
Method Article
Promotor Capture Hallo-c: Hochauflösende, genomweite Profilierung der Promoter Interaktionen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
DNA-regulatorische Elementen, wie z. B. Enhancer, Steuern Genexpression durch Kontaktaufnahme mit körperlich Ziel Gen Promotoren, oft durch weiträumige chromosomalen Interaktionen über genomische Entfernungen. Promotor erfassen Hi-C (PCHi-C) identifiziert signifikante Wechselwirkungen zwischen Promotoren und distalen Regionen ermöglicht die Zuordnung von möglichen regulatorischen Sequenzen, deren Zielgene.
Zusammenfassung
Die dreidimensionale Organisation des Genoms ist mit seiner Funktion verknüpft. Z. B. Steuern regulatorische Elemente wie transkriptionelle Enhancer, den räumlich-zeitlichen Ausdruck ihrer Zielgene durch körperlichen Kontakt, oft erhebliche (in einigen Fällen Hunderte von Kilobases) genomischen Entfernungen überbrücken und unter Umgehung nahe gelegenen Gene. Das menschliche Genom hegt eine geschätzte 1 Million Enhancer, die überwiegende Mehrheit davon haben unbekannte gen Ziele. Ihre Zielgene distalen regulatorische Regionen zuordnen ist daher wichtig zu verstehen, gen-Expressions. Wir entwickelten Promoter erfassen Hi-C (PCHi-C) ermöglichen die genomweite Detektion von distalen Promotor-Interaktion Regionen (PIRs), für alle Veranstalter in einem einzigen Experiment. In PCHi-C sind hochkomplexe Hi-C-Bibliotheken speziell für promotorsequenzen durch in-Lösung Hybrid-Auswahl mit Tausenden von biotinylierte RNA Köder komplementär zu den Enden alle Projektträger-haltigen Einschränkung Fragmente bereichert. Ziel ist es, dann Pulldown promotorsequenzen und ihre häufigen Interaktionspartnern wie Geschmacksverstärker und andere mögliche regulatorische Elemente. Nach hohem Durchsatz gepaart-End Sequenzierung wird ein statistischer Test jeder Veranstalter ligiert Beschränkungsfragment, bedeutende PIRs auf Beschränkung Fragment Ebene zu identifizieren. Wir haben PCHi-C verwendet, um einen Atlas der Langstrecken-Promoter Interaktionen in Dutzenden von Mensch und Maus Zelltypen zu generieren. Diese Veranstalter Interaktom Karten haben zu einem besseren Verständnis der Säugetier-gen-Expressions beigetragen, indem ihre Zielgene vermeintliche regulatorische Regionen zuordnen und bevorzugte räumliche Promotor-Promotor Interaktion Netze offenbart. Diese Information hat auch hohen Relevanz für das Verständnis menschlichen genetischen Krankheit und die Identifizierung von möglichen Krankheitsgenen, durch die Verknüpfung von nicht-kodierenden krankheitsassoziierten Varianten in oder in der Nähe von Sequenzen, deren Zielgene-Sequenz.
Einleitung
Sammeln Beweise legt nahe, dass die dreidimensionale Organisation des Genoms eine wichtige funktionelle Rolle in einer Reihe von atomaren Prozesse spielt, einschließlich gen Aktivierung1,2,3, Unterdrückung4 ,5,6,7,8, Rekombination9,10, DNA-Reparatur11, DNA-Replikation12,13, und zelluläre Seneszenz14. Entfernten Enhancer befinden sich in unmittelbarer räumlicher Nähe an die Projektträger, dass sie15,16,17, regulieren die für ordnungsgemäße Spatio-temporal gen-Expressions erforderlich ist. Enhancer Löschungen zeigen, dass die distale Enhancer für Ziel-gen Transkription18,19,20,21,22und "erzwungene Chromatin looping" zeigt, dass veränderter tethering zwischen Verstärker und ihre Ziel-Promoter in der Hbb -Lokus transkriptionelle Aktivierung23fahren ausreicht. Darüber hinaus können Genom Umgestaltungen, die Gene unter die Kontrolle der ektopische Enhancer bringen unangemessen Genaktivierung und Krankheit24,25,26führen. Zusammen, zeigen diese Beispiele, dass Veranstalter-Enhancer Interaktionen essentiell für gen-Kontrolle sind und erfordern strenge Regulierung um entsprechende Genexpression zu gewährleisten. Der Mensch und Maus Genome sind jeweils auf rund 1 Million Enhancer Hafen geschätzt. Für die überwiegende Mehrheit der diese Enhancer Zielgene sind unbekannt, und die "Rules Of Engagement" zwischen Promotoren und Geschmacksverstärker sind kaum erforscht. Ihre Zielgene transkriptionelle Enhancer zuweisen, bleibt somit eine große Herausforderung bei der Entschlüsselung Säugetier-gen-Expressions.
Unser Verständnis von dreidimensionalen Genom Architektur wurde durch die Einführung der 3 C27 (Chromosom Konformation Capture) und seine Varianten28,29,30,31 revolutioniert . Die mächtigsten dieser Techniken, Hi-C (Hochdurchsatz Chromosom Konformation Capture) soll das gesamte Ensemble der chromosomalen Interaktionen innerhalb einer Zellpopulation zu identifizieren. Hallo-C-Bibliotheken, in der Regel generiert aus Millionen von Zellen, sind sehr komplex, mit einer geschätzten 1011 unabhängige Ligatur Produkte zwischen ~ 4 kb Fragmente im menschlichen Genom32. Als eine Konsequenz, zuverlässige und reproduzierbare Ermittlung von Wechselwirkungen zwischen einzelnen Einschränkung (z. B. diejenigen, die ein Veranstalter oder Enhancer) aus Fragmenten ist Hi-C Daten nicht machbar, wenn Hi-C-Bibliotheken, Ultra-Tiefe Sequenzierung ausgesetzt sind, Das ist keine wirtschaftlich tragfähige Lösung für Labors routinemäßig Hi-C-Bibliotheken vorbereiten. Um dieses Manko zu umgehen, haben wir Promoter erfassen Hi-C bereichern speziell Promotor-haltigen Ligatur Produkte von Hi-C-Bibliotheken entwickelt. Wir konzentrieren auf Promotoren aus zwei Gründen. Zuerst Promotor-Enhancer Kontakte haben gezeigt, dass entscheidend für die richtige Gen Ausdruck Niveaus in zahlreichen Studien werden (siehe Verweise oben), und zweitens wie Promotoren weitgehend invariante zwischen Zelltypen sind, die gleichen Köder Bilderfassungssystem kann verwendet werden, zu befragen die regulatorischen Schaltung über mehrere Zelltypen und Bedingungen. Unser Ansatz beruht auf in-Lösung Hybridisierung von Hi-C-Bibliotheken mit Zehntausenden von biotinylierte RNA 120mers Promotor-haltigen Hi-C Ligatur Produkte und spätere Erfassung auf Streptavidin beschichteten magnetische Beads ergänzen. Dies führt in PCHi-C-Bibliotheken mit viel reduziert Komplexität im Vergleich zu der ursprünglichen Hi-C-Bibliothek, konzentriert sich nur auf die Identifizierung der Fragmente, die Projektträger bei signifikant hohen Frequenzen ligiert werden.
Wir haben PCHi-C in einer Reihe von Mensch und Maus Zelltypen dazu beitragen, ein besseres Verständnis der Gen-Expressions durch Aufdeckung Langstrecken distalen Promotor interagierenden Regionen mit vermeintlichen regulierende Funktion, als auch nicht-zufällige verwendet. Veranstalter-Promotor Kontakte im dreidimensionalen Raum des Kerns. Die Studien haben Hunderttausende von Promoter-Enhancer Kontakte zugeordnet, über zahlreiche Zelle Arten33,34,35,36,37,38, 39, Polycomb repressiven komplexe vermittelte räumliche Genom-Organisation in Maus embryonale Stammzellen7identifiziert, demonstriert umfangreiche Neuverkabelung der Veranstalter Interactomes während zellulare Unterscheidung37, 38 , 39, und verknüpften nicht-kodierende krankheitsassoziierten Sequenz Varianten bis Gen Promotoren35.
PCHi-C ist eine ideal Methode der genomweiten Ensemble von DNA-Sequenzen, die Interaktion mit den Projektträgern zuordnen. Ähnliche Ansätze, wie z. B. erfassen Hi-C kontinuierliche genomische Regionen (siehe Diskussion) sind die Methode der Wahl zur hochauflösenden Interaktion Profile für ausgewählte genomische Regionen zu erhalten. PCHi-C- und Hi-C zu erfassen sind sehr ähnlich aus einer experimentellen Sicht (der einzige Unterschied ist die Wahl der Capture-System), so dass die Ratschläge und Richtlinien wir bieten für beide Ansätze gelten. Hier präsentieren wir Ihnen eine detaillierte Beschreibung der PCHi-C. Wir skizzieren die Begründung und Gestaltung eines PCHi-C-Experiments, bieten eine schrittweise PCHi-C Bibliothek Generation Protokoll und veranschaulichen, wie die Qualität der PCHi-C-Bibliotheken bei verschiedenen Schritte im Protokoll zum qualitativ hochwertigen Daten liefern überwacht werden kann.
Protokoll
(1) Formaldehyd Fixierung
-
Handy-Vorbereitung: beginnen Sie mit einem Minimum von 2 x 107 Zellen pro Versuch.
- Für Zellen in Kultur gezüchtet Aufschwemmen der Zellen in Kulturmedium. Aufschwemmen Sie für ex-Vivo Zellen in 1 x Dulbeccos geändert Eagle Medium (DMEM), mit 10 % (Vol/Vol) fetalen bovine Serum (FBS) ergänzt.
- Entfernen Sie für adhärente Zellen Kulturmedium und 30,625 mL frisches Medium mit 10 % (Vol/Vol) FBS bei Raumtemperatur (RT; 20 – 25 ° C).
- Für Aufhängung Zellen sammeln Sie und Zentrifugieren Sie Zellen bei 400 x g und 20 ° C für 3 min. entfernen überstand und wieder auszusetzen Sie Zelle Pellet in 30,625 mL Medium mit 10 % (Vol/Vol) FBS bei RT
- Verwenden Sie für festen Geweben Trypsin (0,05 % bis 2,5 % Endkonzentration, je nach Zelltyp) oder Dounce Homogenisieren, um eine einzelne Zelle Suspension zu erhalten. Nach diesen zusätzlichen Schritt behandeln Sie Zellen wie Aussetzung Zellen.
-
Hinzugeben Sie 4,375 mL 16 % Methanol-freie Paraformaldehyd (offene Ampulle nur vor dem Gebrauch), eine Endkonzentration von 2 % (Vol/Vol). Fix für 10 min bei RT mit schonendes mischen auf einer Wippe.
Achtung: Paraformaldehyd ist eine gefährliche Chemikalie. Befolgen Sie die entsprechenden Sicherheits- und Gesundheitsvorschriften. - Reaktion durch Zugabe von 5 mL frisch zubereiteten 1 M eiskalte Glycin zu stillen. Mix für 5 min mit sanftes Schaukeln bei RT, und dann für 15 min mit gelegentlichen invertieren auf Eis inkubieren.
-
Waschen und feste Zellen zu sammeln.
- Für adhärente Zellen überstand, fügen Sie 10 mL eiskaltes 1 x PBS pH-Wert 7,4 auf der Platte Wand hinzu und entfernen Sie es. Fügen Sie 1 mL eiskaltes 1 x PBS pH-Wert 7,4, sammeln Sie Zellen mit einer Zelle Schaber und Überführung in ein 50 mL-Tube. Wiederholen Sie zweimal, um so viele Felder wie möglich zu sammeln. Summieren Sie eiskaltem PBS zu 50 mL Endvolumen.
- Für Aussetzung Zellen Zentrifuge Zellen bei 760 X g und 4 ° C für 5 min, überstand zu entfernen und neu Zelle Pellet in 50 mL eiskaltem PBS pH 7.4 auszusetzen.
- Zellen bei 400 x g und 4 ° C für 10 min zentrifugieren und überstand sorgfältig zu entfernen. Die Zelle Pellet kann Snap in flüssigem Stickstoff eingefroren und anschließend für mehrere Monate bei-80 ° C gelagert werden.
(2) Zell-Lyse
- Wieder auszusetzen Zelle Pellet in 50 mL frisch zubereiteten eiskalte Lyse-Puffer (10 mM Tris-HCl pH 8, 0,2 % (Vol/Vol) Igepal CA-630, 10 mM NaCl, und eine Tablette Proteaseinhibitor cocktail) und mischen. Inkubation für 30 min auf Eis, mischen sich gelegentlich durch invertieren. Die Kerne auf 760 x g und 4 ° C für 5 min zentrifugieren und überstand zu entfernen.
3. HindIII Verdauung
- Waschen Sie Zellkerne mit 1,25 X Einschränkung Puffer 2. Wieder auszusetzen Sie Zelle Pellet in 1 mL eiskaltes 1,25 X Einschränkung Puffer 2 und Überführung in eine 1,5 mL-Tube. Drehen Sie die Kerne bei 760 x g und 4 ° C für 5 min und entfernen Sie überstand zu.
- Wieder auszusetzen Sie Zelle Pellet in 1790 µL 1,25 X Einschränkung Puffer 2. Machen Sie 5 Aliquote jeweils 5 Millionen Zellen in 358 µL 1,25 x Einschränkung Puffer 2.
- Fügen Sie 11 µL 10 % (wt/Vol) SDS pro aliquoten und schütteln am 950 Umdrehungen pro Minute (u/min) für 30 min bei 37 ° C in einem Thermomixer. Wenn Zelle Klumpen erscheinen, distanzieren Sie sich von pipettieren, Vermeidung von Luftblasen.
- Fügen Sie 75 µL 10 % Triton x-100 (Vol/Vol) pro aliquoten und schütteln bei 950 u/min und 37 ° C für 15 min in einem Thermomixer. Wenn Zelle Klumpen erscheinen, distanzieren Sie sich von pipettieren, Vermeidung von Luftblasen.
-
Fügen Sie 12 µL 100 U/µL HindIII 100 (von insgesamt 1.200 Einheiten) pro aliquoten und Inkubation bei 37 ° C über Nacht (O/N) unter Schütteln bei 950 u/min in einem Thermomixer.
- Übertragen Sie für die Verdauung-Kontrolle 25 µL der Probe (5 µL aus jeder Aliquote) in einen neuen Schlauch vor der Zugabe des Enzyms (unverdaute Kontrolle) und wiederholen Sie den Vorgang nach Zugabe des Enzyms (verdaute Kontrolle). Inkubieren Sie beide Röhren in der gleichen Weise wie die Hi-C-Bibliothek.
- Am nächsten Morgen fügen Sie 5 µL 100 U/µL HindIII (insgesamt 500 Einheiten) pro aliquoten und Inkubation bei 37 ° C für 2 h unter Schütteln bei 950 u/min in einem Thermomixer.
-
Verdauung-Kontrolle: für die verdaut und unverdaute Steuerelemente (siehe 3.5.1), Crosslink Umkehrung (Schritt 6), Phenol: Chloroform Extraktion und DNA Niederschlag (Schritt 7) durchführen.
- Entwerfen Sie ein paar Zündkapseln, die eine dIII-Website Hinerstrecken. Entwerfen Sie in der gleichen Region ein weiteres Paar Zündkapseln, die eine HindIII Website umfassen nicht. Design-Primer für die quantitative PCR (Q-PCR) mit Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) und die folgenden Parameter:
Primer-Größe: optimale 20 (Min.: 18, Max.: 27); Zündkapsel Tm: Optimale 60 (Min.: 57, Max.: 63); Grundierung CG % Inhalt: Min.: 20, Max.: 80; Größe der Amplifikate: RT-PCR ~ 100 bp (für konventionelle PCR ~ 300 bp); Mispriming Bibliothek: Mensch (menschliche Primer) oder Nagetier und einfache (Maus-Primer). - Durchführen von Q-PCR um 4 mittlere Cts (Schwellenwert-Zyklus) zu erhalten: Ct [D; [H], gewonnen aus der verdauten Probe [D] mit dem Paar Zündkapseln, die umfassen eine HindIII Website [H]; CT [D;-], gewonnen aus der verdauten Probe [D] mit dem Paar Zündkapseln, die eine HindIII Website [-]; umfassen nicht CT [U; [H], gewonnen aus der unverdaute Probe [U] mit dem Paar Zündkapseln, die eine HindIII Website umfassen; CT [U;-], gewonnen aus der unverdaute Probe [U] mit dem Paar Zündkapseln, die eine HindIII Website [-] umfassen nicht. Berechnung des Prozentsatzes der Verdauung als: % Verdauung = 100-100/2(Ct[D,H]-Ct[D,-]) - (Ct[U,H]-Ct[U,-]).
- Entwerfen Sie ein paar Zündkapseln, die eine dIII-Website Hinerstrecken. Entwerfen Sie in der gleichen Region ein weiteres Paar Zündkapseln, die eine HindIII Website umfassen nicht. Design-Primer für die quantitative PCR (Q-PCR) mit Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) und die folgenden Parameter:
4. Biotinylierungen der Beschränkungsfragment Überhänge
- Vorbereitung biotinylierungen-master-Mix: 30,6 µL 10 x Einschränkung Puffer 2, H2O 10.2 µL (Molekulare Biologie Klasse), 7,65 µL 10 mM dCTP 7,65 µL 10 mM dGTP, 7,65 µL 10 mM dTTP, 191.25 µL von 0,4 mM Biotin-14-dATP und 51 µL von 5.000 U/mL DNA-Polymerase ich große () Klenow) Fragment.
- Fügen Sie 60 µL biotinylierungen master-Mix pro aliquoten, Mix und Inkubation bei 37 ° C für 1 h schütteln bei 700 u/min (Thermomixer) für 5 s, 30 s. Legen Sie nach 1 h Aliquote auf Eis
(5) im Zellkern Ligation
- Ligatur-master-Mix vorzubereiten: 510 µL 10 x T4 DNA-Ligase Puffer, 51 µL von 10 mg/mL Rinderserumalbumin (100 X BSA), 1754.4 µL Wasser (Molekulare Biologie Klasse) und 127,5 µL 1 U/µL T4 DNA-Ligase (siehe Tabelle der Materialien).
- 479 µL Ligatur master-Mix pro aliquoten Mix hinzufügen und inkubieren Sie bei 16 ° C für 4 h schütteln bei 700 u/min für 5 s alle 2 min in einem Thermomixer.
- 30 min bei RT inkubieren
6. Crosslink Umkehr
- Kombinieren Sie alle Aliquote in ein 50 mL Zentrifugenröhrchen (geeignet für High-Speed-Zentrifugation).
- Hinzufügen von 62,5 µL RNase A, Mix, 10 mg/mL und 30 min bei 37 ° c inkubieren
- Fügen Sie 300 µL von 10 mg/mL Proteinase K, Mischung, und 30 min bei 37 ° c inkubieren
- Inkubieren Sie Reaktion O/N (oder mindestens 4 Stunden) bei 65 ° C. Am nächsten Morgen 300 µL von 10 mg/mL Proteinase K, Mischung, fügen und 1 h bei 65 ° c inkubieren
(7) DNA-Reinigung
- Fügen Sie 4337.5 µL TLE-Puffer (10 mM Tris-HCl pH 8.0; 0,1 mM EDTA pH 8.0) und mischen.
- Eine neue 50 mL-Tube 1 Volumen (10 mL) Phenol pH 8.0, Wirbel für 10 s und Zentrifuge mit RT und 20.000 X g für 3 min. Transfer 9 mL der oberen (Wasserphase) hinzufügen.
Achtung: Phenol ist eine gefährliche Chemikalie. Befolgen Sie die entsprechenden Sicherheits- und Gesundheitsvorschriften. - Fügen Sie 2 mL TLE-Puffer, um die verbleibenden wässrige Phase, Wirbel für 10 s und Zentrifuge bei RT und 20.000 X g für 3 min. 2,5 mL der wässrigen Phase in das neue Rohr übertragen aus Schritt 7.2, wodurch das Endvolumen 11,5 mL. Röhrchen mit der unteren (Bio) Phase zu verwerfen.
- Eine neue 50 mL-Tube 1 Volumen (11,5 mL) von Phenol: Chloroform: Isoamyl Alkohol (25:24:1), Wirbel für 10 s und Zentrifuge mit RT und 20.000 X g für 3 min. Transfer 11 mL der oberen (Wasserphase) hinzufügen. Wiederholen Sie Schritt 7.3. Die Gesamtstichprobe Lautstärke wird jetzt 13,5 mL.
- Fügen Sie 1,35 mL 3 M Natrium Acetat pH 5,2 und 33,75 mL eiskaltes 100 % Ethanol, Mischung, und bei-80 ° C für 45 min inkubieren oder alternativ über Nacht bei-20 ° C.
- Bei 4 ° C und 20.000 X g für 10 min zentrifugieren, überstand zu entfernen, erneut aussetzen Pellet in 1 mL frisch zubereiteten 70 % (Vol/Vol) Ethanol und übertragen auf einen neuen Schlauch.
- Zentrifugieren bei 4 ° C und bei voller Geschwindigkeit für 3 min in einer Benchtop-Zentrifuge, dann entfernen Sie überstand.
- Neu Pellet in 1 mL Eis kalt 70 % (Vol/Vol) Ethanol und wiederholen Sie Schritt 7,7. Das Pellet bei 37 ° C für 10 min Trocknen und in 650 µL TLE Puffer wieder auszusetzen. Bestimmen Sie die DNA-Ausbeute mithilfe eine Fluoreszenz basierende Assays, um doppelsträngige DNA zu quantifizieren.
Hinweis: Das Protokoll kann hier durch Snap Einfrieren und Lagerung der Probe bei-80 ° C für mehrere Monate oder bei-20 ° C für einen kürzeren Zeitraum angehalten werden.
(8) Qualitätskontrollen
- Monitor-Bibliothek-Integrität und Ligation von DNA Elektrophorese. Run 200 ng der Bibliothek auf einem 0,8 % Agarose/1 X TBE Gel. Die DNA führen als Band mehr als 10 kb.
- Bekannten Zelltyp invariante Short und long - range Interaktionen durch konventionelle PCR zu erkennen. Einsatz 100 ng der Schablone DNA pro PCR-Reaktion. Entwerfen Sie die PCR-Primer, nah und in Richtung der Restriktionsschnittstellen folgen Sie den Anweisungen oben (siehe 3.7.1). Primer-Sequenzen für die Qualitätskontrolle von Maus und Mensch Hi-C-Bibliotheken sind in Tabelle 1aufgeführt.
-
Fill und Ligation Kontrolle: Schneiden Sie die Gel-Bänder mit der Amplifikate aus Steuern 8.2, DNA-Gel-Extrakt und die DNA als Vorlage für 4 einzelnen PCR-Reaktionen mit identischen Grundierung Kombinationen verwenden.
- Reinigen Sie Amplifikate mit einem PCR-Reinigung-Kit und quantifizieren Sie die DNA-Konzentration zu.
- Bereiten Sie vier Verdauung Reaktionen (HindIII [a], NheI [b], HindIII + NheI [c] und kein Enzym [d]) für jede Amplifikate in einem Endvolumen von 15 µL: 500 ng der Amplifikate, 1,5 µL 10 x Einschränkung Puffer 2.1, 0,15 µL von 10 mg/mL Rinderserumalbumin (100 X BSA) , und 0,1 µL (10 Stück) des Enzyms (HindIII [a], NheI [b], HindIII + NheI [c] oder Wasser [d]).
- Für 1 h bei 37 ° C zu verdauen Sie, dann laufen Sie Verdauung Reaktionen auf einem 1,5 % (wt/Vol) Agarose/1 X TBE Gel.
(9) DNA-Fragmentierung
- Übertragen Sie 50,5 µg der Probe in einen neuen Schlauch und TLE Puffer zu einem Endvolumen von 655 µL. Split-Sample in 5 Beschallung Fläschchen (siehe Tabelle der Materialien) indem Sie jedes Fläschchen 130 µL der Bibliothek (10 µg) hinzufügen. Scherkräfte auf eine Größe von ca. 400 bp in einem Ultra-sonikator (siehe Tabelle der Materialien) mit den folgenden Parametern: Einschaltdauer: 10 %; einfallende Spitzenleistung (w): 140; Zyklen pro Burst: 200; Zeit: 55 s.
- Sammeln Sie beschallten Probe in einem frischen 2 mL Röhrchen.
10. Double-sided SPRI-Bead Größenauswahl
- Mix SPRI (feste Phase Reversible Immobilisierung) Perle Lösung gut durch invertieren, 1,85 mL Korn-Lösung zu einem neuen Schlauch übertragen und für 15 min zum RT bringen.
- Das Beispiel 350 µL Wasser (Molekulare Biologie Klasse) hinzufügen (Endvolumen 1 mL).
- Das Beispiel 600 µL SPRI-Perle-Lösung hinzufügen (Gesamtvolumen 1,6 mL; Verhältnis von SPRI Lösung, DNA: 0,6 bis 1), 5 min bei RT inkubieren und spin-Probe in einer Benchtop-Zentrifuge für 2 – 3 s Probe zu sammeln.
- Öffnen Sie den Deckel, legen Sie die Probe auf die magnetische Trennung stehen für 5 min, Transfer klaren überstand in ein neues Rohr und verwerfen Sie Perlen zu.
- SPRI Perlen für den zweiten Schritt der Größe Auswahl konzentrieren: Transfer 930 µL SPRI Perlen in einen neuen Schlauch, legen auf die magnetische Trennung stehen für 5 min und entsorgen klaren überstand. Wieder aussetzen Sie, die Perlen in 310 µL SPRI-Perle-Lösung.
- Die Probe 300 µL konzentrierte SPRI Perlen (Schritt 10.5) hinzufügen (Gesamtvolumen 1,9 mL; Verhältnis SPRI DNA löst jetzt 0,9 bis 1), 5 min bei RT inkubieren und Spin probieren in der Benchtop-Zentrifuge für 2 – 3 s. vorsichtig Öffnen des Deckels , legen Sie das Rohr auf die magnetische Trennung stehen für 5 min und überstand verwerfen.
- Das Probenröhrchen auf Magnetische Trennung Stand 1 mL frisch zubereitete 70 % Ethanol (Vol/Vol) hinzu, 30 s, und verwerfen überstand inkubieren. Zweimal wiederholen.
- Trockenen Perlen bei 37 ° C in einem Thermomixer (Rohr Deckel öffnen) für nicht mehr als 5 min. hinzufügen 300 µL TLE Puffer zur Probe, mischen und 10 min bei Raumtemperatur inkubieren.
- Spin-Probe in einer Benchtop-Zentrifuge für 2 – 3 s, öffnen Sie den Deckel und das Rohr auf die magnetische Trennung stehen für 5 min. Transfer klar überstand in ein neues Rohr und Perlen zu verwerfen.
11. Biotin/Streptavidin Pulldown-Ligation Produkte
- Bereiten Sie Puffer: 1 x TB Puffer (5mM Tris-HCl pH 8.0, 0,5 mM EDTA, 1 M NaCl; 0,05 % Tween 20); 2 x NTB-Puffer (10 mM Tris-HCl pH 8,0; 1 mM EDTA 2 M NaCl); 1 X NTB-Puffer (5 mM Tris-HCl pH 8.0, 0,5 mM EDTA, 1 M NaCl).
- Fügen Sie 200 µL des magnetischen Streptavidin-gekoppelten Beads (siehe Tabelle der Materialien) in einen neuen Schlauch, legen Sie es auf die magnetische Trennung stehen für 1 min und entfernen Sie überstand zu.
-
Waschen Sie Perlen zweimal mit 500 µL 1 X TB Puffer.
- Für jedem Waschschritt während der Biotin Pulldown, Ende Reparatur und Entfernung von Biotin an nicht ligiert DNA enden, dATP Tailing und Adapter Ligatur Schritte, wieder auszusetzen Sie, die Perlen in den entsprechenden Puffer, RT und 15 u/min, 3 min drehen Sie, drehen Sie das Rohr in einer Benchtop-Zentrifuge Legen Sie für 2 – 3 s das Rohr auf die magnetische Trennung stehen für 3 min und entfernen Sie überstand zu.
- Wieder aussetzen Sie Perlen in 300 µL 2 X NTB-Puffer. Perlen und Probe (600 µL Gesamtvolumen) mischen und 15 min auf einem rotierenden Rad bei 3 u/min bei RT inkubieren.
- Perlen auf die magnetische Trennung stehen für 3 min zurückzufordern und den klare überstand zu entfernen. Waschen Sie Perlen zweimal in 500 µL 1 X NTB Puffer, und dann in 200 µL 1 X Ligatur Puffer. Wieder aussetzen Sie, die Perlen in 50 µL 10 X Ligatur Puffer.
12. Reparatur und Entfernung von Biotin an nicht ligiert DNA enden zu beenden
- Kombinieren Sie die Probe (insgesamt 50 µL) mit 50 µL 2,5 mM dNTP-Mix (12,5 µL 10 mM jedes dNTP), 18.1 µL von 3.000 U/mL T4 DNA-Polymerase, 18.1 µL von 10.000 U/mL T4 PNK, 3,7 µL von 5.000 U/mL DNA-Polymerase fragment ich große (Klenow) , und 360.1 µL von H2O.
- Mischen und inkubieren Sie bei 20 ° C für 1 h, schütteln 5 s bei 700 u/min alle 2 min in einem Thermomixer.
- Perlen auf dem Magnetische Trennung Stand zurückfordern, Entfernen des klare Überstands und Perlen in 500 µL 1 x TB Puffer zweimal waschen.
- Waschen Sie Perlen in 500 µL 1 X NTB-Puffer, gefolgt von einem Waschen in 500 µL 1 X TLE.
- Zurückfordern Sie Perlen auf die magnetische Trennung Stand, entfernen Sie den klare überstand zu und erneut aussetzen Sie Perlen in 415 µL 1 X TLE-Puffer.
13. dATP Tailing
- Kombinieren Sie Probe (415 µL) mit 50 µL 10 x Einschränkung Puffer 2, 5 µL 10 mM dATP und 30 µL 5 U/µL Klenow Exo-Minus.
- Mischen und Inkubation bei 37 ° C für 30 min, schütteln 5 s bei 700 u/min alle 2 min in einem Thermomixer.
- Perlen auf dem Magnetische Trennung Stand zurückfordern, Entfernen des klare Überstands und Perlen in 500 µL 1 x TB Puffer zweimal waschen.
- Gewaschen Sie Perlen in 500 µL 1 X NTB-Puffer werden.
14.-Adapter Ligation
- Perlen in 200 µL 1 x Ligatur Reaktion Puffer waschen (siehe Tabelle der Materialien).
- Wieder aussetzen Sie Perlen in 200 µL 1 X Ligatur Reaktion Puffer. Fügen Sie 4 µL DNA Ligase (siehe Tabelle der Materialien) und 16 µL von 15 µM geglüht Pre PE-Adapter (Pre-Tempern der PE-Adapter durch das gleiche Volumina von PE-Adapter 1 und PE 2 (jeweils 30 µM) mischen und Inkubation für ein paar Minuten bei RT). 15 min bei RT inkubieren.
- Perlen auf dem Magnetische Trennung Stand zurückfordern, Entfernen des klare Überstands und Perlen in 500 µL 1 x TB Puffer zweimal waschen.
- Gewaschen Sie Perlen in 500 µL 1 X NTB-Puffer werden. Dann, waschen Perlen in 100 µL 1 x Einschränkung Puffer 2, wieder aussetzen Perlen in 50 µL 1 x Einschränkung Puffer 2 und in einen neuen Schlauch übertragen.
15. Hallo-C-Bibliothek Verstärkung
- Bereiten Sie PCR-master-Mix: 100 µL 5 X Phusion Puffer; 6 µL 25 µM PE PCR Primer 1.0; 6 µL 25 µM PE PCR Primer 2.0; 14 µL dNTP-Mix (10 mM); 6 µL Phusion Polymerase; 318 µL von H2O.
- Mix-PCR-master-Mix mit Perlen (500 µL insgesamt), in 10 Aliquote von 50 µL teilen und verstärken durch PCR mit den folgenden Bedingungen:
30 s bei 98 ° C
7 Zyklen: 10 s bei 98 ° C; 30 s bei 65 ° C; 30 s bei 72 ° C
7 min bei 72 ° C - Sammeln Sie PCR-Reaktionen in einen neuen Schlauch, zurückfordern Sie Perlen auf die magnetische Trennung Stand und Transfer überstand (500 µL) in einen neuen Schlauch.
-
Reinigen Sie die Bibliothek-DNA mit SPRI Perlen.
- Mix SPRI Perlen, 460 µL Perlen in einen neuen Schlauch übertragen und bringen RT für 15 min. hinzufügen 450 µL SPRI-Perlen, die PCR-Reaktionen (Endvolumen 950 µL), 5 min bei RT inkubieren und spin-Probe in einer Benchtop-Zentrifuge für 2 – 3 s Probe zu sammeln.
- Öffnen Sie den Deckel, legen Sie die Probe auf die magnetische Trennung stehen für 5 min und entfernen Sie überstand zu.
- Halten die Perlen auf die magnetische Trennung Stand, fügen Sie 1 mL 70 % igem Ethanol (Vol/Vol hinzu), Probe Rohr auf einer Fläche von Perlen, lassen für 30 s und verwerfen überstand.
- Wiederholen Sie Schritt 15.4.3 zweimal mehr.
- Trocknen Sie Perlen bei 37 ° C in einem Thermomixer (Rohr Deckel öffnen) für nicht mehr als 5 min.
- Die Probe, Mischung, 51 µL TLE Puffer hinzu und inkubieren Sie für 10 min bei 37 ° C, bei 950 u/min in einem Thermomixer schütteln.
- Spin-Probe in einer Benchtop-Zentrifuge für 2 – 3 s, öffnen Sie den Deckel und das Rohr auf die magnetische Trennung stehen für 5 min. Transfer klar überstand in ein neues Rohr und Perlen zu verwerfen.
- Quantifizieren Sie die Konzentration der Hi-C-Bibliothek. Nach 7 Runden von PCR Verstärkung erhalten wir routinemäßig 500 – 1.500 ng von Hi-C-Bibliothek.
16. Hybrid-Lösung Capture
Hinweis: Blocker und Puffer (SHS1-4) die folgenden Lösungen sind aus der SureSelect kit (siehe Tabelle der Materialien).
- Transfer 500 ng zu 1 µg von Hi-C-Bibliothek in ein neues Rohr und verdunsten Probe auf einem Vakuum Konzentrator (siehe Tabelle der Materialien; 45 ° C; Vakuumdruck: Ebene 30,0, Rampe 5) trocknen.
- Wieder aussetzen Sie verdampfte Hi-C-Bibliothek durch Zugabe von 3,6 µL H2O (Molekulare Biologie Klasse), 2,5 µL Blocker 1, 2,5 µL Blocker 2 und 0,6 µL benutzerdefinierte Blocker.
- Übertragen Sie Probe in einen Brunnen eines neuen PCR-Röhrchen-Streifens zu, schließen Sie mit einem PCR-Kappe-Streifen und auf Eis. Bezeichnung als "D" (für DNA-C).
- Vorbereiten den Hybridisierung Puffer: 12,5 µL SHS1-Puffer; 0,5 µL SHS2 Puffer; 5 µL SHS3 Puffer; 6.5 µL SHS4-Puffer.
- Bei 65 ° C für 5 min in einem Thermomixer inkubieren. Transfer in einen Brunnen eines neuen Streifens der PCR-Röhrchen, mit einem PCR-Kappe-Streifen schließen und halten bei RT-Label als "H" (für Hybridisierung Puffer).
- In einen Brunnen eines neuen Streifens der PCR-Röhrchen mix 100 ng/µL biotinylierte 5 µL RNA-Sonden (Store bei-80 ° C und Tauwetter auf Eis kurz vor Gebrauch); 0,5 µL SRNase B (RNase-Inhibitor) und 1,5 µL H2O (Molekulare Biologie Klasse).
- Schließen des PCR Schlauch Streifens mit einer PCR-Kappe-Streifen und Platz auf dem Eis. Bezeichnung als "R" (RNA).
- Richten Sie PCR-Maschine mit den folgenden Parametern:
5 min bei 95 ° C; 25 h bei 65 ° C; Deckel erhitzt; 29 µL Reaktionsvolumen PCR.
Hinweis: Fahren Sie so schnell wie möglich bei allen Eingriffen während die PCR-Maschine ausgeführt wird, um die Probe Verdunstung zu vermeiden. - Legen Sie "D" PCR-Rohr-Streifen in der PCR-Maschine, PCR-Maschine Deckel schließen und Starten der PCR-Reaktion. Wenn das PCR-Programm 65 ° C erreicht, öffnen Sie PCR-Maschine Deckel und "H" PCR-Rohr-Streifen in der PCR-Maschine. Die PCR-Maschine Deckel schließen Sie und inkubieren Sie 3 min. Open der PCR-Maschine Deckel, findet das "R" PCR-Röhrchen Streifen auf der PCR-Maschine, und schließen Sie die PCR-Maschine.
- Nach 2 min Öffnen der PCR-Maschine Deckel und alle PCR-Röhrchen-Strips. Übertragen Sie 13 µL gut "H" zu gut "R", dann alle Volumen von gut "D" in gut "R". Pipettieren nach oben und unten 3 Mal, um die Reaktion zu mischen in der Nähe des PCR-Rohr-Streifens, entfernen Sie das "H" und "D" PCR tube Streifen, und PCR-Maschine Deckel schließen. Inkubieren Sie die Reaktion bei 65 ° C für 24 h.
17. Isolierung der Promoter Fragment-haltigen Ligation Produkte
Hinweis: Die folgenden Schritte sollten mit SureSelect-Adapter-Kit und Bibliothek (siehe Tabelle der Mat) erfolgen.
- Wärmen Sie 1,5 mL Waschpuffer 2 pro Probe bei 65 ° C im Voraus vor.
- Fügen Sie 60 µL Streptavidin-gekoppelten magnetischen Beads (siehe Tabelle der Materialien) in einen neuen Schlauch, legen auf die magnetische Trennung stehen für 1 min und überstand zu entfernen.
- Waschen Sie Perlen dreimal mit 200 µL 1 x Bindung Puffer.
Hinweis: Jedem Waschschritt während der Post-Capture Isolation der Promotor-haltigen Ligatur Produkte wieder auszusetzen Sie Perlen im entsprechenden Puffer, für 3 min bei RT und 15 u/min auf einem rotierenden Rad drehen Sie, drehen Sie sanft das Rohr in eine Benchtop-Zentrifuge für 2 – 3 s zu sammeln Probe, Stelle das Rohr auf die magnetische Trennung stehen für 3 min und entfernen überstand. - Wieder aussetzen Sie Perlen in 200 µL 1 x Bindung Puffer. Öffnen Sie die PCR-Maschine und das PCR-Röhrchen Streifen (während der PCR-Programm noch läuft) und die Hybridisierungsreaktion in das Rohr mit magnetischen Beads zu übertragen. 30 min auf einem rotierenden Rad bei 3 u/min bei RT inkubieren.
- Perlen auf dem Magnetische Trennung Stand zurückzufordern und den klare überstand zu entfernen. Neu suspendieren Sie Perlen in 500 µL Waschpuffer 1, Mischung, und 15 min bei 20 ° C unter Schütteln bei 950 u/min in einem Thermomixer inkubieren.
- Perlen auf die magnetische Trennung stehen für 3 min zurückzufordern und den klare überstand zu entfernen. Neu suspendieren Sie Perlen in 500 µL Waschpuffer 2 mischen Sie und inkubieren Sie 10 min bei 65 ° C unter Schütteln bei 950 u/min in einem Thermomixer. Wiederholen Sie Schritt zwei weitere Male 17,5.
- Zurückfordern Sie Perlen auf die magnetische Trennung Stand, entfernen Sie den klare überstand zu und erneut aussetzen Sie Perlen in 200 µL 1 x Einschränkung Puffer 2. Zurückzufordern Sie Perlen auf dem Magnetische Trennung Stand, entfernen Sie überstand zu und erneut suspendieren Sie Perlen in 30 µL 1 x Einschränkung Puffer 2.
18. PCHi-C-Bibliothek Verstärkung
- Bereiten Sie PCR-master-Mix: 60 µL 5 X PCR Puffer (Phusion-Puffer), 3,6 µL 25 µM PE PCR Primer 1.0, 3,6 µL 25 µM PE PCR Primer 2.0, 8,4 µL dNTP-Mix (10 mM), 3,6 µL Phusion Polymerase und 190.8 µL H2O.
- Master Mix PCR-Mix mit Perlen (300 µL insgesamt), teilen sich in 6 Aliquote von 50 µL und PCR verstärkt mit den folgenden Bedingungen:
30 s bei 98 ° C
4 Zyklen: 10 s bei 98 ° C, 30 s bei 65 ° C, 30 s bei 72 ° C
7 min bei 72 ° C - Sammeln alle PCR Reaktionen in einen neuen Schlauch, zurückfordern die Perlen auf den Magneten und überstand zu übertragen (300 µL; enthält PCHi-C-Bibliothek) in einen neuen Schlauch.
- Reinigen der PCHi-C-Bibliothek mit SPRI Perlen, nach oben unter 15,4 beschrieben.
- Quantifizieren Sie die Konzentration der PCHi-C-Bibliothek.
Ergebnisse
Promotor erfassen Hi-C wurde zur Maus7,34,36,39 und menschlichen33,35,37,38 Hi-C-Bibliotheken für bereichern Veranstalter-Interaktionen. Ein ähnliches Protokoll (benannt HiCap) wurde vom Sandberg Gruppe40beschrieben. Abbildung 1A zeigt den schematischen Workflow für Promoter erfassen Hi-C. In dem hier beschriebenen Protokoll sind Hi-C-Bibliotheken mit im Zellkern Ligatur41, führt zu einer deutlich reduzierten Anzahl von unechten Ligatur Produkte42erzeugt. Für PCHi-C hochkomplexe Maus oder menschlichen Hi-C-Bibliotheken in Lösung Hybridisierung unterliegen und mit 39.021 biotinylierte RNAs komplementär zu 22.225 Maus Promotor-haltigen HindIII Einschränkung Fragmente oder 37.608 biotinylierte RNAs zu erfassen 22.076 menschlichen Promotor-haltigen HindIII Einschränkung Fragmente, Ausrichtung bzw.. Projektträger mit Einschränkung Fragmente kann an einem oder beiden Enden durch individuelle biotinylierte RNAs (Abbildung 1 b) ausgerichtet werden. Wir fanden, dass die Aufnahme der beiden verbesserte Abdeckung der einzelnen Projektträger (Abbildung 1; roh Sequenz liest) fast zwei-Fach, endet, wie erwartet. Also, wann immer möglich (d. h. nicht wiederholende Regionen), wir empfehlen, biotinylierte RNAs komplementär zu beiden Enden ein Beschränkungsfragment erfasst werden.
Um PCHi-C-Bibliothek-Qualität in einem frühen Stadium während der Vorbereitung der Bibliothek zu beurteilen, führen wir zwei Steuerelemente nach DNA-Ligatur und Reinigung, wie zuvor beschrieben31. Die erste ist mit spezifischen Primer-Paaren um Ligatur Produkte wie 3 C27zu verstärken. Wir verwenden Grundierung Paare (Tabelle 1) Zelltyp invariante Langstrecken Ligatur Produkte zu verstärken, wie zwischen das Myc -gen und seine bekannten Enhancer befindet sich ca. 2 Mb entfernt (Abbildung 2A) oder zwischen den Genen der Hist1 Locus () (getrennt durch 1,5 Mb), und zwischen beiden Regionen befindet sich in der Nähe linear ("Short-Range Control").
Die zweite Qualitätskontrolle erfolgt die Effizienz von Biotin Einbau während Klenow-vermittelten Fill Einschränkung Website Überhänge mit Biotin-dATP bestimmen. Erfolgreiche Klenow füllen und anschließende stumpfe Ende Ligatur Ergebnisse in das Verschwinden der ursprünglichen Beschränkung Site zwischen den DNA-Molekülen einer Ligatur Ware und im Falle von HindIII bei der Bildung einer neuen NheI Anerkennung Website (Abb. 2 b ). Das Verhältnis von HindIII NheI verdaute Ligatur Produkt ist eine direkte Auslesen der Biotin-Aufnahme-Effizienz. Eine schlechte Qualität Hi-C-Bibliothek zeigen ein hohes Maß an HindIII Verdauung, während qualitativ hochwertige Bibliotheken verfügen über nahezu vollständigen NheI Verdauung der Ligatur Produkte (Abb. 2 b).
Nach Hi-C Bibliothek Vorbereitung (d. h. nach Biotin-Streptavidin herunterziehen Bildgröße Hi-C Ligatur Produkte, Adapter Ligatur und Pre-Capture-PCR) wird die Integrität und Größe Verteilung der Hi-C-Bibliothek von Bioanalyzer (Abbildung bewertet. 2 C). die gleiche Kontrolle erfolgt am Ende des PCHi-C Bibliothek Vorbereitung (d. h. nach der Hybridisierung Aufnahme der Promotor-haltigen Ligatur Produkte und Post-Capture-PCR). Vergleich der Hi-C und PCHi-C Bioanalyzer profile zeigt, dass erwartungsgemäß Hi-C-Bibliotheken viel konzentrierter als die entsprechenden PCHi-C-Bibliotheken sind, aber die Größenverteilung der Bibliotheken sehr ähnlich ist, angibt, die die Erfassung Schritt in PCHi-C vorstellen keinen Größe Bias (Abbildung 2, D).
Nach gepaart-End Sequenzierung der PCHi-C liest zugeordnet sind, Qualität kontrolliert und mit Hilfe der HiCUP Pipeline43gefiltert. Qualitativ hochwertige PCHi-C-Bibliotheken enthalten zwischen 70-90 % "gültige Paare" (d. h. gepaart-End Sequenz liest zwischen zwei Einschränkung-Fragmente, die nicht auf der linearen genomische Karte; benachbarten sind Abb. 3A, B). Mit Hilfe der im Zellkern Ligatur Protokoll41,42, Lesen der Anteil der Trans Sie Paare (d. h. gepaart-End Sequenz liest zwischen zwei Einschränkung-Fragmente, die auf verschiedenen Chromosomen befinden) in der Regel gering sind, zwischen 5 und 25 % Was die Existenz von Chromosom Territorien und zeigt gute Bibliothek Qualität. Direktvergleich der Prozentsatz von "gültigen Paare" zwischen Hi-C-Bibliotheken und ihre entsprechenden PCHi-C Bibliotheken35, zeigt, dass in allen Fällen den Anteil der gültigen Paare ist höher in der PCHi-C-Bibliotheken (Abb. 3 b). Damit einher geht eine Reduzierung des Anteils der ungültigen "dasselbe fragment interne" liest in PCHi-C (Abbildung 3). Es wird erwartet, wie der Aufnahme Schritt nicht nur für Veranstalter-haltigen Ligatur Produkte bereichert, sondern aufgrund der Position der Erfassung Oligos zur Beschränkung auch für Beschränkung Fragment endet, Fragmente (siehe Abbildung 1 b).
Nach HiCUP Filtern ermitteln wir die Capture-Effizienz. PCHi-C-Bibliotheken enthalten drei Arten des gültigen Reihenfolge lautet nach HiCUP filtern:
(1) Veranstalter: Genom liest (d. h. zwischen erfassten Promoter Fragment und ein nicht-Promotor HindIII Beschränkungsfragment überall im Genom mal gelesen)
(2) Veranstalter: Veranstalter liest (gelesen zwischen zwei erbeuteten promotorfragmente)
(3) Genom: Genom liest (Hintergrund Hi-C Ligatur Produkte wo weder die Ligatur Produktpartner mit einem erbeuteten Promotor Karten). Diese werden vor dem nachgeschalteten Analysen verworfen.
Qualitativ hochwertige PCHi-C-Bibliotheken haben Erfassung Leistungsfähigkeiten (Summe der Kategorien 1 und 2 oben) zwischen 65-90 % (Abbildung 3D). Ein direkter Vergleich zu Hi-C-Bibliotheken zeigt, dass PCHi-C ergibt sich eine ~ 15-fold Bereicherung für Promoter-haltigen Ligatur Produkte (Abbildung 3D), in einigen Fällen 17-fache. Dies ist in der Nähe das hypothetische Maximum (19.6-fold) Bereicherung für PCHi-C, die den Prozentsatz der Genom Einschränkung Fragmente abhängig ist von der Capture-System abgedeckt. Größere Bereicherung kann erreicht werden, indem Sie entwerfen Capture-Systeme gezielt weniger Beschränkung Fragmente44,45,46.
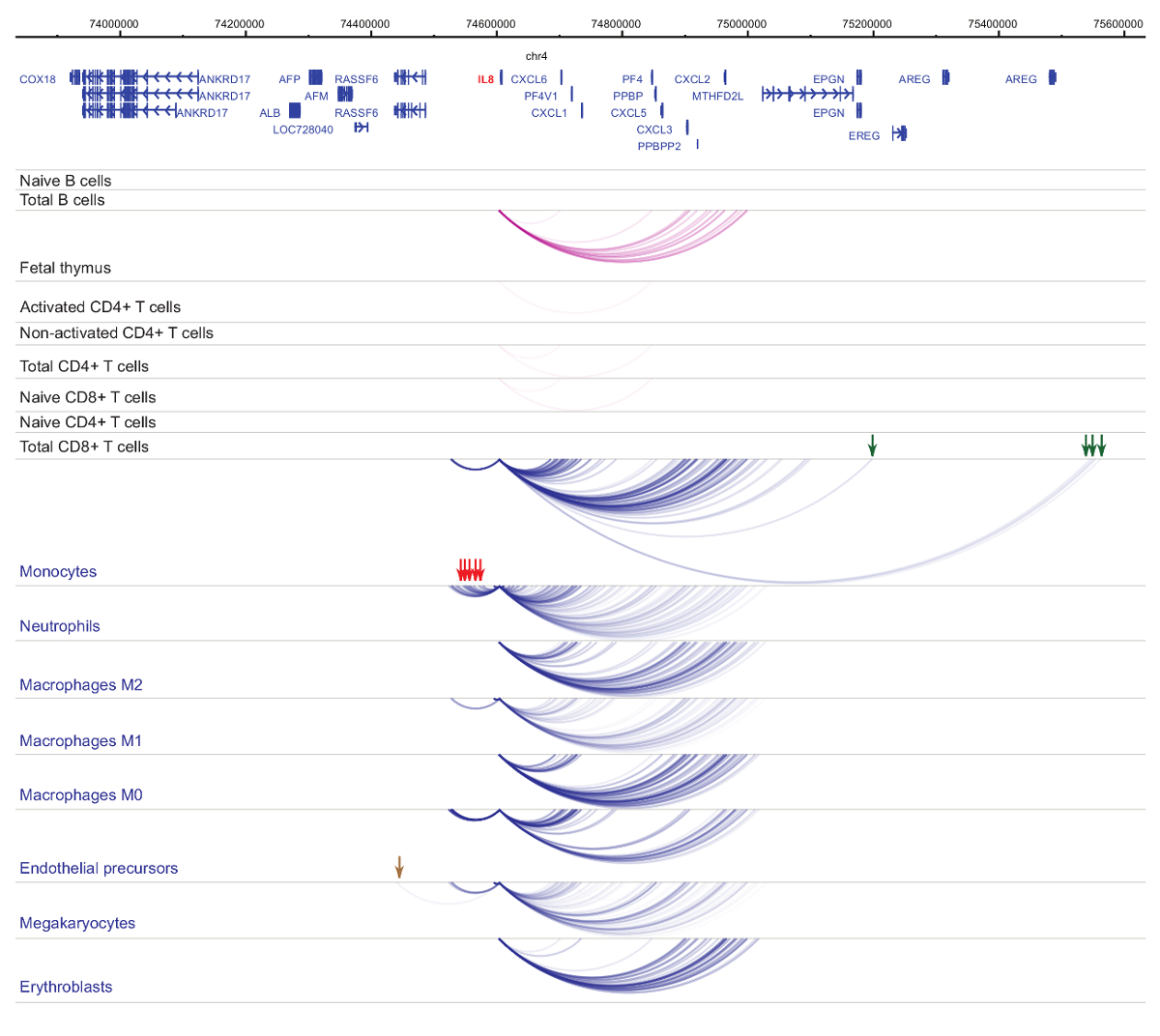
Analyse der Veranstalter Interactomes zeigt Zelle Art und Abstammung-Spezifität33,34,35, mit ausgeprägten Veränderungen während der Zelldifferenzierung37,38,39 . Abbildungen 4 und 5 zeigen Beispiele für Linie Spezifität und Differenzierung Dynamik bei bestimmten Promotoren. Z. B. ALAD ist konstitutiv in allen Zellen exprimiert, aber seinen Ausdruck ist hochreguliert in Erythroblasts47. Der Projektträger ALAD Kontakte mehrere distale Fragmente in allen hämatopoetischen Zellen und engagiert sich in zusätzliche Interaktionen speziell in Erythroblasts (Abbildung 4). IL-8 zeigt keine statistisch signifikanten Wechselwirkungen in B-Zellen, sehr wenige Interaktionen in T-Zellen, sondern Dutzende von Interaktionen in den Zellen der myeloischen Linie, einschließlich Zelltyp spezifische Wechselwirkungen in Monozyten, Neutrophilen und Megakaryozyten) ( Abbildung 5). Diese Beispiele zeigen, wie PCHi-C verwendet werden kann, um Zelltyp spezifische Interactomes zu entwirren und Promotor-Interaktion Regionen mit regulatorischen Potenzial zu identifizieren.

Abbildung 1 : Promoter erfassen Hi-C Begründung und Capture Köder Design. (A) schematische Workflow der PCHi-C. Im Kern Ligatur Hi-C41,42 (I) gefolgt von in-Lösung Hybridisierung mit biotinylierte RNA Köder (II) gezielt die Einschränkung Fragmente von allen menschlichen (hier abgebildet) oder Maus Gen Promotoren (III). (B) Köder Design für PCHi-C. Biotinylierte RNA Erfassung Köder (rote gebogene Linien) sollen an den Enden der Promotor-haltigen Einschränkung Fragmente (grau; beachten Sie, dass die promotorsequenzen selbst (rot) nur durch die RNA-Capture-Köder ausgerichtet sind, wenn sie an Einschränkung befinden Fragment-enden). Ligatur-Produkte bestehend aus Promotor-haltigen Einschränkung Fragmente (grau) und deren Interaktion Einschränkung Bruchstücke (gelb und grün) sind isolierte durch Sequenz-Komplementarität Hybridisierung zwischen Köder RNA und DNA-Ziel, und die anschließende Biotin-Streptavidin Pulldown, siehe A. (C) Vergleich der PCHi-C Capture Effizienz für Promoter-haltigen Einschränkung Fragmente gezielt durch eine RNA Köder Aufnahme Sonde Vs zwei RNA Köder erfassen Sensoren (siehe Schema b). Bitte klicken Sie hier für eine größere Version dieser Figur.
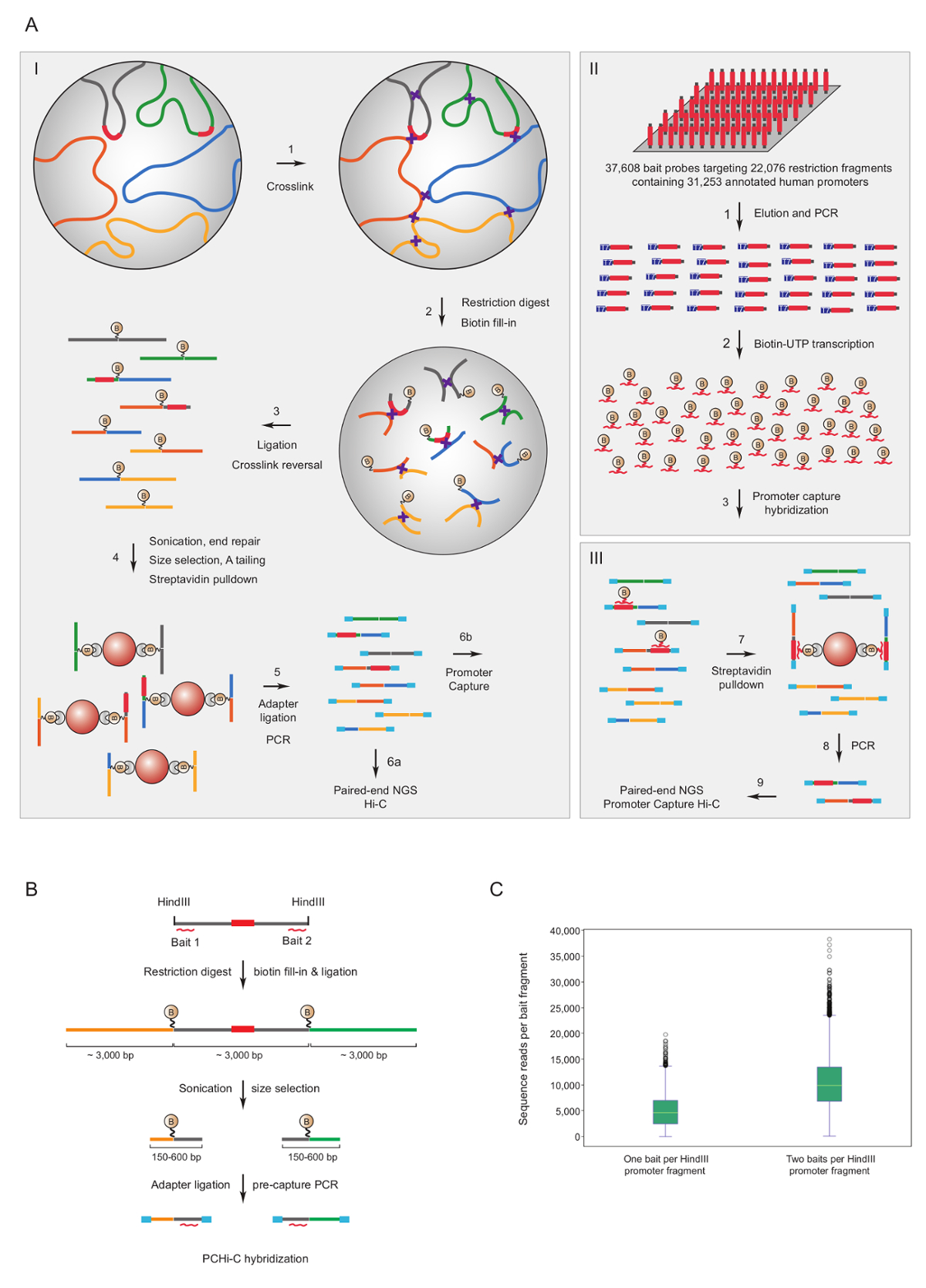{kind=link}

Abbildung 2 : PCHi-C Pre-Sequenzierung Qualitätskontrollen. (A) Links, schematische räumliche Gegenüberstellung zwischen Promotor und PIR, was zu einem Hi-C Ligatur Produkt bestehend aus einem Promotor-haltigen Beschränkungsfragment (grau; promotorsequenz in rot) und ein PIR Beschränkungsfragment (gelb). Recht, DNA-gel-Elektrophorese zeigt Beispiele von Hi-C-Ligatur-Produkte verstärkt mit spezifischen Primer-Paaren (wie im Schaltplan auf der linken Seite dargestellt). (B) Links, repräsentative Beispiele für HindIII, NheI und HindIII/NheI Einschränkung verdaut Hi-C Ligatur Produkte (PCR-Produkte in A gezeigt). Rechts, schematische Darstellung der DNA-Sequenz nach Hi-C Ligatur nach erfolglosen (oben) oder erfolgreiche (unten) dNTP Klenow Fill Einschränkung Kreuzungen und nachfolgende Ligatur. (C) Vertreter Hi-C Bioanalyzer Bibliotheksprofil (1/5 Verdünnung). (D) Vertreter PCHi-C Bibliothek Bioanalyzer Profil (keine Verdünnung). Bitte klicken Sie hier für eine größere Version dieser Figur.
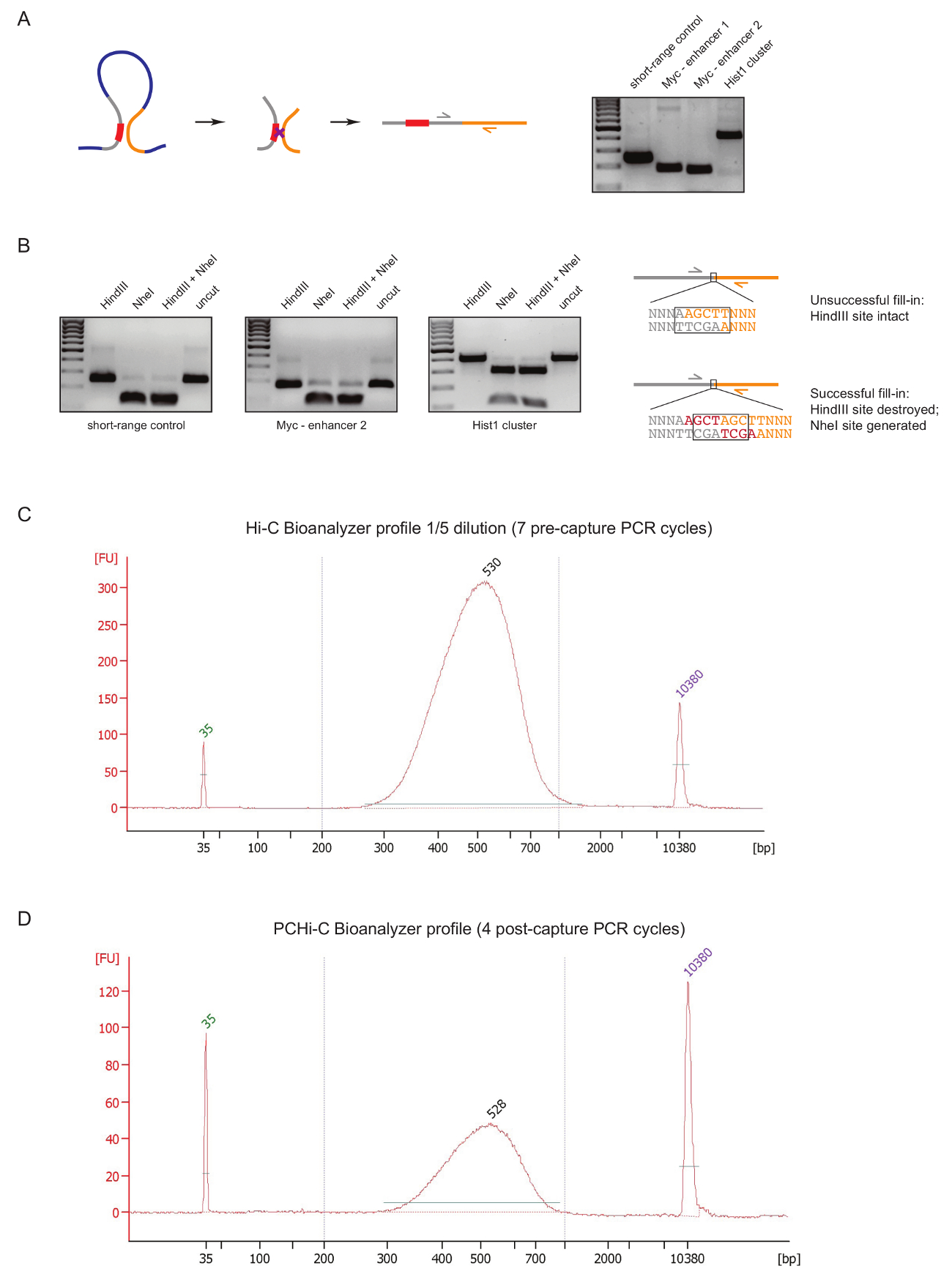{kind=link}

Abbildung 3 : PCHi-C Post-Sequenzierung Qualitätskontrollen. (A) Vergleich der prozentualen gültigen Reihenfolge lesen Paare nach HiCUP43 Verarbeitung in PCHi-C gegen entsprechende Hi-C-Bibliotheken (Daten aus Javierre Et Al., 2016-35). (B) Vertreter HiCUP PCHi-C Ergebnis zeigt gültige lesen Paare und andere Sequenz-Kategorien, die vor dem nachgeschalteten Analysen (Daten aus Javierre Et Al., 201635) verworfen werden. (C) Vergleich der Prozentsatz "interne dasselbe Fragment" liest nach HiCUP Verarbeitung in PCHi-C gegen entsprechende Hi-C-Bibliotheken (Daten aus Javierre Et Al., 2016-35). (D) Vergleich der prozentualen Sequenz liest mit angehaltenem promotorfragmente (Capture Effizienz) in PCHi-C gegen entsprechende Hi-C-Bibliotheken (Daten aus Javierre Et Al., 2016-35). Bitte klicken Sie hier für eine größere Version dieser Figur.
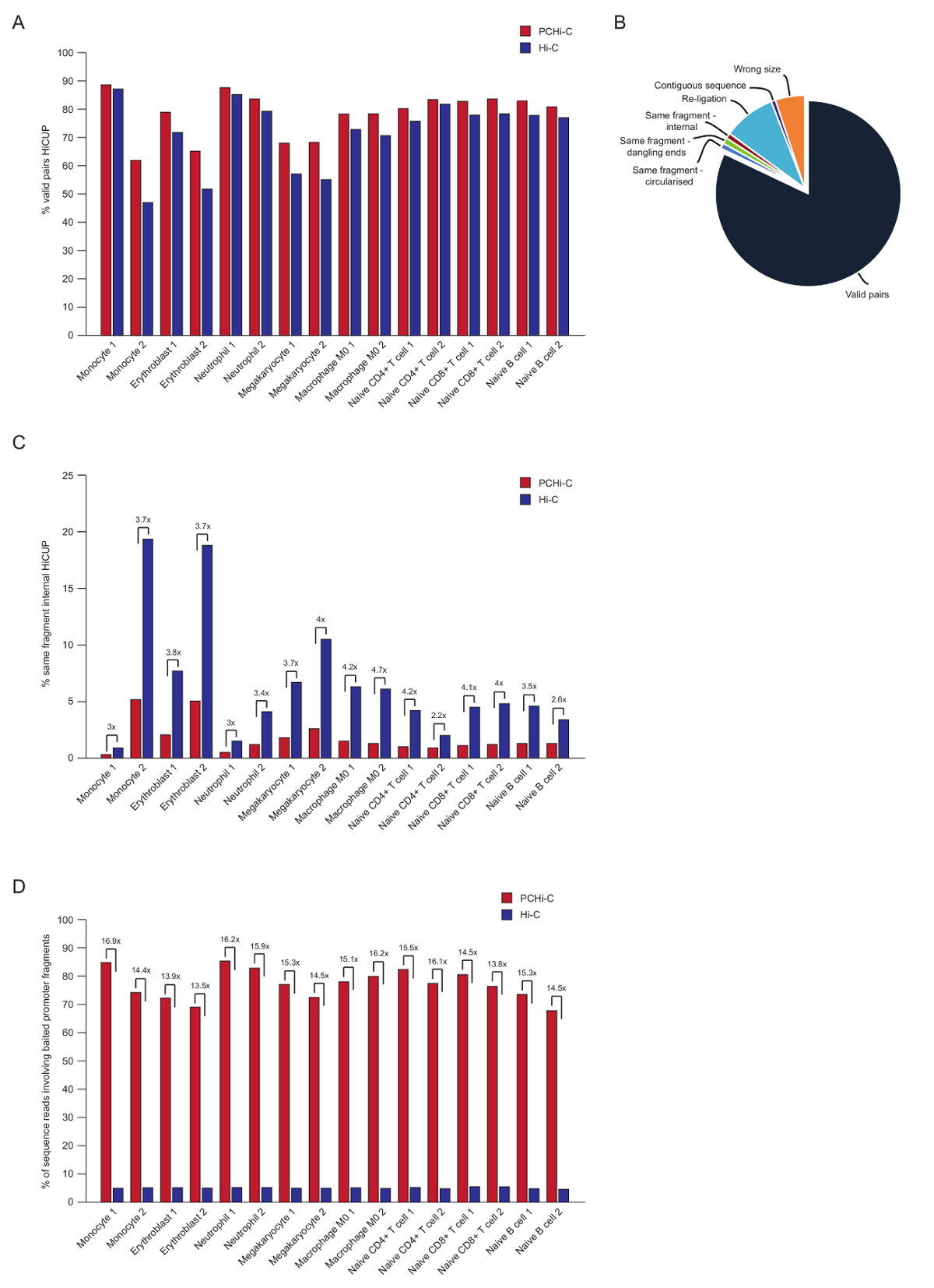{kind=link}

Abbildung 4: ALAD PCHi-C-Profil in humanen hämatopoetischen Zellen. Promotor Interaktionen myeloiden Zelltypen sind als blaue Bögen und Projektträger Interaktionen des lymphatischen Zelltypen sind als Lila Bögen angezeigt. Erythroblast-spezifische Interaktionen werden durch rote Pfeile (Daten aus Javierre Et Al., 201635) angezeigt. Bitte klicken Sie hier für eine größere Version dieser Figur.
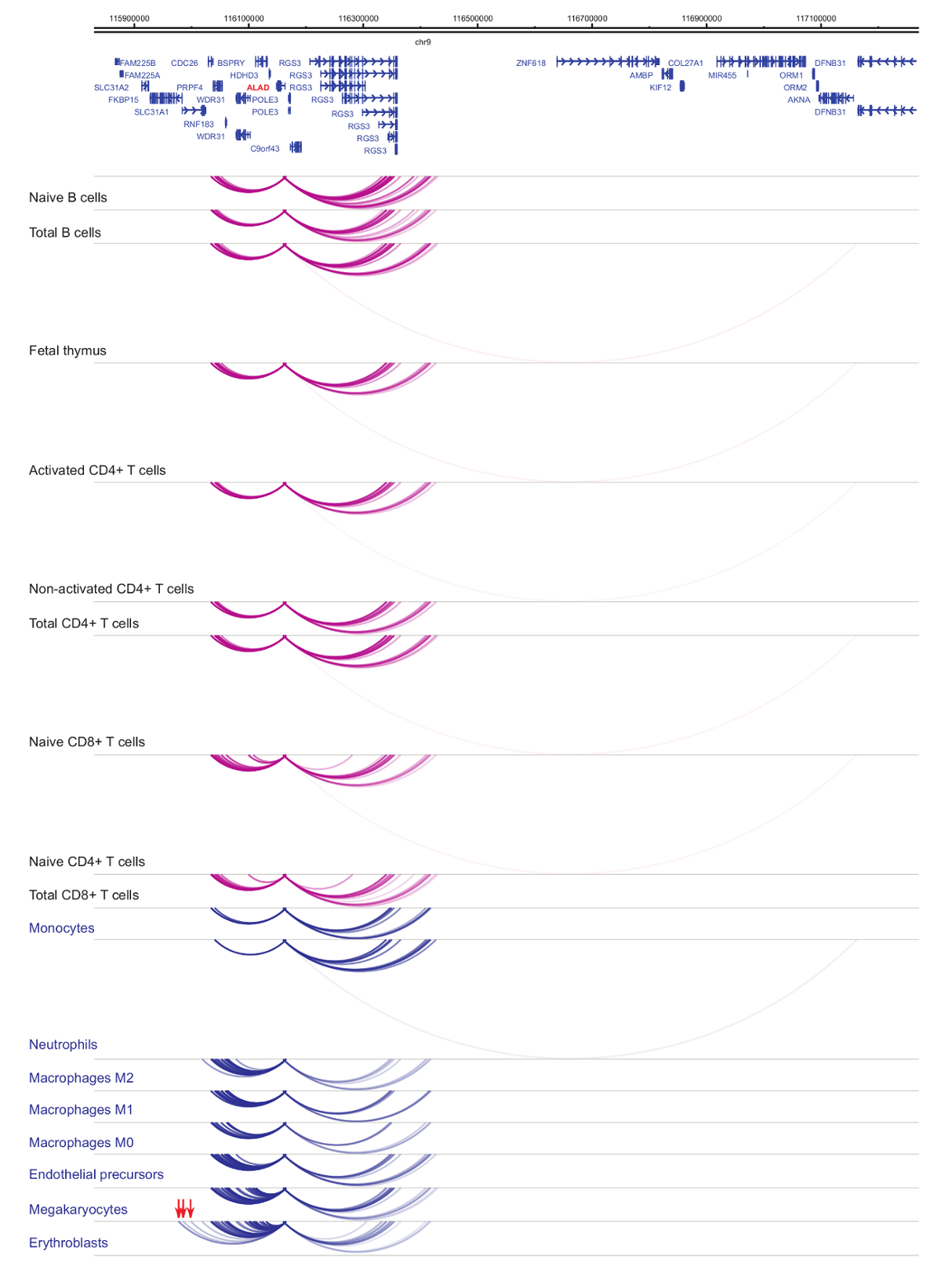{kind=link}

Abbildung 5 : IL8 PCHi-C-Profil in humanen hämatopoetischen Zellen. Promotor Interaktionen myeloiden Zelltypen sind als blaue Bögen und Projektträger Interaktionen des lymphatischen Zelltypen sind als Lila Bögen angezeigt. Monocyte-spezifische Interaktionen werden durch grüne Pfeile angedeutet, Neutrophilen-spezifischen Interaktionen sind durch rote Pfeile gekennzeichnet und eine Megakaryocyte-spezifische Interaktion wird durch einen braunen Pfeil (Daten aus Javierre Et Al., 201635) angezeigt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Menschlichen | ||||||||
| Name | Sequenz | Chromosom | Strang | Start GRCh38/hg38 | Ende GRCh38/hg38 | Primer-Kombinationen, 3 C Interaktionen und Biotin-Aufnahme zu testen | ||
| HS AHF64 Dekker | GCATGCATTAGCCTCTGCTGTTCTCTGAAATC | 11 | + | 116803960 | 116803991 | Verwenden Sie in Kombination mit hs AHF66 Dekker | ||
| HS AHF66 Dekker | CTGTCCAAGTACATTCCTGTTCACAAACCC | 11 | + | 116810219 | 116810248 | Verwenden Sie in Kombination mit hs AHF64 Dekker | ||
| HS-MYC-locus | GGAGAACCGGTAATGGCAAA | 8 | - | 127733814 | 127733833 | Verwenden Sie in Kombination mit hs MYC +1820 oder hs MYC-538 | ||
| HS MYC +1820 | AAAATGCCCATTTCCTTCTCC | 8 | + | 129554527 | 129554547 | Verwenden Sie in Kombination mit hs MYC locus | ||
| HS MYC-538 | TGCCTGATGGATAGTGCTTTC | 8 | - | 127195696 | 127195716 | Verwenden Sie in Kombination mit hs MYC locus | ||
| HS HIST1 F | AAGCAGGAAAAGGCATAGCA | 6 | + | 26207174 | 26207193 | Verwenden Sie in Kombination mit hs HIST1 R | ||
| HS HIST1 R | TCTTGGGTTGTGGGACTTTC | 6 | + | 27771575 | 27771594 | Verwenden Sie in Kombination mit hs HIST1 F | ||
| Maus | ||||||||
| Sequenz | Chromosom | Strang | Start GRCm38/mm10 | Ende GRCm38/mm10 | Primer-Kombinationen, 3 C Interaktionen und Biotin-Aufnahme zu testen | |||
| TCATGAGTTCCCCACATCTTTG | 8 | + | 84841090 | 84841111 | Verwenden Sie in Kombination mit mm Calr2 | |||
| CTGTGGGCACCAGATGTGTAAAT | 8 | + | 84848519 | 84848541 | Verwenden Sie in Kombination mit mm Calr1 | |||
| TATCAAGGGTGCCCGTCACCTTCAGC | 6 | + | 125163098 | 125163123 | Verwenden Sie in Kombination mit Gapdh4 Dekker | |||
| GGGCTTTTATAGCACGGTTATAAAGT | 6 | + | 125163774 | 125163799 | Verwenden Sie in Kombination mit Gapdh3 Dekker | |||
| GGAGGAGGGAAAAGGAGTGATT | 6 | + | 52212829 | 52212850 | Verwenden Sie in Kombination mit mm Hoxa13 | |||
| CAGGCATTATTTGCTGAGAACG | 6 | - | 52253490 | 52253511 | Verwenden Sie in Kombination mit mm Hoxa7 | |||
| GGGTAATGGTGTCACTAACTGG | 13 | + | 23571284 | 23571305 | Verwenden Sie in Kombination mit Hist1h3e oder mm Hist1h4i | |||
| GGGTTTGATGAGTTGGTGAAG | 13 | + | 23566541 | 23566561 | Verwenden Sie in Kombination mit mm Hist1h2ae | |||
| TTGGGCCAAAGCCTATATGA | 13 | + | 22043085 | 22043104 | Verwenden Sie in Kombination mit mm Hist1h2ae | |||
Tabelle 1: Primer-Sequenzen für die Qualitätskontrolle von Mensch und Maus Hi-C-Bibliotheken.
Diskussion
Modularer Aufbau der Promoter erfassen Hi-C
Promotor erfassen Hi-C dient zur speziell Hi-C-Bibliotheken für Interaktionen mit Promotoren bereichern. Diese Interaktionen umfassen nur eine Teilmenge der Ligatur Produkte in Hi-C-Bibliothek.
Capture Hi-C kann leicht geändert werden, um Hi-C-Bibliotheken für genomische Region oder Regionen von Interesse zu bereichern, indem Sie ändern die Capture-System. Capture Regionen können kontinuierliche genomic Segmente44,45,46,48, Enhancer, die wurden identifiziert in PCHi-C ('Reverse erfassen Hi-C'35) oder DNase ich überempfindlich Websites49 . Die Größe des Capture-System ist abhängig von den experimentellen Bereich einstellbar. Z. B. Dryden Et Al. Zielen Sie auf 519 Köder Fragmente in drei gen-Wüsten Brust Krebs44zugeordnet. Die Capture-System von Martin Et Al. richtet sich an beide kontinuierliche genomic Segmente ("Capture Region": 211 genomische Regionen insgesamt; 2.131 Einschränkung Fragmente) und Promotoren (3.857 Gen Promotoren)45ausgewählt.
SureSelect Bibliotheken sind in verschiedenen Größen erhältlich: 1 kb bis 499 kb (5.190 – 4.806), 500 kb bis 2,9 Mb (5.190 – 4.816) und 3 Mb bis 5,9 Mb (5.190 – 4.831). Da jede einzelne Aufnahme Biotin-RNA 120 Nukleotide lang ist, diese Systeme zu erfassen maximal 4.158 beherbergen, 24.166 und 49.166 einzelnen Sonden, bzw. zu erfassen. Dies entspricht bzw. 2.079, 12.083 und 24.583 gezielte Einschränkung Fragmente (Beachten Sie, dass die Zahlen für Einschränkung Fragmente Untergrenzen basiert auf der Annahme, dass zwei einzelne Aufnahme Sonden für jede Beschränkung gestaltet werden können Fragment – in Wirklichkeit durch repetitive Sequenzen werden dies nicht der Fall für jede Beschränkung fragment (siehe auch Abbildung 1 b, C), was zu einer höheren Anzahl von Aktionsleisten Einschränkung Fragmente für eine Konstante Anzahl von verfügbaren Erfassung Sonden ).
Das hier beschriebene Protokoll basiert auf der Verwendung von ein Restriktionsenzym mit einer 6 bp Anerkennung Website langreichweitige Wechselwirkungen aufzudecken. Mit einem Restriktionsenzym mit einer 4 bp Anerkennung Website für höhere Auflösung mehr proximalen Interaktionen ist auch möglich40,49.
Einschränkungen der PCHi-C
Eine inhärente Einschränkung alle Chromosom Konformation Erfassung Assays ist, dass ihre Auflösung durch das Restriktionsenzym verwendet für die Bibliothek-Generation bestimmt wird. Interaktionen, die zwischen DNA-Elementen befindet sich auf der gleichen Beschränkungsfragment auftreten sind für "C-Type" Assays unsichtbar. Darüber hinaus in PCHi-C, in einigen Fällen mehr als eine Transkription Startsite befinden sich auf der gleichen Veranstalter-haltigen Beschränkungsfragment und PIRs in einigen Fällen Hafen beide aktiv und repressiven Histon-Markierungen, so dass es schwierig zu lokalisieren, die regulatorischen Elemente vermitteln die Wechselwirkungen und die regulatorischen Ausgabe des Projektträgers Interaktionen zu prognostizieren. Mit Restriktionsenzymen mit 4 bp Anerkennung Sites mildert dieses Problem aber geht zu Lasten der stark zunehmende Komplexität der Hi-C-Bibliothek (Hi-C-Bibliotheken generiert mit 4 bp Anerkennung Website Restriktionsenzyme sind mindestens 100 Mal komplexer als Hi-C Bibliotheken mit 6 bp Anerkennung Website Restriktionsenzymen generiert), und die damit verbundenen Kosten für die Sequenzierung der nächsten Generation.
Eine weitere Einschränkung ist, dass die aktuelle PCHi-C Protokoll verlangt, dass Millionen von Zellen als Ausgangsmaterial, Analyse der Projektträger Wechselwirkungen in seltene Zelltypen entgegensteht. Eine modifizierte Version des PCHi-C ermöglichen das Verhör der Veranstalter Kontakte in Zell-Populationen mit 10.000 bis 100.000 Zellen (z. B. während der frühen Embryonalentwicklung oder Blutstammzellen) wäre daher eine wertvolle Ergänzung zur Erfassung Hallo-C Toolbox.
Schließlich, wie alle Methoden, die auf Formaldehyd Fixierung verlassen, zeichnet PCHi-C nur Interaktionen, die "eingefroren werden" zum Zeitpunkt der Fixierung. So sind um die Kinetik und Dynamik der Projektträger Interaktionen zu untersuchen, Methoden wie Höchstauflösung live Zelle Mikroskopie neben PCHi-C. erforderlich
Methoden, um räumliche Chromosom Organisation mit hoher Auflösung zu sezieren
Die große Komplexität der chromosomalen Interaktion Bibliotheken verbietet die zuverlässige Identifizierung von Produkten der Interaktion zwischen zwei besonderen Beschränkung Fragmente mit statistischer Signifikanz. Um dieses Problem zu umgehen, wurde Sequenz Capture verwendet, entweder Hi-C33,34,40,44 oder 3 C-50,-51 -Bibliotheken für spezifische Wechselwirkungen zu bereichern. Der große Vorteil der Verwendung von Hi-C-Bibliotheken über 3C-Bibliotheken für die anreicherungsschritt ist, dass Hi-C, im Gegensatz zu 3C, ein anreicherungsschritt für echte Ligatur Produkte enthält. Infolgedessen ist der Prozentsatz der gültigen liest in PCHi-C-Bibliotheken etwa 10-fach höher als im Capture-C Bibliotheken50, enthielt ca. 5 – 8 % gültig nach HiCUP Filtern liest. Sahlen Et Al. haben direkt gegenüber Capture-C, HiCap, die wie PCHi-C Hi-C-Bibliotheken für Capture Bereicherung, im Gegensatz zu Capture-C nutzt die 3 C-Bibliotheken verwendet. In Übereinstimmung mit unseren Erkenntnissen, fanden sie, dass Capture-C-Bibliotheken vor allem UN aufgespaltenen Fragmente40zusammengesetzt sind. Darüber hinaus hatte HiCap Bibliotheken eine höhere Komplexität als Capture-C Bibliotheken40.
Eine Variante von Capture-C, genannt der nächsten Generation Capture-C52 (NG Capture-C) verwendet eine Oligo pro Beschränkung Fragment Ende, als vorher festgelegten PCHi-C33,34, anstelle von überlappenden Sonden verwendet im original Capture-C Protokoll50. Dadurch erhöht sich den Prozentsatz der gültigen liest im Vergleich zu Capture-C bescheiden, aber NG Capture-C beschäftigt zwei sequentielle runden Capture Bereicherung und eine relativ hohe Anzahl von PCR-Zyklen (20 bis 24 Zyklen insgesamt im Vergleich zu 11 Zyklen in der Regel für PCHi-C), die zwangsläufig führt zu höheren Zahlen der Reihenfolge Duplikate und geringere Komplexität der Bibliothek. In Studie Experimente bei der Optimierung der PCHi-C, fanden wir, dass der Anteil der einzigartigen (d. h. nicht dupliziert) Paare betrug nur rund 15 % lesen, wenn wir 19 PCR-Zyklen verwendet (13 Zyklen Pre-capture + 6 Zyklen Post-Datenerfassung; nicht dargestellt), jedoch Optimierung auf eine niedrigere Zahl von PCR-Zyklen, in der Regel ergibt 75 – 90 % einzigartige lesen Sie Paaren. So, Verringerung der Zahl der PCR-Zyklen deutlich erhöht die Menge an informativen Sequenzdaten.
Eine neue Methode verbindet ChIP mit Hi-C auf chromosomale Wechselwirkungen vermittelt durch ein bestimmtes Protein des Interesses (HiChIP53) konzentrieren. Im Vergleich zu ChIA PET54, die auf eine ähnliche Begründung beruht, enthält HiChIP Daten eine höhere Anzahl von informativen Sequenz liest, damit mehr Zuversicht Interaktion aufrufen53. Es wird sehr interessant sein, die entsprechenden HiChIP direkt zu vergleichen und Datensätze erfassen Hi-C einmal sie verfügbar (z. B. HiChIP mit einem Antikörper gegen die Cohesin Einheit Smc1a53 mit Capture Hi-C für alle Smc1a Beschränkung gebunden werden (Fragmente) nebeneinander. Eine inhärente Unterschied zwischen diesen beiden Ansätzen ist, dass Capture Hi-C nicht auf Chromatin Immunopräzipitation setzt, und daher in der Lage ist, Verhören chromosomale Interaktionen unabhängig von Protein-Belegung. Dies ermöglicht Vergleich von 3D Genom-Organisation in dem Vorhandensein oder Fehlen von bestimmten Faktor Bindung, wie verwendet wurde, um PRC1 als schlüsselregulator der Maus ESC räumliche Genom Architektur7identifizieren.
PCHi-C und GWAS
Genomweite Assoziationsstudien (GWAS) haben ergeben, dass mehr als 95 % der krankheitsassoziierten Sequenzvarianten befinden sich in nicht-kodierenden Regionen des Genoms, oft bei großen Entfernungen zu Protein-kodierenden Gene55. GWAS-Varianten sind oft fand in unmittelbarer Nähe zum DNase ich überempfindlich Websites, das ist ein Markenzeichen von Sequenzen mit potenziellen Regulierungstätigkeit. PCHi-C- und Hi-C erfassen intensiv genutzt, Promotoren GWAS Risiko Loci in Brust Krebs44, Darmkrebs48und Autoimmunerkrankung35,45,46verwickelt zu verknüpfen. Ein PCHi-C Studie über 17 verschiedene menschliche blutbildenden Zelle Arten fand SNPs zugeordnete Autoimmunerkrankung in PIRs in lymphoiden Zellen angereichert wurden während Sequenzvarianten, die Blutplättchen und roten Blutkörperchen bestimmte Merkmale zugeordnet überwiegend in gefunden wurden die Makrophagen und Erythroblasts, bzw.35,56. So Gewebetyp spezifischen Promotor Interactomes aufgedeckt durch PCHi-C kann helfen, um zu verstehen, die Funktion der nicht-kodierenden krankheitsassoziierten Varianten-Sequenz und identifizieren neue mögliche Krankheitsgene für eine therapeutische Intervention.
Merkmale der Promotor-Interaktion Regionen
Einige Linien des Beweises Promotor Interactomes mit Gen-Expressions verknüpfen. Zunächst mehrere PCHi-C-Studien haben gezeigt, dass genomische Regionen Interaktion mit Promotoren (hoch) ausgedrückten Gene in Zusammenhang mit Enhancer Aktivität, z. B. H3K27 Acetylierung und p300 Bindung33,34 Mark angereichert sind , 37. fanden wir eine positive Korrelation zwischen Gen-Ausdruck-Ebene und die Anzahl der interagierenden Enhancer, was darauf hindeutet, dass additive Effekte der Geschmacksverstärker führen zu erhöhte Genexpression Ebenen34,35. Zweitens: natürlich vorkommende Ausdruck, die quantitative Trait Loci (eQTLs) in PIRs angereichert sind, die die gleichen Gene verbunden sind, deren Ausdruck die eQTLs35betroffen ist. Drittens fand durch die Integration von Reise-57 und PCHi-C Daten, Cairns Et Al. , dass Reise-Reporter-Gene zuordnen PIRs in Maus WSR stärker Reporter Genexpression als Reporter-Gene bei Integration Websites in nicht-Promotor-wechselwirkenden Regionen zeigen 58, darauf hinweist, dass PIRs transkriptionelle Regulierungstätigkeit besitzen. Zusammen, zufolge diese Feststellungen Promotor Interactomes aufgedeckt durch PCHi-C in verschiedenen Maus und menschlichen Zelltypen wichtige regulatorische Module für gen-Expressions beinhalten.
Es ist erwähnenswert, dass Enhancer repräsentieren nur einen kleinen Bruchteil (~ 20 %) von allen PIRs von PCHi-C33,34aufgedeckt. Anderen PIRs konnten strukturelle oder topologische Rollen, anstatt direkte transkriptionelle regulatorische Funktionen. Allerdings gibt es auch Hinweise darauf, dass PCHi-C DNA-Elemente mit regulatorischer Funktion entdecken kann, die nicht klassischen Enhancer Marken Hafen zu tun. In einer menschlichen lymphatischen Zelllinie wurde der BRD7 Veranstalter gefunden, mit einer Region frei von Enhancer Marken interagieren, die gezeigt wurde, um Enhancer Aktivität im Reporter-gen Assays33besitzen. Regulatorische Elemente mit ähnlichen Eigenschaften möglicherweise häufiger als derzeit angenommen. Ein CRISPR-basierten Bildschirm für regulatorische DNA-Elemente identifiziert unmarkierte regulatorische Elemente (Massnahmen), die Genexpression zu steuern, sondern sind frei von Enhancer kennzeichnet z. B.59.
In anderen Fällen wurden die PIRs Chromatin Marken zugeordnet transkriptionellen Repression Hafen gezeigt. PIRs und interagierenden Promotoren durch PRC1 in Maus WSR gebunden waren in ein räumlich ausgedehntes verdrängten Gene tragen engagiert die repressiven H3K27me37markieren. In menschlichen Lymphoblastoid Zellen unterdrückt eine entfernte Element interagiert mit dem BCL6 -Promotor Transgen Reporter-Gen Ausdruck33, was darauf hindeutet, dass es funktionieren kann, um BCL6 Transkription in ihrem systemeigenen Kontext zu unterdrücken.
PIRs angereichert für Belegung des Chromatin-Isolator-Proteins CTCF in menschlichen WSR und NEC37 noch eine weitere Klasse von PIRs darstellen kann. Diesen Ergebnissen zufolge gemeinsam PIRs beherbergen eine Sammlung von regulatorischen Genaktivitäten noch zu funktional charakterisiert werden.
Offenlegungen
Autoren haben nichts preisgeben.
Danksagungen
Wir danken Valeriya Malysheva für kritische Lektüre des Manuskripts und Hilfe von Experten mit Abbildung 1. Diese Arbeit wurde vom Medical Research Council, UK (MR/L007150/1) und die UK Biotechnologie und biologische Wissenschaften Research Council, UK (BB/J004480/1) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 16% (vol/vol) paraformaldehyde solution | Agar Scientific | R1026 | |
| Dulbecco's Modified Eagle Medium (DMEM) 1x | Life Technologies | 41965-039 | |
| Fetal bovine serum (FBS) sterile filtered | Sigma | F9665 | |
| Low-retention filter tips | Starlab | S1180-3810, S1180-1810, S1180-8810 and S1182-1830 | |
| 10x PBS pH 7.4 | Life Technologies | 70011-036 | |
| Molecular biology grade water | Sigma-Aldrich | W4502 | |
| 1 M Tris-HCl pH 8.0 | Life Technologies | 15568-025 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 5 M NaCl | Life Technologies | 24740-011 | |
| Protease inhibitor cocktail (EDTA-free) | Roche Diagnostics | 11873580001 | |
| Restriction buffer 2 (10x NEBuffer 2) | New England Biolabs | B7002 | |
| DNA LoBind tube, 1.5 mL | Eppendorf | 0030 108.051 | |
| DNA LoBind tube, 2 mL | Eppendorf | 30108078 | |
| 20% (wt/vol) SDS | Bio-Rad Laboratories | 161-0418 | |
| 20% (vol/vol) Triton X-100 | Sigma-Aldrich | T8787 | |
| HindIII, 100 U/uL | New England Biolabs | R0104 | |
| 10 mM dCTP | Life Technologies | 18253-013 | |
| 10 mM dGTP | Life Technologies | 18254-011 | |
| 10 mM dTTP | Life Technologies | 18255-018 | |
| 0.4 mM Biotin-14-dATP | Life Technologies | 19524-016 | |
| DNA polymerase I large (Klenow) fragment 5000 units/mL | New England Biolabs | M0210 | |
| 10x T4 DNA ligase reaction buffer | New England Biolabs | B0202 | |
| 100x 10mg/ml Bovine Serum Albumin | New England Biolabs | B9001 | |
| T4 DNA ligase, 1 U/μL | Invitrogen | 15224-025 | |
| RNase A | Roche | 10109142001 | |
| Proteinase K, recombinant, PCR grade | Roche | 3115836001 | |
| 20 000×g 50 ml centrifuge tube | VWR | 525-0156 | |
| 0.5 M EDTA pH 8.0 | Life Technologies | 15575-020 | |
| Phenol pH 8.0 | Sigma | P4557 | |
| Phenol: Chloroform: Isoamyl Alcohol 25:24:1 | Sigma | P3803 | |
| Sodium acetate pH 5.2 | Sigma | S7899 | |
| Quant-iT PicoGreen | Invitrogen | P7589 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| Restriction buffer 2.1 (10x NEBuffer 2.1) | New England Biolabs | B7202 | |
| NheI, 100U/uL | New England Biolabs | R0131 | |
| Micro TUBE AFA Fiber Pre-slit snap cap 6x16mm vials | Covaris | 520045 | For sonication |
| SPRI beads (Agencourt AMPure XP) | Beckman Coulter | A63881 | |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| Tween 20 | Sigma | P9416 | |
| 10 mM dATP | Life Technologies | 18252-015 | |
| T4 DNA polymerase 3000 units/mL | New England Biolabs | M0203 | |
| T4 PNK 10000 units/mL | New England Biolabs | M0201 | |
| Klenow exo minus 5000 units/mL | New England Biolabs | M0212 | |
| Quick ligation reaction buffer | New England Biolabs | B6058 | |
| NEB DNA Quick ligase | New England Biolabs | M2200 | |
| PE adapter 1.0 (5'-P-GATCGGAAGAGCGGTTCAGC AGGAATGCCGAG-3') | Illumina | ||
| PE adapter 2.0 (5'-ACACTCTTTCCCTACACGACGCT CTTCCGATCT-3') | Illumina | ||
| NEB Phusion PCR kit | New England Biolabs | M0530 | |
| PE PCR primer 1.0 (5'-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTAC ACGACGCTCTTCCGATCT-3') | Illumina | ||
| PE PCR primer 2.0 (5'-CAAGCAGAAGACGGCATACGA GATCGGTCTCGGCATTCCT GCTGAACCGCTCTTCCGATCT-3') | Illumina | ||
| PCR strips | Agilent Technologies | 410022 and 401425 | |
| SureSelect SSEL TE Reagent ILM PE full adaptor kit | Agilent Technologies | 931108 | |
| SureSelect custom 3-5.9 Mb library | Agilent Technologies | 5190-4831 | custom design mouse or human PCHi-C system |
| Dynabeads MyOne Streptavidin T1 beads | Invitrogen | 65601 | |
| E220 high-performance focused ultra-sonicator | Corvaris | E220 |
Referenzen
- Osborne, C. S., et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nature Genetics. 36, 1065-1071 (2004).
- Schoenfelder, S., et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature Genetics. 42, 53-61 (2010).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501, 227-231 (2013).
- Bantignies, F., et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 144, 214-226 (2011).
- Engreitz, J. M., et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 341, 1237973(2013).
- Denholtz, M., et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 13, 602-616 (2013).
- Schoenfelder, S., et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics. 47, 1179-1186 (2015).
- Kundu, S., et al. Polycomb Repressive Complex 1 generates discrete compacted domains that change during differentiation. Molecular Cell. 65, 432-446 (2017).
- Skok, J. A., Gisler, R., Novatchkova, M., Farmer, D., de Laat, W., Busslinger, M. Reversible contraction by looping of the Tcra and Tcrb loci in rearranging thymocytes. Nature Immunology. 8, 378-387 (2007).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148, 908-921 (2012).
- Aymard, F., et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology. 24, 353-361 (2017).
- Ryba, T., et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research. 20, 761-770 (2010).
- Pope, B. D., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 515, 402-405 (2014).
- Chandra, T., et al. Global reorganization of the nuclear landscape in senescent cells. Cell Reports. 10, 471-483 (2015).
- Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., Fraser, P. Long-range chromatin regulatory interactions in vivo. Nature Genetics. 32, 623-626 (2002).
- Tolhuis, B., Palstra, R. J., Splinter, E., Grosveld, F., de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Molecular Cell. 10, 1453-1465 (2002).
- Amano, T., Sagai, T., Tanabe, H., Mizushina, Y., Nakazawa, H., Shiroishi, T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Developmental Cell. 16, 47-57 (2009).
- Zuniga, A., et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes & Development. 18, 1553-1564 (2004).
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132, 797-803 (2005).
- D'Haene, B., et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 5, e1000522(2009).
- Sur, I. K., et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 338, 1360-1363 (2012).
- Herranz, D., et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature Medicine. 20, 1130-1137 (2014).
- Deng, W., et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 149, 1233-1244 (2012).
- Groschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157, 369-381 (2014).
- Lupianez, D. G., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 161, 1012-1025 (2015).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538, 265-269 (2016).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295, 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38, 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genetics. 38, 1341-1347 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research. 16, 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289-293 (2009).
- Belton, J. M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., Dekker, J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 58, 268-276 (2012).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47, 598-606 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582-597 (2015).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167, 1369-1384 (2016).
- Wilson, N. K., et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 127, e12-e23 (2016).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife. 6, (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49, 1522-1528 (2017).
- Siersbaek, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66, 420-435 (2017).
- Sahlen, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Nagano, T., et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64 (2013).
- Nagano, T., Varnai, C., Schoenfelder, S., Javierre, B. M., Wingett, S. W., Fraser, P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology. 16, 175(2015).
- Wingett, S., et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Res. 4, 1310(2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24, 1854-1868 (2014).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- McGovern, A., et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol.ogy. 17, 212(2016).
- Hodge, D., et al. A global role for EKLF in definitive and primitive erythropoiesis. Blood. 107, 3359-3370 (2006).
- Jager, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter Interactions between two states of pluripotency. Cell Stem Cell. 17, 748-757 (2015).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46, 205-212 (2014).
- Kolovos, P., et al. Targeted Chromatin Capture (T2C): A novel high-resolution high-throughput method to detect genomic interactions and regulatory elements. Epigenetics Chromatin. 7, 10(2014).
- Davies, J. O., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13, 74-80 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13, 919-922 (2016).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462, 58-64 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337, 1190-1195 (2012).
- Petersen, R., et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat. Commun. 8, 16058(2017).
- Akhtar, W., et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 154, 914-927 (2013).
- Cairns, J., et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biology. 17, 127(2016).
- Rajagopal, N., et al. High-throughput mapping of regulatory DNA. Nature Biotechnology. 34, 167-174 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten