20 | Sigma | P9416 | |
| 10 mM dATP | Life Technologies | 18252-015 | |
| T4 DNA polymerase 3000 units/mL | New England Biolabs | M0203 | |
| T4 PNK 10000 units/mL | New England Biolabs | M0201 | |
| Klenow exo minus 5000 units/mL | New England Biolabs | M0212 | |
| Quick ligation reaction buffer | New England Biolabs | B6058 | |
| NEB DNA Quick ligase | New England Biolabs | M2200 | |
| PE adapter 1.0 (5'-P-GATCGGAAGAGCGGTTCAGC AGGAATGCCGAG-3') | Illumina | ||
| PE adapter 2.0 (5'-ACACTCTTTCCCTACACGACGCT CTTCCGATCT-3') | Illumina | ||
| NEB Phusion PCR kit | New England Biolabs | M0530 | |
| PE PCR primer 1.0 (5'-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTAC ACGACGCTCTTCCGATCT-3') | Illumina | ||
| PE PCR primer 2.0 (5'-CAAGCAGAAGACGGCATACGA GATCGGTCTCGGCATTCCT GCTGAACCGCTCTTCCGATCT-3') | Illumina | ||
| PCR strips | Agilent Technologies | 410022 and 401425 | |
| SureSelect SSEL TE Reagent ILM PE full adaptor kit | Agilent Technologies | 931108 | |
| SureSelect custom 3-5.9 Mb library | Agilent Technologies | 5190-4831 | custom design mouse or human PCHi-C system |
| Dynabeads MyOne Streptavidin T1 beads | Invitrogen | 65601 | |
| E220 high-performance focused ultra-sonicator | Corvaris | E220 |
Riferimenti
- Osborne, C. S., et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nature Genetics. 36, 1065-1071 (2004).
- Schoenfelder, S., et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature Genetics. 42, 53-61 (2010).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501, 227-231 (2013).
- Bantignies, F., et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 144, 214-226 (2011).
- Engreitz, J. M., et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 341, 1237973(2013).
- Denholtz, M., et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 13, 602-616 (2013).
- Schoenfelder, S., et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics. 47, 1179-1186 (2015).
- Kundu, S., et al. Polycomb Repressive Complex 1 generates discrete compacted domains that change during differentiation. Molecular Cell. 65, 432-446 (2017).
- Skok, J. A., Gisler, R., Novatchkova, M., Farmer, D., de Laat, W., Busslinger, M. Reversible contraction by looping of the Tcra and Tcrb loci in rearranging thymocytes. Nature Immunology. 8, 378-387 (2007).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148, 908-921 (2012).
- Aymard, F., et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology. 24, 353-361 (2017).
- Ryba, T., et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research. 20, 761-770 (2010).
- Pope, B. D., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 515, 402-405 (2014).
- Chandra, T., et al. Global reorganization of the nuclear landscape in senescent cells. Cell Reports. 10, 471-483 (2015).
- Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., Fraser, P. Long-range chromatin regulatory interactions in vivo. Nature Genetics. 32, 623-626 (2002).
- Tolhuis, B., Palstra, R. J., Splinter, E., Grosveld, F., de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Molecular Cell. 10, 1453-1465 (2002).
- Amano, T., Sagai, T., Tanabe, H., Mizushina, Y., Nakazawa, H., Shiroishi, T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Developmental Cell. 16, 47-57 (2009).
- Zuniga, A., et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes & Development. 18, 1553-1564 (2004).
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132, 797-803 (2005).
- D'Haene, B., et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 5, e1000522(2009).
- Sur, I. K., et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 338, 1360-1363 (2012).
- Herranz, D., et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature Medicine. 20, 1130-1137 (2014).
- Deng, W., et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 149, 1233-1244 (2012).
- Groschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157, 369-381 (2014).
- Lupianez, D. G., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 161, 1012-1025 (2015).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538, 265-269 (2016).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295, 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38, 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genetics. 38, 1341-1347 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research. 16, 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289-293 (2009).
- Belton, J. M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., Dekker, J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 58, 268-276 (2012).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47, 598-606 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582-597 (2015).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167, 1369-1384 (2016).
- Wilson, N. K., et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 127, e12-e23 (2016).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife. 6, (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49, 1522-1528 (2017).
- Siersbaek, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66, 420-435 (2017).
- Sahlen, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Nagano, T., et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64 (2013).
- Nagano, T., Varnai, C., Schoenfelder, S., Javierre, B. M., Wingett, S. W., Fraser, P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology. 16, 175(2015).
- Wingett, S., et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Res. 4, 1310(2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24, 1854-1868 (2014).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- McGovern, A., et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol.ogy. 17, 212(2016).
- Hodge, D., et al. A global role for EKLF in definitive and primitive erythropoiesis. Blood. 107, 3359-3370 (2006).
- Jager, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter Interactions between two states of pluripotency. Cell Stem Cell. 17, 748-757 (2015).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46, 205-212 (2014).
- Kolovos, P., et al. Targeted Chromatin Capture (T2C): A novel high-resolution high-throughput method to detect genomic interactions and regulatory elements. Epigenetics Chromatin. 7, 10(2014).
- Davies, J. O., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13, 74-80 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13, 919-922 (2016).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462, 58-64 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337, 1190-1195 (2012).
- Petersen, R., et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat. Commun. 8, 16058(2017).
- Akhtar, W., et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 154, 914-927 (2013).
- Cairns, J., et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biology. 17, 127(2016).
- Rajagopal, N., et al. High-throughput mapping of regulatory DNA. Nature Biotechnology. 34, 167-174 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati