
Method Article
Promotor captura Oi-c: Alta resolução, todo o genoma caracterização das interações do promotor
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Elementos reguladores de DNA, como potenciadores, controlam a expressão gênica fisicamente contactando promotores de genes alvo, muitas vezes através de interações cromossômicas de longo alcance, abrangendo grandes distâncias genômicas. Promotor de capturar Hi-C (PCHi-C) identifica interações significativas entre promotores e regiões distais, permitindo a atribuição de potenciais sequências reguladoras de seus genes-alvo.
Resumo
A organização tridimensional do genoma está ligada à sua função. Por exemplo, elementos reguladores tais como potenciadores transcriptional controlam a expressão espaço-temporal dos seus genes alvo através do contato físico, muitas vezes ponte genômica distâncias consideráveis (em alguns casos centenas de kilobases) e ignorando genes nas proximidades. O genoma humano abriga uma potenciadores de 1 milhão estimado, a grande maioria dos quais têm desconhecido alvos de gene. Atribuição de regiões reguladoras distais para seus genes-alvo, portanto, é crucial entender controle de expressão do gene. Nós desenvolvemos o promotor capturar Hi-C (PCHi-C) para permitir a detecção de todo o genoma de regiões de promotor-interagindo distais (PIRs), para todos os promotores em uma única experiência. No PCHi-C, altamente complexo Hi-C bibliotecas especificamente são enriquecidas para sequências de promotor através de seleção em solução híbrida com milhares de iscas biotinilado RNA complementares até o fim de todos os fragmentos de restrição que contêm promotor. O objectivo é então suspenso promotor sequências e seus parceiros de interacção frequente como potenciadores e outros elementos reguladores potenciais. Após o sequenciamento de emparelhado-fim do elevado-throughput, um teste estatístico é aplicado para cada fragmento de restrição promotor-ligados para identificar PIRs significativos no nível de fragmento de restrição. Nós usamos PCHi-C para gerar um atlas de interações de longo alcance promotor em dezenas de humanos e tipos de células de rato. Estes mapas de interactome promotor têm contribuído para uma maior compreensão do controle da expressão de gene mamíferos atribuindo putativos regiões reguladoras de seus genes-alvo e revelando as redes de interação espacial preferencial do promotor-promotor. Esta informação também tem alta relevância para a compreensão de doenças genéticas humanas e a identificação de potenciais genes de doença, vinculando não-codificantes associada a doença sequenciar variantes em ou perto de sequências de controle para seus genes-alvo.
Introdução
Acumular evidência sugere que a organização tridimensional do genoma desempenha um papel funcional importante em uma variedade de processos nucleares, incluindo gene ativação1,2,3, repressão4 ,5,6,7,8, recombinação9,10, reparação de DNA11, DNA replicação12,13, e senescência celular14. Potenciadores de distantes encontram-se em estreita proximidade espacial com os promotores regulam15,16,17, que é essencial para o controle da expressão de gene espácio-temporal adequada. Exclusões de potenciador mostram que potenciadores distais são essenciais para alvo gene transcrição18,19,20,21,22e 'forçado a cromatina loop' demonstra que amarrar projetado entre um intensificador e seu promotor alvo no locus do Hbb é suficiente para conduzir a ativação transcricional23. Além disso, rearranjos do genoma que trazem genes sob o controle de potenciadores de uma gravidez ectópica podem resultar na ativação do gene inapropriado e doença24,25,26. Juntos, estes exemplos ilustram que atrapalha-promotor interações são essenciais para o controle do gene e exigem o Regulamento apertado para garantir a expressão do gene apropriado. O ser humano e genomas do mouse cada estima-se que abrigam potenciadores de ao redor 1 milhão. Para a maioria destes potenciadores, genes-alvo são desconhecidos, e as 'regras' entre promotores e realçadores são mal compreendidas. Atribuir transcriptional potenciadores de seus genes-alvo, portanto, permanece um grande desafio em decifrar controle de expressão de genes de mamíferos.
Nossa compreensão da arquitetura do genoma tridimensional foi revolucionada pela introdução de 3C27 (captura de conformação do cromossomo) e suas variantes28,29,30,31 . A mais poderosa destas técnicas, Hi-C (captura de conformação de cromossomo alto throughput) é projetada para identificar todo o conjunto de interações cromossômicas dentro de uma população celular. Oi-C bibliotecas, geralmente geradas a partir de milhões de células, são altamente complexas, com um estimado 1011 produtos independente da ligadura entre fragmentos de ~ 4 kb no genoma humano32. Como consequência, reprodutível e confiável identificação das interações entre restrição individual de fragmentos (tais como aqueles que contêm um promotor ou potenciador) de dados Hi-C não são viáveis a menos Hi-C bibliotecas são submetidas a sequenciamento ultraprofundas, que não é uma solução economicamente viável para os laboratórios preparando Hi-C bibliotecas rotineiramente. Para contornar esta lacuna, nós desenvolvemos o promotor capturar Hi-C para enriquecer especificamente promotor-contendo produtos de ligadura de bibliotecas Hi-C. Enfocamos os promotores por duas razões. Primeiro, contatos atrapalha-promotor foram mostrados para ser crucial para os níveis de expressão de gene apropriado em numerosos estudos (ver referências acima), e em segundo lugar, como promotores são em grande parte invariável entre tipos de células, o mesmo sistema de captura de isca pode ser usado para interrogar o circuito regulador em vários tipos de células e condições. Nossa abordagem baseia-se na solução de hibridização das bibliotecas Hi-C com dezenas de milhares de 120mers biotinilado RNA complementar ao promotor-contendo produtos de ligadura Hi-C e subsequente captura em grânulos magnéticos streptavidin-revestido. Isso resulta em bibliotecas PCHi-C, com muito menor complexidade comparada à biblioteca de Hi-C original, focando apenas a identificação de fragmentos que são ligados para promotores em significativamente altas frequências.
Nós usamos PCHi-C em um número de humanos e tipos de células de rato para contribuir para uma melhor compreensão do controle de expressão do gene por descobrindo regiões de interação promotora distal de longo alcance com função reguladora putativa, bem como não-aleatória contatos do promotor-promotor no espaço tridimensional do núcleo. Os estudos mapeou centenas de milhares de contatos atrapalha-promotor através de numerosas células tipos33,34,35,36,37,38, 39, identificado organização mediada por Polycomb repressivo complexo espacial do genoma em células-tronco embrionárias de rato7, demonstrada em larga escala a religação do promotor interactomes durante a diferenciação celular37, 38 , 39e vinculados não-codificantes associada a doença de sequências variantes de genes promotores35.
PCHi-C é um método ideal para mapear o genoma-largo conjunto de sequências de DNA, interagindo com os promotores. Abordagens relacionadas, tais como capturar Hi-C de contínuas regiões genômicas (ver discussão) são o método de escolha para obter perfis de interação de alta resolução para regiões genômicas selecionadas. PCHi-C e Hi-C capturar são extremamente semelhantes do experimental ponto de vista (a única diferença é a escolha do sistema de captura), para que os conselhos e orientações que nós fornecemos são aplicáveis a ambas as abordagens. Aqui, apresentamos uma descrição detalhada do PCHi-C. Podemos delinear a lógica e o projeto de um experimento de PCHi-C, fornecer um protocolo de geração de biblioteca PCHi-C passo a passo e ilustrar como a qualidade das bibliotecas PCHi-C pode ser monitorizada em várias etapas no protocolo para produzir dados de alta qualidade.
Protocolo
1. fixação de formaldeído
-
Preparação de células: comece com um mínimo de 2 x 107 células por experiência.
- Para as células crescidas em cultura, Ressuspender as células em meio de cultura. Para as células ex vivo , resuspenda em 1 x de Dulbecco modificado águia médio (DMEM), suplementado com 10% (vol/vol) de soro fetal bovino (FBS).
- Para as células aderentes, remover o meio de cultura e adicionar 30,625 mL de meio fresco com 10% (vol/vol) FBS à temperatura ambiente (RT; 20-25 ° C).
- Para células de suspensão, coletar e centrifugar as células em 400 x g e 20 ° C por 3 min. remover sobrenadante e ressuspender centrifugado em 30,625 mL do meio com 10% (vol/vol) FBS no RT
- Para tecidos sólidos, use tripsina (0,05% para 2,5% concentração final, dependendo do tipo de célula) ou homogenizacao homogeneização para obter uma suspensão de célula única. Após esta etapa adicional, trate as células como as células de suspensão.
-
Adicione 4,375 mL de 16% metanol livre paraformaldeído (ampola aberta apenas antes do uso) a uma concentração final de 2% (vol/vol). Correção para 10 min a RT com suave mistura sobre um roqueiro.
Cuidado: Paraformaldehyde é um produto químico perigoso. Siga os regulamentos de saúde e segurança adequados. - Saciar a reação pela adição de 5 mL de recém-preparado 1m gelada glicina. Misture por 5 min com balanço suave no RT e então incubar no gelo por 15 min com inversão ocasional.
-
Lave e recolher pilhas fixas.
- Para as células aderentes, remover o sobrenadante, adicionar 10 mL de gelado 1 x pH PBS 7.4 na parede placa e removê-lo. Adicionar 1 mL de gelado 1 x pH PBS 7.4, coletar células usando um raspador de célula e transferir para um tubo de 50 mL. Repita duas vezes para recolher o maior número de células possível. PBS gelado somam volume final de 50 mL.
- Para células de suspensão, células de centrífuga a 760 x g 4 ° C por 5 min, remover o sobrenadante e ressuspender centrifugado em 50 mL de pH de PBS gelado 7,4.
- Centrifugar as células em 400 x g e 4 ° C por 10 min e retire com cuidado o sobrenadante. O centrifugado pode ser snap congelado em nitrogênio líquido e posteriormente armazenados a-80 ° C durante vários meses.
2. lise celular
- Ressuspender o pellet de célula em 50 mL de tampão de Lise gelada recém-preparado (pH 10 mM Tris-HCl 8, 0,2% (vol/vol) Igepal CA-630, 10 mM de NaCl e um comprimido inibidor de protease cocktail) e misture. Incubar no gelo por 30 min, misture ocasionalmente por inversão. Centrifugue os núcleos em 760 x g e 4 ° C por 5 min e retirar o sobrenadante.
3. hindIII digestão
- Lave o núcleo de células com tampão de restrição 1.25 x 2. Ressuspender o pellet de células em 1 mL de tampão de restrição x 1.25 gelada 2 e transferir para um tubo de 1,5 mL. Girar os núcleos em 760 x g e 4 ° C por 5 min e retirar o sobrenadante.
- Re-suspenda centrifugado em 1790 µ l de tampão de restrição 1.25 x 2. Fazer 5 alíquotas, cada uma contendo 5 milhões de células em µ l 358 de 1,25 x buffer de restrição 2.
- Adicione 11 µ l de 10% (wt/vol) SDS por alíquota e agitar a 950 revoluções por minuto (rpm) por 30 min a 37 ° C em um thermomixer. Se aparecerem grumos de células, dissocia pipetando, evitando bolhas.
- Adicione 75 µ l de 10% Triton X-100 (vol/vol) por alíquota e agitar a 950 rpm e 37 ° C por 15 min em um thermomixer. Se aparecerem grumos de células, dissocia pipetando, evitando bolhas.
-
Adicionar 12 µ l de 100 U / µ l HindIII 100 (1.200 unidades no total) por alíquota e incubar a 37 ° C durante a noite (O/N) agitando a 950 rpm em um thermomixer.
- Para o controle da digestão, 25 µ l de amostra (5 µ l de cada alíquota) em um novo tubo de transferência antes de adicionar a enzima (controle não digerido) e repita o mesmo procedimento, após a adição da enzima (controle digerido). Incube ambos os tubos da mesma maneira que a biblioteca de Hi-C.
- Na manhã seguinte, adicionar 5 µ l de 100 U / µ l HindIII (500 unidades no total) por alíquota e incubar a 37 ° C por 2 h, agitando a 950 rpm em um thermomixer.
-
Controle da digestão: para os controles digeridos e não digeridos (ver 3.5.1), realizar reversão crosslink (etapa 6), extração de fenol: clorofórmio e precipitação do DNA (etapa 7).
- Desenha um par de primers que abrangem um site de dIII Hin. Na mesma região, desenha um outro par de primers que não abrangem um site de dIII Hin. Projetar primers para PCR quantitativo (Q-PCR) usando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) e os seguintes parâmetros:
Tamanho da primeira demão: 20 ideal (min.: 18, máx.: 27); Primeira demão Tm: Ideal 60 (min.: 57, máx: 63); % De teor de cartilha CG: min.: 20, máx: 80; Amplicon tamanho: RT-PCR ~ 100 bp (para PCR convencional ~ 300 bp); Mispriming biblioteca: humanos (primeiras demão humanas) ou roedores e simples (primeiras demão do mouse). - Executar Q-PCR para obter 4 Cts médios (ciclo de limiar): Ct [D; H., obtido a partir da amostra digerida [D] com o par de primers que abrangem um site de dIII Hin[H]; CT [D;-], obtidos a partir da amostra digerida [D] com o par de primers que não abrangem um site de dIII Hin[-]; CT [U; H., obtido a partir da amostra não digerida [U] com o par de primers que abrangem um site de dIII Hin; CT [U;-], obtidos a partir da amostra não digerida [U] com o par de primers que não abrangem um site de dIII Hin[-]. Calcular a porcentagem de digestão como: digestão % = 100-100/2(Ct[D,H]-Ct[D,-]) - (Ct[U,H]-Ct[U,-]).
- Desenha um par de primers que abrangem um site de dIII Hin. Na mesma região, desenha um outro par de primers que não abrangem um site de dIII Hin. Projetar primers para PCR quantitativo (Q-PCR) usando Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) e os seguintes parâmetros:
4. Biotinylation de saliências de fragmento de restrição
- Preparar a mistura de mestre biotinylation: 30.6 µ l de 10x buffer de restrição 2, 10,2 µ l de H2O (grau de biologia molecular), 7.65 µ l de 10mm dCTP, 7.65 µ l de 10 mM dGTP, 7.65 µ l de 10mm dTTP, 191.25 µ l de 0,4 mM biotina-14-dATP e 51 µ l de 5.000 U/mL de DNA polimerase eu (grande Fragmento de Klenow).
- Adicione 60 µ l de mistura de mestre biotinylation por alíquota, mistura e incubar a 37 ° C, durante 1 h, agitando a 700 rpm (thermomixer) por 5 s, a cada 30 s. Após 1 h, coloca alíquotas no gelo.
5. no núcleo-ligadura
- Preparar a mistura de mestre da ligadura: 510 µ l de 10x buffer de T4 DNA ligase, 51 µ l de 10 mg/mL de albumina de soro bovino (BSA 100x), 1754.4 µ l de água (grau de biologia molecular) e 127.5 µ l de 1 ligase do DNA de T4 U / µ l (veja a Tabela de materiais).
- Adicionar 479 µ l do mix de mestre da ligadura por alíquota mistura e incubar a 16 ° C por 4 h a tremer a 700 rpm por 5 s cada 2 min em um thermomixer.
- Incubar 30 min em RT.
6. Crosslink reversão
- Combine todas as alíquotas em um tubo de centrífuga de 50 mL (apropriado para centrifugação de alta velocidade).
- Adicione 62,5 µ l de 10 mg/mL RNase A, mistura e incube por 30 min a 37 ° C.
- Acrescente 300 µ l de Proteinase K, mistura, de 10 mg/mL e incube por 30 min a 37 ° C.
- Incubar a reação O/N (ou pelo menos 4 h) a 65 ° C. Na manhã seguinte, adicionar 300 µ l de Proteinase K, mistura, de 10 mg/mL e incubar durante 1 h a 65 ° C.
7. purificação de DNA
- Adicione 4337.5 µ l de tampão TLE (10 mM Tris-HCl pH 8.0; pH de 0,1 mM EDTA 8.0) e misture.
- Adicione 1 volume (10 mL) fenol pH 8.0, vórtice durante 10 s e centrifugar em RT e 20.000 x g durante 3 min. transferir 9 mL da fase superior (aquosa) para um novo tubo de 50 mL.
Atenção: O fenol é um produto químico perigoso. Siga os regulamentos de saúde e segurança adequados. - Adicionar 2 mL de tampão TLE à fase aquosa restante, vórtice para 10 s e centrifugar RT e 20.000 x g durante 3 min. 2,5 mL da fase aquosa para o novo tubo de transferência de passo 7.2, fazendo o volume final 11,5 mL. Descarte o tubo contendo a fase inferior (orgânica).
- Adicione 1 volume (11,5 mL) de álcool isoamílico: fenol: clorofórmio (25:24:1), vortex durante 10 s e centrifugar em RT e 20.000 x g durante 3 min. transferir 11 mL da fase superior (aquosa) para um novo tubo de 50 mL. Repita o passo 7.3. O volume total da amostra será agora 13,5 mL.
- Adicionar 1,35 mL de 3M de sódio acetato pH 5.2 e 33,75 mL de etanol a 100% fria como o gelo, mistura e incubar a-80 ° C por 45 min ou em alternativa durante a noite a-20 ° C.
- Centrifugar a 4 ° C e 20.000 x g durante 10 minutos, retire o sobrenadante, Ressuspender o sedimento em 1 mL de etanol a 70% (vol/vol) recém-preparado e transfira para um tubo novo.
- Centrifugar a 4 ° C e a toda a velocidade para 3 min em uma centrífuga de bancada e, em seguida, remover o sobrenadante.
- Ressuspender o pellet em 1 mL de gelo frio 70% (vol/vol) etanol e repita a etapa 7,7. Secar a pelota a 37 ° C por 10 min e ressuspender em 650 µ l de tampão TLE. Determine o rendimento do ADN usando um ensaio baseado em fluorescência para quantificar o DNA de cadeia dupla.
Nota: O protocolo pode ser pausado aqui por snap, congelação e armazenamento de amostra a-80 ° C durante vários meses, ou a-20 ° C durante um período mais curto de tempo.
8. controle de qualidade
- Monitorar a integridade de biblioteca e ligadura por electroforese de DNA. Executar 200 ng de biblioteca em um gel de x TBE de 1/agarose 0,8%. O DNA deve ser executado como uma banda mais de 10 kb.
- Detecta a célula-tipo conhecido invariável curto e longo - range interações por PCR convencional. Uso 100 ng do modelo de DNA por reação de PCR. Desenha os primers PCR perto e para os locais de restrição, seguindo as instruções acima (ver 3.7.1). Sequências da primeira demão, controle de qualidade de rato e humanos Hi-C bibliotecas estão listadas na tabela 1.
-
Controle de preenchimento e ligadura: recorte as bandas de gel contendo os amplicons de controlar 8.2, gel-extrato de DNA e usar o ADN como modelo para 4 individuais reações de PCR com combinações idênticas da primeira demão.
- Purificar amplicons usando um kit de purificação de PCR e quantificar a concentração de DNA.
- Prepare quatro reações digestão (HindIII [a], NheI [b], HindIII + NheI [c] e nenhuma enzima [d]) para cada amplicon em um volume final de 15 µ l: 500 ng de amplicons, 1,5 µ l de 10x buffer de restrição 2.1, 0,15 µ l de 10 mg/mL de albumina de soro bovino (BSA 100x) e 0,1 µ l (10 unidades) da enzima (HindIII [a], [b] NheI, HindIII + NheI [c] ou água [d]).
- Digerir durante 1 h a 37 ° C e, em seguida, execute as reações de digestão em um gel de x TBE de 1/agarose 1,5% (wt/vol).
9. fragmentação de DNA
- Transferência de 50,5 µ g da amostra em um tubo novo e adicionar tampão TLE até um volume final da amostra 655 µ l. Split em 5 frascos de sonication (ver Tabela de materiais), adicionando 130 µ l de biblioteca (10 µ g) para cada frasco. Tesoura para um tamanho de ~ 400 bp em um ultrasonicador (ver Tabela de materiais) usando os seguintes parâmetros: fator de serviço: 10%; pico de potência incidente (w): 140; ciclos por explosão: 200; tempo: 55 s.
- Colete amostra de lisados em um tubo 2 mL.
10. seleção de tamanho verso SPRI-grânulo
- Solução de grânulo SPRI Mix (imobilização reversível da fase sólida) bem invertendo, 1,85 mL da solução de talão de transferência para um novo tubo e traga a RT por 15 min.
- Adicionar 350 µ l de água (grau de biologia molecular) para a amostra (volume final 1 mL).
- Adicionar 600 µ l da solução de grânulo SPRI à amostra (total de volume de 1,6 mL; relação da solução SPRI para DNA: 0,6 a 1), incubar durante 5 min à RT e girar a amostra em uma centrífuga de bancada para 2-3 s coletar a amostra.
- Abra a tampa, coloque a amostra no stand separação magnética por 5 min, clara sobrenadante para um novo tubo de transferência e descarte de grânulos.
- Concentre-se grânulos SPRI para a segunda etapa de seleção de tamanho: 930 transferir µ l de SPRI contas para um novo tubo, coloque no stand separação magnética por 5 min e descarte sobrenadante claro. Re-suspenda as contas em 310 µ l da solução de grânulo SPRI.
- Adicionar 300 µ l de grânulos SPRI concentrados (etapa 10,5) para a amostra (total de volume de 1,9 mL; relação SPRI solução de DNA é agora 0,9 a 1), incubar a RT por 5 min e rotação da amostra em uma centrífuga de bancada para 2-3 s. abra cuidadosamente a tampa , colocar o tubo no stand separação magnética por 5 min e descartar o sobrenadante.
- Adicionar 1 mL de etanol a 70% (vol/vol) recentemente preparada para o tubo de amostra no stand separação magnética, incube durante 30 s e o sobrenadante de descarte. Repita duas vezes.
- Grânulos de secos a 37 ° C, em um thermomixer (tampa aberta do tubo), não mais que 5 min. Adicionar 300 µ l de tampão TLE à amostra, misturar e incubar durante 10 minutos à temperatura ambiente.
- Girar a amostra em uma centrífuga de bancada para 2-3 s, abra a tampa e lugar o tubo sobre a separação magnética suportar 5 min. transferência limpar o sobrenadante para um tubo novo e descartar os grânulos.
11. biotina/estreptavidina suspenso de ligadura produtos
- Preparar os amortecedores: TB tampão 1x (pH 5mM Tris-HCl 8.0; 0,5 mM EDTA; 1 M NaCl 0,05% Tween 20); 2 x buffer de BNP (10 mM Tris-HCl pH 8.0; 1 mM EDTA; 2 M NaCl); 1 x NTB amortecedor (5 mM Tris-HCl pH 8.0; 0,5 mM EDTA; 1 M NaCl).
- Adicionar 200 µ l de magnéticos grânulos streptavidin-acoplado (ver Tabela de materiais) para um novo tubo, coloque-o sobre o carrinho de separação magnética para 1 min e remover o sobrenadante.
-
Lave grânulos duas vezes com 500 µ l de tampão de 1x TB.
- Para cada etapa de lavagem durante a biotina suspenso, reparação final e remoção de biotina em extremidades de DNA não-ligados, dATP rejeito e etapas de ligadura do adaptador, re-suspender as contas no buffer correspondente, girar na RT e 15 rpm por 3 min, girar o tubo em uma centrífuga de bancada para s de 2 – 3, colocar o tubo no stand separação magnética por 3 min e retirar o sobrenadante.
- Re-suspenda contas em 300 µ l de tampão de x NTB 2. Mistura de grânulos e amostra (volume total de 600 µ l) e incubar a RT por 15 min em uma roda rotativa a 3 rpm.
- Recuperar os grânulos no stand separação magnética por 3 min e remover o sobrenadante claro. Lave os grânulos duas vezes em 500 µ l de tampão de x NTB 1 primeiro e em seguida em 200 µ l de tampão de ligadura 1 x. Re-suspenda as contas em 50 µ l de tampão de ligadura x 10.
12. fim de reparação e remoção de biotina em extremidades de DNA não-ligados
- Combine a amostra (50 µ l no total) com 50 µ l de 2.5 mM dNTP mix (12,5 µ l de 10 mM de cada dNTP), 18.1 µ l de 3.000 U/mL T4 DNA Polymerase, 18.1 µ l de 10.000 U/mL T4 PNK, 3,7 µ l de 5.000 U/mL de DNA polimerase que grande (Klenow) fragmento e 360.1 µ l de H2O.
- Misturar e incubar a 20 ° C, durante 1 h, agitando 5 s a cada 2 min em um thermomixer de 700 rpm.
- Recuperar os grânulos no stand separação magnética, remover o sobrenadante claro e lave os grânulos duas vezes em 500 µ l de tampão de TB 1x.
- Lave os grânulos em 500 µ l de 1 buffer de x NTB, seguido por uma lavagem em 500 µ l de 1 x TLE.
- Recuperar os grânulos no stand separação magnética, remover o sobrenadante claro e ressuspender grânulos em 415 µ l de tampão de x TLE 1.
13. dATP seguindo
- Combine a amostra (415 µ l) com 50 µ l de tampão de restrição 2, 5 µ l de 10 mM dATP e 30 µ l de 5 U / µ l Klenow exo-menos de 10x.
- Misturar e incubar a 37 ° C por 30 min, agitando 5 s a cada 2 min em um thermomixer de 700 rpm.
- Recuperar os grânulos no stand separação magnética, remover o sobrenadante claro e lave os grânulos duas vezes em 500 µ l de tampão de TB 1x.
- Lave os grânulos em 500 µ l de tampão de x NTB 1.
14. adaptador ligadura
- Lave os grânulos em 200 µ l de tampão de reação da ligadura 1x (ver Tabela de materiais).
- Re-suspenda contas em 200 µ l de 1 amortecedor da reação da ligadura x. Adicione 4 µ l de DNA ligase (ver Tabela de materiais) e 16 µ l de 15 µM pre-recozido adaptadores de PE (pre-recozem os adaptadores PE misturando volumes iguais de PE adaptador 1 e PE adaptador 2 (ambos em 30 µM) e incubar durante alguns minutos a RT). Incube a RT por 15 min.
- Recuperar os grânulos no stand separação magnética, remover o sobrenadante claro e lave os grânulos duas vezes em 500 µ l de tampão de TB 1x.
- Lave os grânulos em 500 µ l de tampão de x NTB 1. Em seguida, lave grânulos em 100 µ l de tampão de restrição 2, 1x Ressuspender grânulos em 50 µ l de tampão de restrição 2 1x e transferir para um tubo novo.
15. Oi-C biblioteca amplificação
- Preparar a mistura de mestre PCR: 100 µ l de tampão de x Phusion 5; 6 µ l de primer de PCR PE 25 µM 1.0; 6 µ l de primer de PCR PE 25 µM 2.0; 14 µ l do dNTP mix (10 mM cada); 6 µ l da Phusion polimerase; 318 µ l de H2O.
- Mistura de mestre de PCR Mix com os grânulos (500 µ l no total), divida em 10 alíquotas de 50 µ l e amplificar pelo PCR usando as seguintes condições:
30 anos a 98 ° C
7 ciclos de: 10 s a 98 ° C; 30 s a 65 ° C; 30 s a 72 ° C
7 min a 72 ° C - Recolher as reações de PCR para um novo tubo, recuperar contas no carrinho de separação magnética e transferência sobrenadante (500 µ l) para um tubo novo.
-
Purifica o DNA de biblioteca usando SPRI grânulos.
- Grânulos de SPRI Mix, transferir 460 µ l de grânulos em um novo tubo e trazer a RT por 15 min. Adicionar 450 µ l de grânulos SPRI para as reações de PCR (volume final 950 µ l), incubar durante 5 min à RT e girar a amostra em uma centrífuga de bancada para 2-3 s coletar a amostra.
- Abra a tampa, coloque a amostra no stand separação magnética por 5 min e retirar o sobrenadante.
- Mantendo os grânulos no stand separação magnética, adicione 1 mL de etanol a 70% (vol/vol) ao tubo da amostra sobre uma área clara de grânulos, deixar por 30 s e o sobrenadante de descarte.
- Repita a etapa 15.4.3 mais duas vezes.
- Secar contas a 37 ° C, em um thermomixer (tampa tubo aberta), por não mais que 5 min.
- Adicionar 51 µ l de tampão TLE à amostra, mistura e incubar durante 10 minutos a 37 ° C, agitando a 950 rpm em um thermomixer.
- Girar a amostra em uma centrífuga de bancada para 2-3 s, abra a tampa e lugar o tubo sobre a separação magnética suportar 5 min. transferência limpar o sobrenadante para um tubo novo e descartar os grânulos.
- Quantificar a concentração de Hi-C biblioteca. Após 7 rodadas de amplificação por PCR, obtemos rotineiramente 500 – 1.500 ng de biblioteca Hi-C.
16. híbrido em solução captura
Nota: Bloqueador e buffer (SHS1-4) soluções abaixo são do SureSelect kit (veja a Tabela de materiais).
- Transferência de 500 ng de 1 µ g de Hi-C biblioteca para um novo tubo e evaporar a amostra em um concentrador de vácuo (ver Tabela de materiais; 45 ° C; pressão do vácuo: nível 30,0, rampa 5) até secar.
- Re-suspenda evaporada Hi-C biblioteca adicionando 3,6 µ l de H2O (grau de biologia molecular), 2,5 µ l de bloqueador 1, 2,5 µ l de bloqueador 2 e 0,6 µ l de bloqueador personalizado.
- Transferir a amostra para um poço de uma nova faixa de tubo PCR, feche com uma tira de tampão PCR e colocar no gelo. Rótulo como "D" (para DNA C).
- Preparar o tampão de hibridização: 12,5 µ l de tampão de SHS1; 0,5 µ l de tampão de SHS2; 5 µ l de tampão de SHS3; 6.5 µ l de tampão de SHS4.
- Incube a 65 ° C por 5 min em um thermomixer. Transferir para um poço de uma nova faixa de tubo PCR, feche com uma tira de tampão PCR e manter no RT. Label como "H" (para tampão de Hibridização).
- Num poço de uma nova faixa de tubo PCR, misturar 5 µ l de 100 ng / µ l biotinilado sondas de RNA (loja a-80 ° C e degelo no gelo imediatamente antes da utilização); 0,5 µ l de SRNase B (inibidor de RNase) e 1,5 µ l de H2O (grau de biologia molecular).
- Feche a tira do tubo PCR com uma tira de tampão PCR e lugar no gelo. Rótulo como "R" (RNA).
- Configure a máquina PCR, usando os seguintes parâmetros:
5 min a 95 ° C; h 25 a 65 ° C; tampa aquecida; Volume de reação de PCR de 29 µ l.
Nota: Proceda tão rapidamente quanto possível durante todos os procedimentos, enquanto a máquina PCR está em execução para evitar a evaporação da amostra. - Coloque a fita de tubo de PCR "D" na máquina de PCR, feche a tampa da máquina do PCR e iniciar a reação de PCR. Quando o programa PCR atinge 65 ° C, abra a tampa da máquina do PCR e coloque a fita de tubo de PCR "H" na máquina de PCR. Feche a tampa da máquina do PCR e incubar durante 3 min. abrir a tampa de máquina PCR, lugar o tubo de "R" PCR strip na máquina de PCR e feche a máquina PCR.
- Depois de 2 min, abra a tampa da máquina do PCR e todas as tiras de tubo PCR. Transferi 13 µ l bem "h" em bem "R", então todo volume do bem "D" para "R" bem. Pipeta e descer 3 vezes para misturar a reação, feche a tira do tubo do PCR, remover o "H" e "D" PCR tubo tiras e fechem a tampa do máquina PCR. Incube a reação a 65 ° C por 24 h.
17. isolamento de promotor fragmento contendo produtos de ligadura
Nota: As etapas a seguir são recomendadas para ser feito com o kit de adaptador de SureSelect e biblioteca (ver Tabela de Materals).
- Pré-aquecer 1,5 mL de tampão de lavagem 2 por amostra a 65 ° C com antecedência.
- Adicionar 60 µ l de grânulos magnéticos streptavidin-acoplado (ver Tabela de materiais) para um novo tubo, coloque sobre o carrinho de separação magnética para 1 min e remover o sobrenadante.
- Lave grânulos três vezes com 200 µ l de tampão de ligação 1x.
Nota: Para cada etapa de lavagem durante o isolamento de pós-captura de promotor-contendo produtos de ligadura, re-suspender contas no buffer correspondente, rodar por 3 min em RT e 15 rpm em uma roda giratória, girar suavemente o tubo em uma centrífuga de bancada para 2-3 s coletar amostra, lugar o tubo sobre a separação magnética carrinho por 3 min e remover sobrenadante. - Re-suspenda contas em 200 µ l de tampão de ligação 1x. Abra a máquina PCR e a tira de tubo PCR (enquanto ainda está sendo executado o programa PCR) e transferir a reação de hibridização para o tubo com os grânulos magnéticos. Incube a RT por 30 min em uma roda rotativa a 3 rpm.
- Recuperar os grânulos no stand separação magnética e remover o sobrenadante claro. Re-suspender contas em 500 µ l de tampão de lavagem 1, mistura e incube por 15 min a 20 ° C e agitando a 950 rpm em um thermomixer.
- Recuperar os grânulos no stand separação magnética por 3 min e remover o sobrenadante claro. Re-suspender contas em 500 µ l de tampão de lavagem 2, misturar e incubar a 10 min a 65 ° C e agitando a 950 rpm em um thermomixer. Repita a etapa 17,5 mais duas vezes.
- Recuperar os grânulos no stand separação magnética, remover o sobrenadante claro e ressuspender grânulos em 200 µ l de tampão de restrição 2 1x. Recuperar os grânulos no stand separação magnética, remover o sobrenadante e ressuspender grânulos em 30 µ l de tampão de restrição 2 1x.
18. PCHi-C biblioteca amplificação
- Preparar a mistura de mestre PCR: 60 µ l de 5 x PCR buffer (buffer de Phusion), 3,6 µ l de 25 µM PE PCR primer 1.0, 3,6 µ l de 25 µM PE PCR primer 2.0, 8.4 µ l de dNTP mix (10 mM cada), 3,6 µ l de polymerase Phusion e 190.8 µ l de H2O.
- Mistura de mestre de PCR Mix com os grânulos (300 µ l no total), dividir em 6 alíquotas de 50 µ l e PCR-amplificar usando as seguintes condições:
30 s a 98 ° C
4 ciclos de: 10 s a 98 ° C, 30 s a 65 ° C, 30 s a 72 ° C
7 min a 72 ° C - Recolher todas as reações de PCR em um novo tubo, recupera as contas sobre o ímã e transferir o sobrenadante (300 µ l; contém biblioteca PCHi-C) em um tubo novo.
- Purifica a biblioteca PCHi-C usando grânulos SPRI, seguindo os passos descritos abaixo dos 15,4.
- Quantificar a concentração da biblioteca PCHi-C.
Resultados
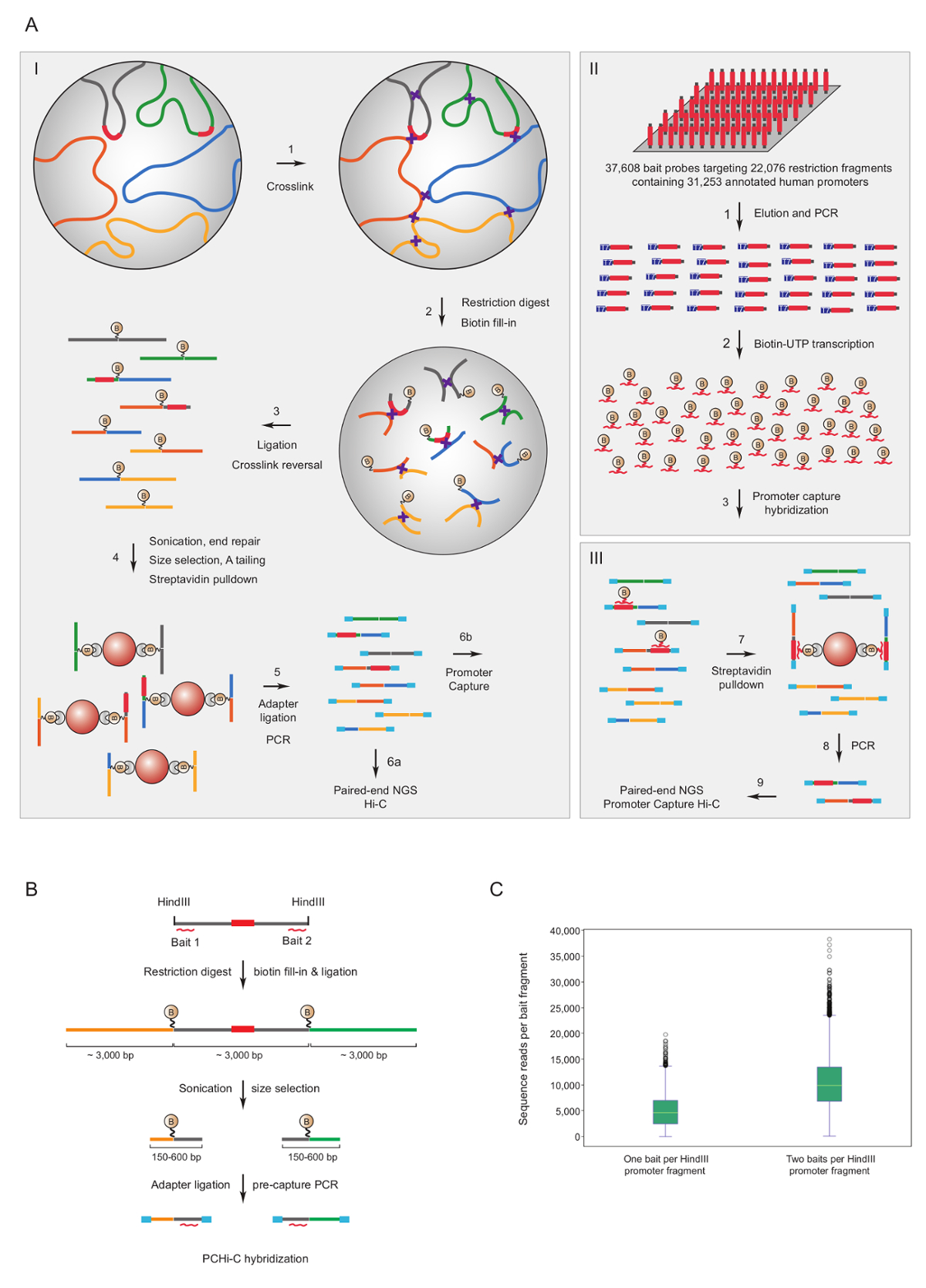
Promotor de capturar Hi-C tem sido usada para enriquecer o rato7,34,36,39 e humano33,35,37,38 Hi-C bibliotecas para interações do promotor. Um protocolo semelhante (chamado HiCap) tem sido descrito por Sandberg grupo40. A figura 1A mostra o fluxo de trabalho esquemático para promotor capturar Hi-C. No protocolo descrito aqui, Hi-C bibliotecas são geradas usando a ligadura no núcleo41, que resulta em um número significativamente reduzido de ligadura espúrias produtos42. Para PCHi-C, mouse altamente complexo ou bibliotecas de Hi-C humanas estão sujeitos a na solução de hibridização e capturar usando 39.021 biotinilado RNAs complementares para 22.225 rato contendo promotor HindIII fragmentos da limitação ou 37.608 biotinilado RNAs segmentação 22.076 humano contendo promotor HindIII fragmentos da limitação, respectivamente. Promotor, contendo fragmentos de restrição pode ser direcionado em uma ou ambas as extremidades por individual biotinilado RNAs (figura 1B). Nós achamos que a captura de ambos termina melhorada a cobertura de cada um dos promotores (Figura 1; sequência primas leituras) quase duas vezes, como esperado. Assim, sempre que possível (ou seja, nas regiões não repetitivas), aconselhamos usar biotinilado RNAs complementares para ambas as extremidades de um fragmento de restrição para ser capturado.
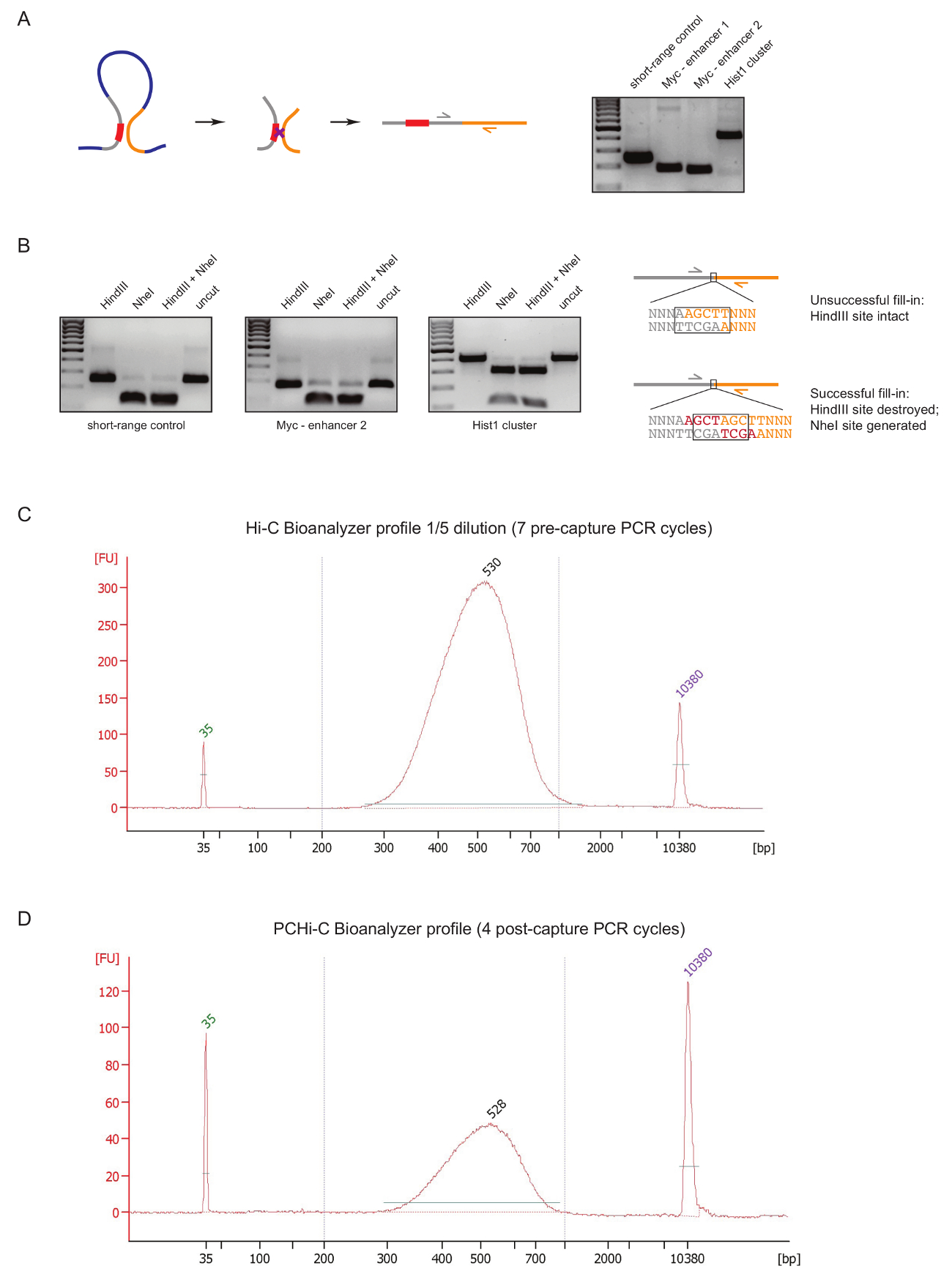
Para avaliar a qualidade de biblioteca PCHi-C na fase inicial durante a preparação da biblioteca, realizamos dois controles após ligadura do ADN e purificação, como descrito anteriormente,31. A primeira é usar pares específicos da primeira demão para amplificar produtos ligadura como 3C27. Nós usamos pares da primeira demão (tabela 1) para amplificar produtos de invariável da ligadura de longo alcance do tipo de célula, tal como entre o gene Myc e seus conhecidos potenciadores localizado aproximadamente 2MB distância (Figura 2A) ou entre genes do Hist1 locus ( separados por 1,5 Mb) e entre duas regiões situadas nas proximidades linear (controle de curto alcance).
O segundo controle de qualidade é realizado para determinar a eficiência da incorporação de biotina durante o preenchimento de Klenow-mediada da restrição local saliências com biotina-dATP. Preenchimento de Klenow sucesso e resultados subsequentes sem corte-extremidade de ligadura no desaparecimento do site original restrição entre as moléculas de DNA de um produto de ligadura e no caso de HindIII na formação de um novo site de reconhecimento NheI (Figura 2B ). A relação entre o HindIII NheI produto de ligadura digerido é uma leitura direta da eficiência de incorporação de biotina. Uma biblioteca de Hi-C de má qualidade vai mostrar um elevado nível de digestão HindIII, Considerando que qualidade de bibliotecas têm quase completa NheI digestão dos produtos da ligadura (Figura 2B).
Após a preparação de biblioteca de Hi-C (ou seja, depois de estreptavidina-biotina puxar para baixo de produtos de ligadura Hi-C tamanho selecionados, ligadura de adaptador e pre-captura PCR), a distribuição de tamanho e integridade da biblioteca Hi-C é avaliada pelo Bioanalyzer (Figura 2 C). O mesmo controle é realizado no final da preparação de biblioteca PCHi-C (ou seja, após a captura de hibridação do promotor-contendo produtos de ligadura e pós-captura PCR). Comparação dos perfis Hi-C e PCHi-C Bioanalyzer mostra que, como esperado, bibliotecas Hi-C são muito mais concentradas do que as bibliotecas PCHi-C correspondentes, mas a distribuição de tamanho das bibliotecas é altamente similar, indicando que a captura de intervir PCHi-C não introduz um viés de tamanho (Figura 2, D).
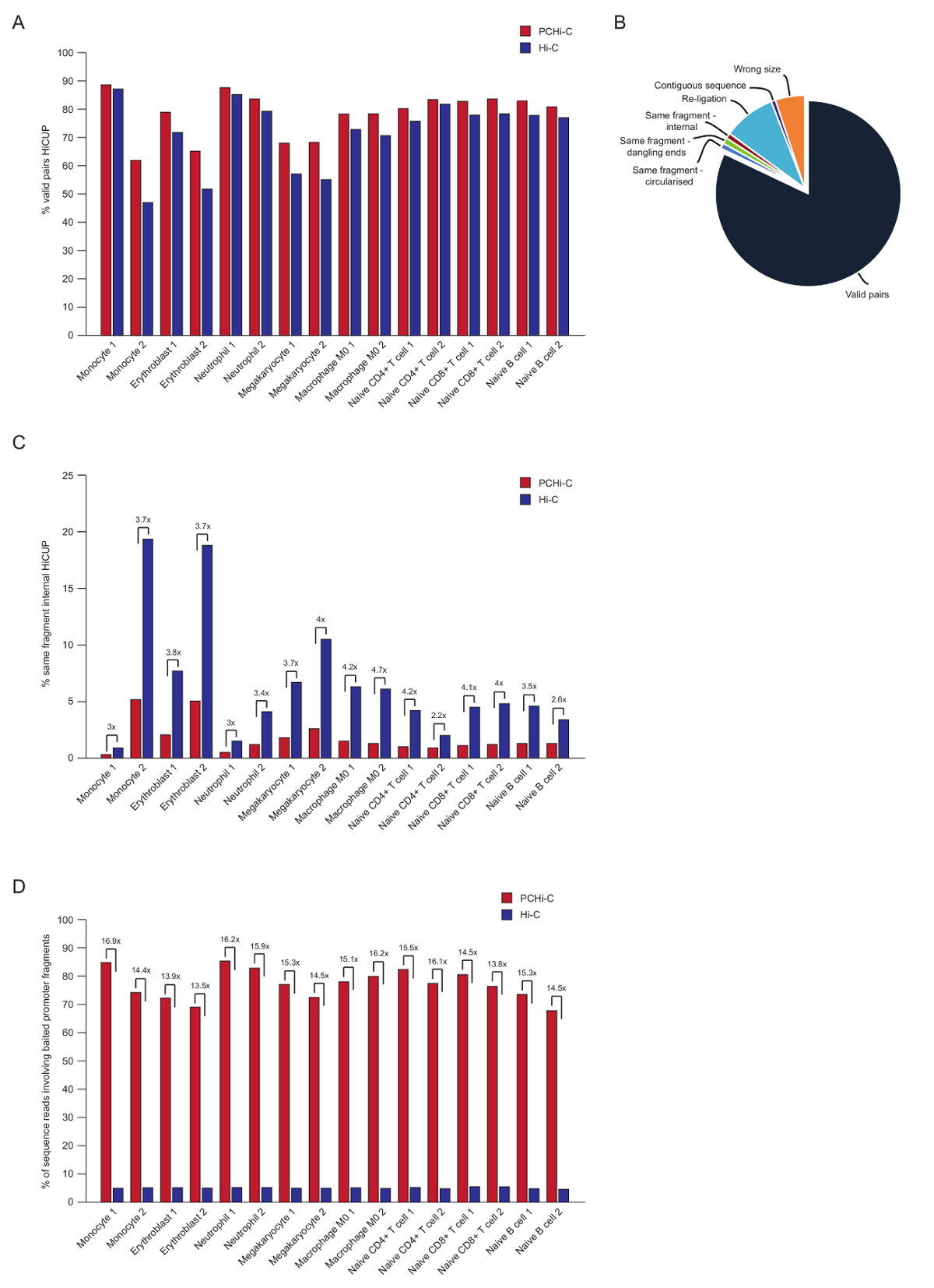
Após o sequenciamento de ponta emparelhado, o lê PCHi-C é mapeado, qualidade controlada e filtrada usando o pipeline de HiCUP43. Alta qualidade PCHi-C bibliotecas contêm entre 70-90% 'válido pares' (ou seja, emparelhado-sequência de leituras entre dois fragmentos de restrição que não são vizinhas no mapa genoma linear; Figura 3A, B). Usando a ligadura no núcleo protocolo41,42, a percentagem de trans ler pares (i.e., emparelhado-sequência de leituras entre dois fragmentos de restrição que estão localizados em diferentes cromossomos) são geralmente baixos, entre 5 e 25%, refletindo a existência de territórios cromossómicos e indicando a qualidade de boa biblioteca. Comparação direta da percentagem de 'pares válidos' entre bibliotecas Hi-C e seus correspondentes PCHi-C bibliotecas35, mostra que em todos os casos, a porcentagem de pares válidos é maior nas bibliotecas PCHi-C (Figura 3B). Isto é acompanhado por uma redução da percentagem de leituras não válido 'mesmo fragmento interno' em PCHi-C (Figura 3). Isso é esperado, como a etapa de captura não só enriquece para promotor-contendo produtos de ligadura, mas também para fins de fragmento de restrição, devido a posição da captura de oligos relativa à restrição de fragmentos (ver figura 1B).
Após a filtragem de HiCUP, podemos determinar a eficiência de captura. PCHi-C bibliotecas contêm três tipos de leituras de sequência válida após filtragem de HiCUP:
1.) promotor: genoma lê (ou seja, leituras entre um fragmento de promotor capturados e um fragmento de restrição HindIII não-promotor em qualquer lugar no genoma)
2.) promotor: promotor lê (leituras entre dois fragmentos capturados promotor)
3.) genoma: genoma lê (produtos de ligadura de fundo Hi-C onde nenhum dos parceiros da ligadura produto mapeia para um promotor capturado). Estas são descartadas antes da análise a jusante.
Alta qualidade PCHi-C bibliotecas têm eficiências de captura (soma das categorias 1 e 2 acima) entre 65-90% (Figura 3D). Uma comparação direta com bibliotecas Hi-C mostra que PCHi-C resulta em uma ~ 15-fold enriquecimento para promotor-produtos que contenham ligadura (Figura 3D), em alguns casos 17-fold. Isso é perto o hipotético máximo (19.6-fold) enriquecimento para PCHi-C, que depende da percentagem de fragmentos de restrição do genoma abrangidas pelo sistema de captura. Maior enriquecimento pode ser alcançado através da concepção de sistemas de captura direcionamento menos restrição fragmentos44,,45,46.
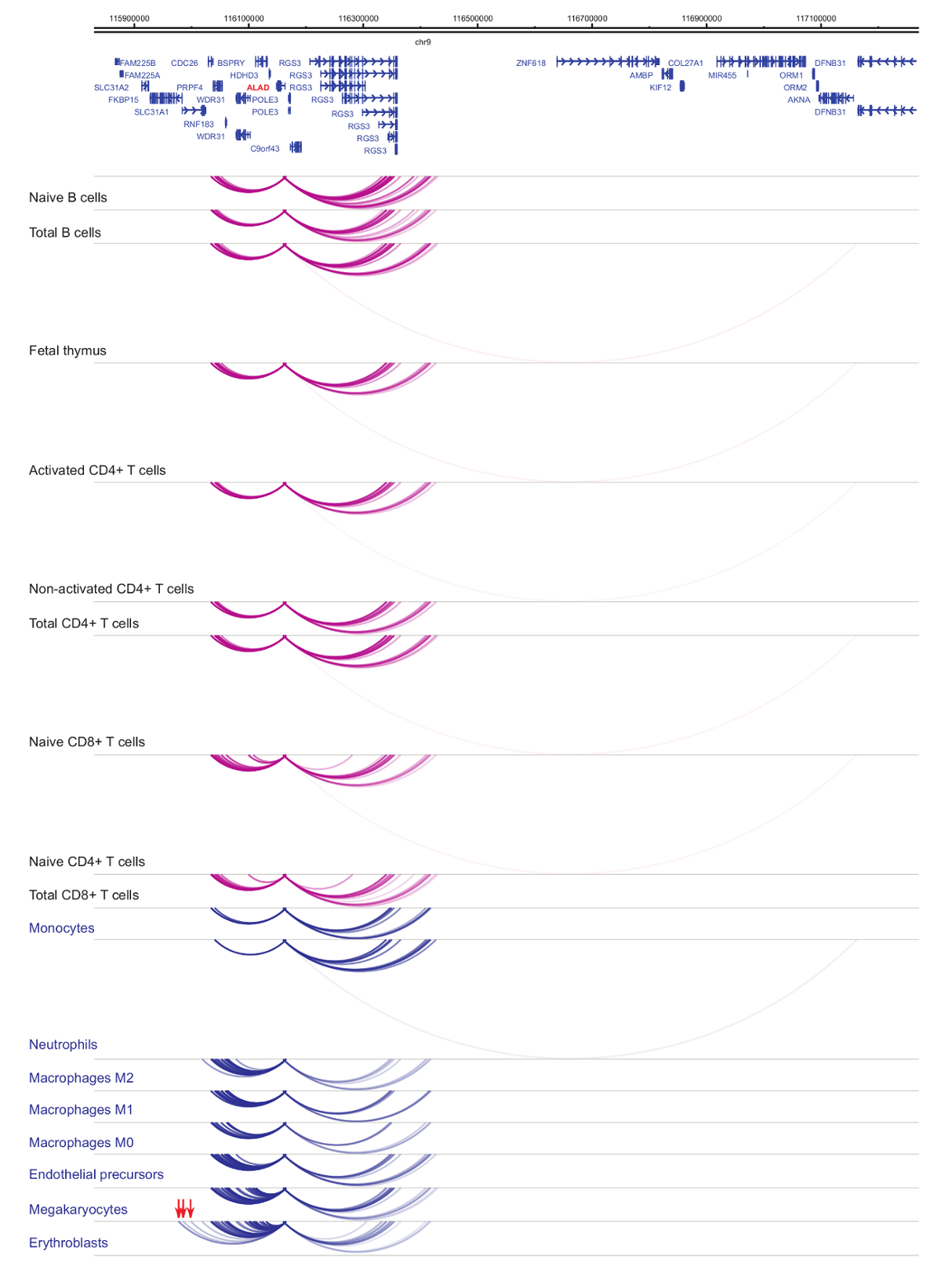
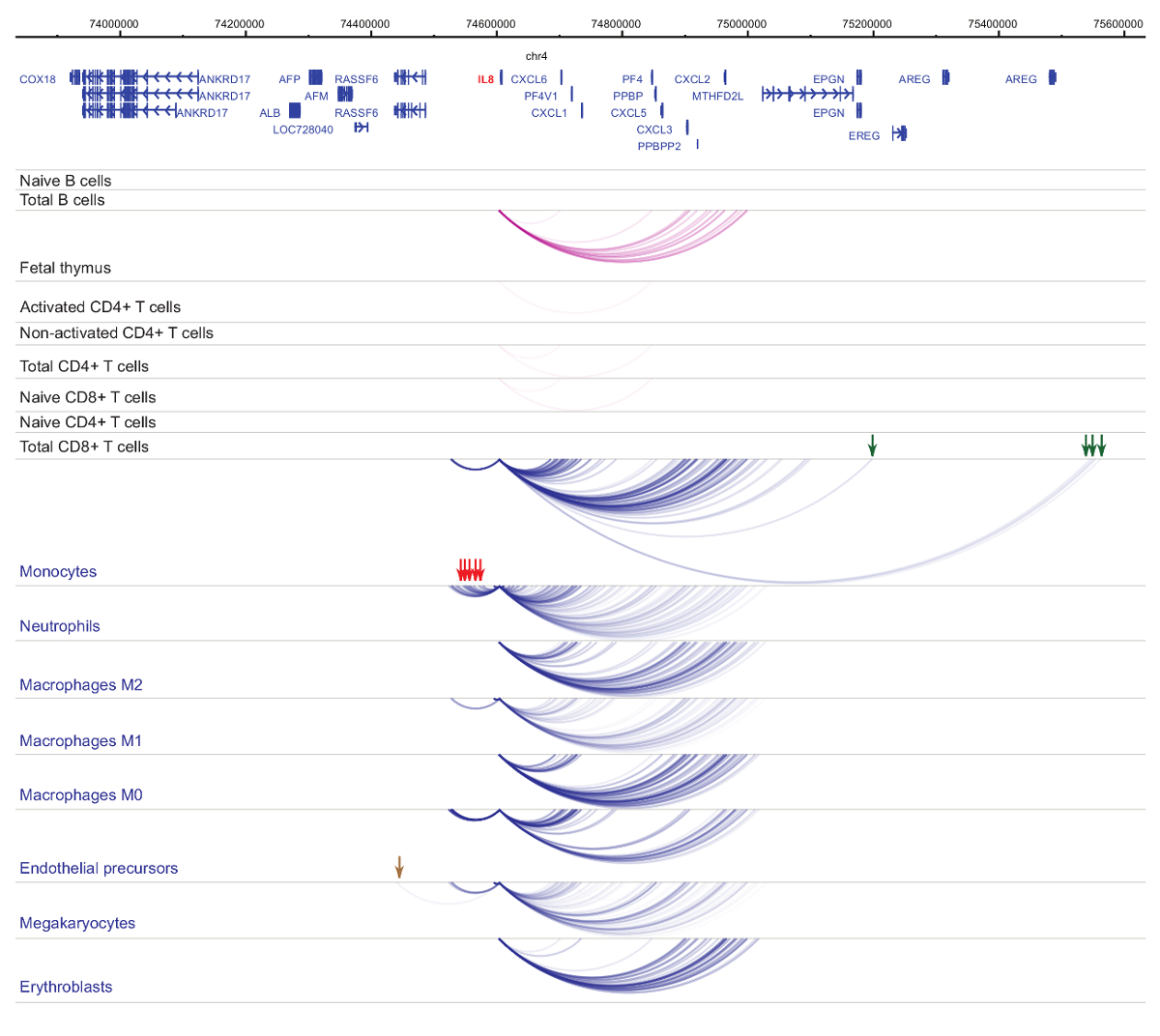
Análise do promotor interactomes demonstra a célula tipo e especificidade linhagem33,34,35, com alterações pronunciadas durante a diferenciação celular37,38,39 . Figuras 4 e 5 mostram exemplos de dinâmica de especificidade e diferenciação de linhagem em promotores específicos. Por exemplo, ALAD é constitutivamente expressa em todas as células, mas a sua expressão é upregulated em eritroblastos47. O promotor ALAD entra em contato com vários fragmentos distais em todas as células hematopoiéticas e se engaja em interações adicionais especificamente em eritroblastos (Figura 4). IL-8 mostra interações não estatisticamente significantes em células B, poucas interações em células T, mas dezenas de interações em células da linhagem mieloide, incluindo interações específicas do tipo de célula em (monócitos, neutrófilos e megacariócitos A Figura 5). Estes exemplos demonstram como PCHi-C pode ser usado para desvendar o tipo de célula específica interactomes e identificar regiões promotor-interagindo com potencial regulamentar.

Figura 1 : Promotor capturar Hi-C lógica e captura de isca design. (A) fluxo de trabalho esquemático do PCHi-C. Ligadura em núcleo Hi-C41,42 (I) é seguida na solução de hibridização com iscas biotinilado RNA (II) visando os fragmentos de restrição de humanos (retratado aqui) ou (III) de promotores de genes de rato. (B) projeto de isca para PCHi-C. Iscas de captura de RNA biotinilado (vermelhas linhas curvas) destinam-se contra as extremidades do promotor, contendo fragmentos de restrição (cinza; note que as sequências de promotor se (vermelho) só são alvo de iscas de captura o RNA se eles estão localizados em restrição fragmento de extremidades). Produtos de ligadura consistindo de promotor contendo fragmentos da limitação (cinzento) e suas interação fragmentos de restrição (amarelo e verde) são isoladas através de hibridação de sequência-complementaridade entre isca de RNA e DNA alvo e subsequentes estreptavidina-biotina pulldown, conforme mostrado na eficiência de captura a. (C) comparação de PCHi-C para promotor, contendo fragmentos de restrição alvo de um RNA isca captura sonda vs duas sondas de captura de isca de RNA (ver esquema em B). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2 : Controles de qualidade pré-sequenciamento PCHi-C. (A) deixou, esquemático da justaposição espacial entre o promotor e o PIR, resultando em um produto de ligadura de Hi-C consiste de um fragmento de restrição contendo promotor (cinza; sequência de promotor em vermelho) e um fragmento de restrição de PIR (amarelo). Certo, DNA gel de electroforese, mostrando exemplos de produtos de Hi-C ligadura amplificados usando pares da primeira demão específicas (como descrito no esquema da esquerda). (B) à esquerda, exemplos representativos de restrição HindIII, NheI e HindIII/NheI digere de produtos de ligadura Hi-C (produtos PCR apresentados na). Bem, esquema de DNA sequência após ligadura da Hi-C após vencida (topo) ou sucesso (parte inferior) do dNTP Klenow de preenchimento de junções de restrição e posterior ligadura. (C) representante Hi-C biblioteca bioanalyzer perfil (diluição de 1/5). (D) representante PCHi-biblioteca bioanalyzer perfil C (sem diluição). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3 : PCHi-C pós-sequenciamento controles de qualidade. (A) comparação de percentual válido sequência ler pares após HiCUP43 processamento no PCHi-C vs correspondente bibliotecas Hi-C (dados de Javierre et al, 201635). (B) o representante HiCUP PCHi-C resultado mostrando válido ler pares e outras categorias de sequência que são descartadas antes da análise a jusante (dados de Javierre et al, 201635). (C) comparação de percentual 'mesmo fragmento interno' lê depois HiCUP processamento no PCHi-C vs correspondente bibliotecas Hi-C (dados de Javierre et al, 201635). (D) a comparação de sequência de porcentagem lê envolvendo fragmentos de anzóis promotor (eficiência de captura) no PCHi-C vs correspondente bibliotecas Hi-C (dados de Javierre et al, 201635). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: ALAD perfil PCHi-C em células hematopoiéticas humanas. Interações promotor de tipos de células mieloides são mostradas como arcos azuis e interações promotor de tipos de células linfoides são mostradas como arcos roxos. Interações específicas eritroblasto são indicadas por setas vermelhas (dados de Javierre et al, 201635). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5 : IL8 Perfil PCHi-C em células hematopoiéticas humanas. Interações promotor de tipos de células mieloides são mostradas como arcos azuis e interações promotor de tipos de células linfoides são mostradas como arcos roxos. Interações específicas de monócitos são indicadas pelas setas verdes, interações específicas de neutrófilos são indicadas por setas vermelhas e uma interação específica megacariócito é indicada por uma seta marrom (dados de Javierre et al, 201635). Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Humana | ||||||||
| Nome | Sequência de | Cromossomo | Strand | Iniciar GRCh38/hg38 | Final GRCh38/hg38 | Combinações de cartilha para testar interações 3C e incorporação de biotina | ||
| HS AHF64 Dekker | GCATGCATTAGCCTCTGCTGTTCTCTGAAATC | 11 | + | 116803960 | 116803991 | usar em combinação com hs AHF66 Dekker | ||
| HS AHF66 Dekker | CTGTCCAAGTACATTCCTGTTCACAAACCC | 11 | + | 116810219 | 116810248 | usar em combinação com hs AHF64 Dekker | ||
| locus MYC HS | GGAGAACCGGTAATGGCAAA | 8 | - | 127733814 | 127733833 | uso em combinação com hs MYC +1820 ou hs MYC-538 | ||
| HS MYC +1820 | AAAATGCCCATTTCCTTCTCC | 8 | + | 129554527 | 129554547 | usar em combinação com o locus MYC hs | ||
| HS MYC-538 | TGCCTGATGGATAGTGCTTTC | 8 | - | 127195696 | 127195716 | usar em combinação com o locus MYC hs | ||
| HS HIST1 F | AAGCAGGAAAAGGCATAGCA | 6 | + | 26207174 | 26207193 | usar em combinação com hs HIST1 R | ||
| HS HIST1 R | TCTTGGGTTGTGGGACTTTC | 6 | + | 27771575 | 27771594 | usar em combinação com hs F HIST1 | ||
| Mouse | ||||||||
| Sequência de | Cromossomo | Strand | Iniciar GRCm38/mm10 | Final GRCm38/mm10 | Combinações de cartilha para testar interações 3C e incorporação de biotina | |||
| TCATGAGTTCCCCACATCTTTG | 8 | + | 84841090 | 84841111 | usar em combinação com mm Calr2 | |||
| CTGTGGGCACCAGATGTGTAAAT | 8 | + | 84848519 | 84848541 | usar em combinação com mm Calr1 | |||
| TATCAAGGGTGCCCGTCACCTTCAGC | 6 | + | 125163098 | 125163123 | usar em combinação com Gapdh4 Dekker | |||
| GGGCTTTTATAGCACGGTTATAAAGT | 6 | + | 125163774 | 125163799 | usar em combinação com Gapdh3 Dekker | |||
| GGAGGAGGGAAAAGGAGTGATT | 6 | + | 52212829 | 52212850 | usar em combinação com mm Hoxa13 | |||
| CAGGCATTATTTGCTGAGAACG | 6 | - | 52253490 | 52253511 | usar em combinação com mm Hoxa7 | |||
| GGGTAATGGTGTCACTAACTGG | 13 | + | 23571284 | 23571305 | usar em combinação com Hist1h3e ou mm Hist1h4i | |||
| GGGTTTGATGAGTTGGTGAAG | 13 | + | 23566541 | 23566561 | usar em combinação com mm Hist1h2ae | |||
| TTGGGCCAAAGCCTATATGA | 13 | + | 22043085 | 22043104 | usar em combinação com mm Hist1h2ae | |||
Tabela 1: Sequências de Primer para controle de qualidade de humanos e bibliotecas de rato Hi-C.
Discussão
Design modular do promotor capturar Hi-C
Promotor capturar Hi-C destina-se especificamente a enriquecer bibliotecas Hi-C para interações envolvendo promotores. Essas interações compõem apenas um subconjunto de produtos da ligadura presentes em uma biblioteca de Hi-C.
Captura Hi-C pode ser facilmente modificado para enriquecer as bibliotecas de Hi-C para qualquer região genômica ou regiões de interesse, alterando o sistema de captura. Regiões de captura podem ser contínua segmentos genômicos44,45,46,48, potenciadores que foram identificadas no PCHi-C (' reverter capturar Hi-C'35), ou DNase eu hipersensíveis sites49 . O tamanho do sistema de captura pode ser ajustado dependendo do escopo experimental. Por exemplo, Dryden et al. 519 fragmentos de isca em três desertos de gene associados com câncer de mama44-alvo. O sistema de captura por Martin et al. destinos de ambos os segmentos genômicos contínuos ('Região Capture': 211 regiões genômicas no total; 2.131 fragmentos de restrição) e selecionado os promotores (3.857 promotores de gene)45.
SureSelect bibliotecas estão disponíveis em escalas diferentes tamanho: 1 kb para 499 kb (5.190 – 4.806), de 500 kb para 2,9 Mb (5.190-4.816) e 3 Mb de 5.9 Mb (5.190 – 4.831). Como cada captura individual biotina-RNA é 120 nucleotides longos, estes capturar sistemas acomodar um máximo de 4.158, 24.166 e 49.166 individuais de captura de sondas, respectivamente. Isso corresponde a 2.079, 12.083 e 24.583 fragmentos de restrição do alvo, respectivamente (Observe que os números de fragmentos de restrição são limites inferiores com base no pressuposto de que as duas sondas de captura individuais podem ser projetadas para cada restrição fragmento — na realidade devido à sequências repetitivas, isto não será o caso para cada restrição do fragmento (ver também figura 1B, C), resultando em um maior número de fragmentos de restrição targetable por um número constante de captura disponíveis sondas ).
O protocolo descrito aqui é baseado na utilização de uma enzima de restrição com um site de reconhecimento bp 6 para descobrir a interações de longo alcance. Usar uma enzima de restrição com um site de reconhecimento bp 4 para maior resolução de interações mais proximais é também possível40,49.
Limitações do PCHi-C
Uma limitação inerente de todos os ensaios de captura de conformação cromossomo é que sua resolução é determinada pela enzima de restrição, utilizada para a geração de biblioteca. Interações que ocorrem entre elementos de DNA localizados no fragmento de restrição mesmo são invisíveis para os ensaios de 'tipo C'. Além disso, no PCHi-C, em alguns casos mais de um site de início de transcrição pode ser localizado no mesmo promotor contendo fragmento de restrição, e PIRs em alguns casos abrigam as duas marcas de histona ativo e repressivo, tornando-se difícil identificar que regulamentar elementos de mediam as interações e prever a saída regulamentar das interações do promotor. Usando enzimas de restrição com 4 locais de reconhecimento do bp atenua esta questão, mas vem à custa de muito maior complexidade de biblioteca Hi-C (Hi-C bibliotecas geradas com 4 bp reconhecimento local enzimas de restrição são pelo menos 100 vezes mais complexas do que o Hi-C bibliotecas geradas com 6 bp reconhecimento local enzimas de restrição) e os custos associados para sequenciamento de próxima geração.
Outra limitação é que o atual protocolo PCHi-C requer milhões de células como material, impedindo a análise de interações de promotor em tipos de células raras de partida. Uma versão modificada do PCHi-C para habilitar o interrogatório de contatos de promotor em populações de células com 10.000 a 100.000 células (por exemplo durante o desenvolvimento embrionário precoce ou células-tronco hematopoiéticas), portanto, seria uma adição valiosa à captura Oi-C caixa de ferramentas.
Finalmente, como todos os métodos que se baseiam na fixação de formaldeído, PCHi-C registra apenas as interações que são 'congeladas' no ponto de fixação de tempo. Assim, para estudar a cinética e dinâmica de interações de promotor, são necessários métodos tais como a microscopia de célula viva de super-resolução ao lado PCHi-C.
Métodos para dissecar a organização espacial de cromossomo em alta resolução
A vasta complexidade de bibliotecas cromossômicas interação proíbe a identificação fiável de produtos da interação entre dois fragmentos de restrição específica, com significância estatística. Para contornar este problema, captura de sequência tem sido usada para enriquecer ou Hi-C33,34,40,44 ou 3 C50,51 bibliotecas para interações específicas. A grande vantagem do uso de bibliotecas de bibliotecas mais 3C Hi-C para a etapa de enriquecimento é que Hi-C, ao contrário de 3C, inclui uma etapa de enriquecimento para produtos genuínos da ligadura. Como consequência, a percentagem de leituras válidas em bibliotecas PCHi-C é de aproximadamente 10-fold superior em captura-C bibliotecas50, que continha cerca de 5 – 8% válido lê depois da filtragem de HiCUP. Sahlen et al compararam diretamente captura-C para HiCap, que como PCHi-C utiliza bibliotecas Hi-C para enriquecimento de captura, em contraste com captura-C, que usa bibliotecas C 3. Consistente com nossos achados, encontraram que captura-C bibliotecas são compostas principalmente de fragmentos un-ligados40. Além disso, as bibliotecas HiCap tinham uma complexidade maior do que a captura-C bibliotecas40.
Uma variante de captura-C, chamado geração captura-C52 NG captura-C usa um oligo por fim do fragmento de restrição, como estabelecido anteriormente em33,PCHi-C34, em vez de sobreposição de sondas utilizadas no original Protocolo de captura-C50. Isso aumenta a porcentagem de leituras válidas em relação ao captura-C modestamente, mas NG captura-C emprega duas rodadas sequenciais de enriquecimento de captura, e os ciclos de um número relativamente elevado de PCR (ciclos de 20 a 24 no total, em comparação com 11 ciclos normalmente para PCHi-C), que inevitavelmente resulta em números mais altos de duplicatas de sequência e baixa complexidade de biblioteca. Em julgamento experiências durante a otimização do PCHi-C, verificou-se que a percentagem de exclusivo (ou seja, não duplicado) ler pares rondou apenas 15% quando usamos 19 ciclos PCR (13 ciclos pre-capture + 6 ciclos pós-captura; dados não mostrados), no entanto otimização para um menor número de ciclos PCR, normalmente produz 75-90% pares leitura únicas. Assim, reduzir substancialmente o número de ciclos de PCR aumenta a quantidade de dados da sequência informativa.
Um recente método combina ChIP com Hi-C para focar cromossômicas interações mediadas por uma proteína específica de interesse (HiChIP53). Em comparação com ChIA-PET54, que é baseado em uma lógica semelhante, HiChIP dados contém um número maior de leituras de sequência informativa, permitindo a interação de maior confiança, chamando53. Será muito interessante comparar diretamente o correspondente HiChIP e conjuntos de dados Hi-C capturar uma vez eles se tornam disponíveis (por exemplo, usando um anticorpo contra a Smc1a de unidade de cohesin de HiChIP53 com captura Hi-C para todos os Smc1a vinculado a restrição fragmentos) lado a lado. Uma diferença inerente entre essas duas abordagens é que capturar Hi-C não depende da imunoprecipitação da cromatina e, portanto, é capaz de interrogar cromossômicas interações independentemente da ocupação de proteína. Isso permite a comparação de organização do genoma 3D na presença ou ausência de ligação do fator específico, como tem sido usado para identificar o PRC1 como um regulador chave do mouse ESC genoma espacial arquitetura7.
PCHi-C e GWAS
Estudos de associação de genoma-larga (GWAS) revelaram que maior que 95% das doenças associadas variantes de sequência estão localizadas em regiões não-codificantes do genoma, muitas vezes a grandes distâncias de genes codificantes de proteínas55. Variantes GWAS são frequentemente encontrados em estreita proximidade com DNase I sites hipersensíveis, que é uma marca registrada de sequências com potencial actividade regulamentar. PCHi-C e capturar Hi-C têm sido amplamente utilizados para vincular os promotores a loci de risco GWAS implicados no câncer de mama44, câncer colorretal48e doença auto-imune35,,45,46. Um PCHi-C estudar em 17 diferentes humana células hematopoiéticas tipos encontrados SNPs associadas à doença auto-imune foram enriquecidos em PIRs em células linfoides, Considerando que variantes de sequência associadas com traços específicos de plaquetas e glóbulos vermelhos foram encontradas predominantemente em os macrófagos e eritroblastos, respectivamente de35,56. Assim, promotor específico tipo de tecido, interactomes descoberto pelo PCHi-C pode ajudar a entender a função do não-codificantes associada a doença variantes de sequências e identificar novos genes de doença potencial para intervenção terapêutica.
Características das regiões do promotor-interagindo
Várias linhas de evidência link promotor interactomes para controle de expressão do gene. Primeiro, vários estudos PCHi-C demonstraram que regiões genômicas, interagindo com os promotores de genes expressos (altamente) são enriquecidas em marcas associadas com atividade potenciador, como H3K27 acetilação e p300 vinculação33,34 , 37. encontramos uma correlação positiva entre o nível de expressão do gene e o número de realçadores interagindo, sugerindo que os efeitos aditivos de resultado de realçadores na expressão aumentada do gene níveis34,35. Em segundo lugar, de ocorrência natural expressão loci característica quantitativa (eQTLs) é enriquecidos em PIRs que estão conectados para os mesmos genes cuja expressão é afetado pelo eQTLs35. Em terceiro lugar, integrando viagem57 e dados PCHi-C, Cairns et al encontraram que viagem repórter genes mapeamento para PIRs em rato CES mostram repórter mais forte expressão do gene do que genes repórter em locais de integração em regiões não-promotor-interagindo 58, indicando que o PIRs possuem actividade regulamentar transcriptional. Juntos, estes achados sugerem que o promotor interactomes descobertos pelo PCHi-C em vários mouse e tipos de células humanas incluem chaves reguladoras módulos para controle de expressão de gene.
É interessante notar que os realçadores representam apenas uma pequena fração (~ 20%) de todos os PIRs descobertos por PCHi-C33,34. Outros PIRs poderiam ter funções estruturais ou topológicas ao invés de funções reguladoras transcriptional diretas. No entanto, há também evidências que PCHi-C pode descobrir elementos de DNA com função reguladora que não abriga marcas potenciador clássica. Em uma linhagem de células linfoides humana, o promotor de BRD7 foi encontrado para interagir com uma região desprovida de marcas de potenciador que foi demonstrada possuir atividade potenciador de ensaios de gene repórter33. Elementos reguladores com características semelhantes podem ser mais abundantes do que atualmente apreciada. Por exemplo, uma tela baseada em CRISPR para regulamentar DNA elementos identificados não marcadas elementos reguladores (UREs) que controlam a expressão gênica, mas são desprovidas de potenciador marca59.
Em outros casos, PIRs foram mostrados para marcas de cromatina associadas com repressão transcriptional do porto. PIRs e promotores interagindo vinculados pelo PRC1 no rato CES estavam envolvidos em uma extensa rede espacial dos genes reprimidos tendo que o repressivo marca H3K27me37. Em células humanas lymphoblastoid, um elemento distante interagindo com o promotor BCL6 reprimida transgene repórter gene expressão33, sugerindo que ele pode funcionar para repress a transcrição BCL6 em seu contexto nativo.
PIRs enriquecidos para ocupação da proteína do isolador de cromatina CTCF humana CES e CNE37 pode representar ainda outra classe de PIRs. Coletivamente, estes resultados sugerem que o PIRs abrigam uma coleção de atividades reguladoras do gene ainda para ser funcionalmente caracterizada.
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
Agradecemos Valeriya Malysheva pela leitura crítica do manuscrito e ajuda especializada com a Figura 1. Este trabalho foi apoiado pelo Conselho de pesquisa médica, UK (Sr/L007150/1) e UK biotecnologia e Conselho de pesquisa de ciências biológicas, UK (BB/J004480/1).
Materiais
| Name | Company | Catalog Number | Comments |
| 16% (vol/vol) paraformaldehyde solution | Agar Scientific | R1026 | |
| Dulbecco's Modified Eagle Medium (DMEM) 1x | Life Technologies | 41965-039 | |
| Fetal bovine serum (FBS) sterile filtered | Sigma | F9665 | |
| Low-retention filter tips | Starlab | S1180-3810, S1180-1810, S1180-8810 and S1182-1830 | |
| 10x PBS pH 7.4 | Life Technologies | 70011-036 | |
| Molecular biology grade water | Sigma-Aldrich | W4502 | |
| 1 M Tris-HCl pH 8.0 | Life Technologies | 15568-025 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 5 M NaCl | Life Technologies | 24740-011 | |
| Protease inhibitor cocktail (EDTA-free) | Roche Diagnostics | 11873580001 | |
| Restriction buffer 2 (10x NEBuffer 2) | New England Biolabs | B7002 | |
| DNA LoBind tube, 1.5 mL | Eppendorf | 0030 108.051 | |
| DNA LoBind tube, 2 mL | Eppendorf | 30108078 | |
| 20% (wt/vol) SDS | Bio-Rad Laboratories | 161-0418 | |
| 20% (vol/vol) Triton X-100 | Sigma-Aldrich | T8787 | |
| HindIII, 100 U/uL | New England Biolabs | R0104 | |
| 10 mM dCTP | Life Technologies | 18253-013 | |
| 10 mM dGTP | Life Technologies | 18254-011 | |
| 10 mM dTTP | Life Technologies | 18255-018 | |
| 0.4 mM Biotin-14-dATP | Life Technologies | 19524-016 | |
| DNA polymerase I large (Klenow) fragment 5000 units/mL | New England Biolabs | M0210 | |
| 10x T4 DNA ligase reaction buffer | New England Biolabs | B0202 | |
| 100x 10mg/ml Bovine Serum Albumin | New England Biolabs | B9001 | |
| T4 DNA ligase, 1 U/μL | Invitrogen | 15224-025 | |
| RNase A | Roche | 10109142001 | |
| Proteinase K, recombinant, PCR grade | Roche | 3115836001 | |
| 20 000×g 50 ml centrifuge tube | VWR | 525-0156 | |
| 0.5 M EDTA pH 8.0 | Life Technologies | 15575-020 | |
| Phenol pH 8.0 | Sigma | P4557 | |
| Phenol: Chloroform: Isoamyl Alcohol 25:24:1 | Sigma | P3803 | |
| Sodium acetate pH 5.2 | Sigma | S7899 | |
| Quant-iT PicoGreen | Invitrogen | P7589 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| Restriction buffer 2.1 (10x NEBuffer 2.1) | New England Biolabs | B7202 | |
| NheI, 100U/uL | New England Biolabs | R0131 | |
| Micro TUBE AFA Fiber Pre-slit snap cap 6x16mm vials | Covaris | 520045 | For sonication |
| SPRI beads (Agencourt AMPure XP) | Beckman Coulter | A63881 | |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| Tween 20 | Sigma | P9416 | |
| 10 mM dATP | Life Technologies | 18252-015 | |
| T4 DNA polymerase 3000 units/mL | New England Biolabs | M0203 | |
| T4 PNK 10000 units/mL | New England Biolabs | M0201 | |
| Klenow exo minus 5000 units/mL | New England Biolabs | M0212 | |
| Quick ligation reaction buffer | New England Biolabs | B6058 | |
| NEB DNA Quick ligase | New England Biolabs | M2200 | |
| PE adapter 1.0 (5'-P-GATCGGAAGAGCGGTTCAGC AGGAATGCCGAG-3') | Illumina | ||
| PE adapter 2.0 (5'-ACACTCTTTCCCTACACGACGCT CTTCCGATCT-3') | Illumina | ||
| NEB Phusion PCR kit | New England Biolabs | M0530 | |
| PE PCR primer 1.0 (5'-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTAC ACGACGCTCTTCCGATCT-3') | Illumina | ||
| PE PCR primer 2.0 (5'-CAAGCAGAAGACGGCATACGA GATCGGTCTCGGCATTCCT GCTGAACCGCTCTTCCGATCT-3') | Illumina | ||
| PCR strips | Agilent Technologies | 410022 and 401425 | |
| SureSelect SSEL TE Reagent ILM PE full adaptor kit | Agilent Technologies | 931108 | |
| SureSelect custom 3-5.9 Mb library | Agilent Technologies | 5190-4831 | custom design mouse or human PCHi-C system |
| Dynabeads MyOne Streptavidin T1 beads | Invitrogen | 65601 | |
| E220 high-performance focused ultra-sonicator | Corvaris | E220 |
Referências
- Osborne, C. S., et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nature Genetics. 36, 1065-1071 (2004).
- Schoenfelder, S., et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature Genetics. 42, 53-61 (2010).
- de Wit, E., et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 501, 227-231 (2013).
- Bantignies, F., et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 144, 214-226 (2011).
- Engreitz, J. M., et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 341, 1237973(2013).
- Denholtz, M., et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 13, 602-616 (2013).
- Schoenfelder, S., et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics. 47, 1179-1186 (2015).
- Kundu, S., et al. Polycomb Repressive Complex 1 generates discrete compacted domains that change during differentiation. Molecular Cell. 65, 432-446 (2017).
- Skok, J. A., Gisler, R., Novatchkova, M., Farmer, D., de Laat, W., Busslinger, M. Reversible contraction by looping of the Tcra and Tcrb loci in rearranging thymocytes. Nature Immunology. 8, 378-387 (2007).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148, 908-921 (2012).
- Aymard, F., et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology. 24, 353-361 (2017).
- Ryba, T., et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research. 20, 761-770 (2010).
- Pope, B. D., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 515, 402-405 (2014).
- Chandra, T., et al. Global reorganization of the nuclear landscape in senescent cells. Cell Reports. 10, 471-483 (2015).
- Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., Fraser, P. Long-range chromatin regulatory interactions in vivo. Nature Genetics. 32, 623-626 (2002).
- Tolhuis, B., Palstra, R. J., Splinter, E., Grosveld, F., de Laat, W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Molecular Cell. 10, 1453-1465 (2002).
- Amano, T., Sagai, T., Tanabe, H., Mizushina, Y., Nakazawa, H., Shiroishi, T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Developmental Cell. 16, 47-57 (2009).
- Zuniga, A., et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes & Development. 18, 1553-1564 (2004).
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132, 797-803 (2005).
- D'Haene, B., et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 5, e1000522(2009).
- Sur, I. K., et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 338, 1360-1363 (2012).
- Herranz, D., et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature Medicine. 20, 1130-1137 (2014).
- Deng, W., et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 149, 1233-1244 (2012).
- Groschel, S., et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157, 369-381 (2014).
- Lupianez, D. G., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 161, 1012-1025 (2015).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538, 265-269 (2016).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295, 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38, 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genetics. 38, 1341-1347 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research. 16, 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326, 289-293 (2009).
- Belton, J. M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., Dekker, J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 58, 268-276 (2012).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47, 598-606 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582-597 (2015).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167, 1369-1384 (2016).
- Wilson, N. K., et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 127, e12-e23 (2016).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife. 6, (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49, 1522-1528 (2017).
- Siersbaek, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66, 420-435 (2017).
- Sahlen, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Nagano, T., et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64 (2013).
- Nagano, T., Varnai, C., Schoenfelder, S., Javierre, B. M., Wingett, S. W., Fraser, P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology. 16, 175(2015).
- Wingett, S., et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Res. 4, 1310(2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24, 1854-1868 (2014).
- Martin, P., et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nature Communications. 6, 10069(2015).
- McGovern, A., et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol.ogy. 17, 212(2016).
- Hodge, D., et al. A global role for EKLF in definitive and primitive erythropoiesis. Blood. 107, 3359-3370 (2006).
- Jager, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter Interactions between two states of pluripotency. Cell Stem Cell. 17, 748-757 (2015).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46, 205-212 (2014).
- Kolovos, P., et al. Targeted Chromatin Capture (T2C): A novel high-resolution high-throughput method to detect genomic interactions and regulatory elements. Epigenetics Chromatin. 7, 10(2014).
- Davies, J. O., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13, 74-80 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13, 919-922 (2016).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462, 58-64 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337, 1190-1195 (2012).
- Petersen, R., et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat. Commun. 8, 16058(2017).
- Akhtar, W., et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 154, 914-927 (2013).
- Cairns, J., et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biology. 17, 127(2016).
- Rajagopal, N., et al. High-throughput mapping of regulatory DNA. Nature Biotechnology. 34, 167-174 (2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados