
Method Article
Análisis de proteómica basada en espectrometría de masa usando la base de datos OpenProt para develar nuevas proteínas traducción de marcos de lectura abierto no-canónico
En este artículo
Resumen
OpenProt es una base de datos de libre acceso que aplica un modelo policistrónico de genomas eucariotas. Aquí, presentamos un protocolo para el uso de bases de datos de OpenProt al interrogar a conjuntos de datos de espectrometría de masas. OpenProt base de datos para el análisis de experimentos de la proteómica permite para el descubrimiento de la novela y proteínas previamente indetectables.
Resumen
Anotación del genoma es fundamental para la investigación de Proteómica de hoy como dibuja los contornos del paisaje proteómicos. Los modelos tradicionales de libre lectura anotación de marco (ORF) imponer dos criterios arbitrarios: una longitud mínima de 100 codones y una sola ORF por transcripción. Sin embargo, un número creciente de estudios Informe expresión de proteínas de supuestamente no codificantes regiones, desafiar la exactitud de las anotaciones de genoma actual. Estas novela se encontraron proteínas codificadas dentro no-codificación RNAs 5' y 3' regiones sin traducir (UTRs) de mRNAs o superposición de una secuencia de codificación conocida (CDS) en una alternativa ORF. OpenProt es la primera base de datos que aplica un modelo policistrónico de genomas eucarióticos, permitiendo la anotación de ORFs múltiples por transcripción. OpenProt es libremente accesible y ofrece descargas personalizadas de secuencias de la proteína a través de 10 especies. Con OpenProt la base de datos para los experimentos de la proteómica permite el descubrimiento de nuevas proteínas y pone de relieve la naturaleza policistrónico de genes eucarióticos. El tamaño de la base de datos OpenProt (todos predicen proteínas) es importante y necesita ser tomado en cuenta para el análisis. Sin embargo, con ajustes de tasa (FDR) apropiadas descubrimiento falsas o el uso de una base de datos de OpenProt restringida, los usuarios beneficiarán de una visión más realista del paisaje de proteómica. En general, OpenProt es una herramienta disponible gratuitamente que fomentará la proteómica descubrimientos.
Introducción
En las últimas décadas, espectrometría de masas (MS-) basado en proteómica se ha convertido en la técnica oro descifrar proteomas de las células eucariotas1,2,3,4,5. Este método se basa en las anotaciones de genoma actual para generar una base de secuencia de proteínas de referencia que describe el alcance de posibilidades6,7,8. Sin embargo, las anotaciones del genoma sostener criterios arbitrarios para la anotación de ORF, como una longitud mínima de 100 codones y una sola ORF por transcripción9,10. Un número creciente de estudios cuestionar el actual modelo de anotación e informe de descubrimientos de ORFs unannotated funcionales de genomas eucariotas8,11,12,13, 14. Estas nuevas proteínas se encuentran codificados en supuestamente no-codificación RNAs, en 5' o 3' no traducidas (UTR) de regiones de mRNAs o superposición de la secuencia de codificación canónica (cCDS) en un marco alternativo. Aunque la mayoría de estos descubrimientos ha sido fortuita, demuestran las reservas de anotaciones de genoma actual y la naturaleza policistrónico de genes eucarióticos8.
Aquí, destacamos el uso de bases de datos OpenProt para proteómica basada en la MS. OpenProt es la primera base de datos para mantener un modelo de anotación policistrónico de transcriptomas eucariotas. Está disponible en www.openprot.org15. Una proporción de estos predijo que ORFS sería aleatoria y funcional, razón por la cual OpenProt acumula evidencia experimental y funcional para aumentar la confianza. La evidencia experimental son expresión de la proteína (de MS) y traducción en evidencia (por ribosome profiling)15. Pruebas funcionales incluyen homología de proteínas (con un en-paranoico como enfoque) y de predicción funcional dominio15.
OpenProt ofrece la posibilidad de descargar varias bases de datos, que contienen sólo proteínas bien soportadas a bases de datos a medida. Aquí, presentamos una tubería para el uso de bases de datos de OpenProt y ofrecerá información sobre la base de datos para elegir teniendo en cuenta el objetivo experimental. La tubería de proteomics análisis presentada aquí es apoyada por el marco de la galaxia libre acceso y fácil de usar, pero las bases de datos pueden funcionar con cualquier flujo de trabajo de17,16,18. También presentaremos cómo utilizar el sitio web OpenProt para reunir más información sobre nuevas proteínas detectadas por las bases de datos MS. Using OpenProt proporcionará una visión más exhaustiva del paisaje proteómicos y fomentará los descubrimientos proteómica y biomarcadores en una manera más sistemática que los métodos actuales.
Este protocolo destaca el uso de bases de datos de OpenProt15 cuando interrogando a conjuntos de datos de MS; no revisará el diseño del experimento sí mismo, que ha sido completamente revisado en otra parte20,21,22. En un esfuerzo por permanecer completamente de código abierto, el protocolo es libremente disponible (Suplementario S1 Material–S4). Para facilitar la lectura, todos los términos utilizados en OpenProt y por este medio a lo largo de este protocolo se definen en la tabla 1.
Protocolo
1. descargar base de datos de OpenProt
Nota: Las bases de datos personalizadas en base a datos de RNA-seq por ejemplo también se pueden obtener y el procedimiento se detalla en la sección segunda del presente Protocolo. Si se necesita una base de datos personalizada, por favor vaya a la sección siguiente.
- Ir al sitio web de OpenProt: www.openprot.org y abrir la página de descargas con el enlace en el menú superior de la página.
- Haga clic en las especies de interés basándose en los datos experimentales analizados.
- Haga clic en el tipo de proteína deseado.
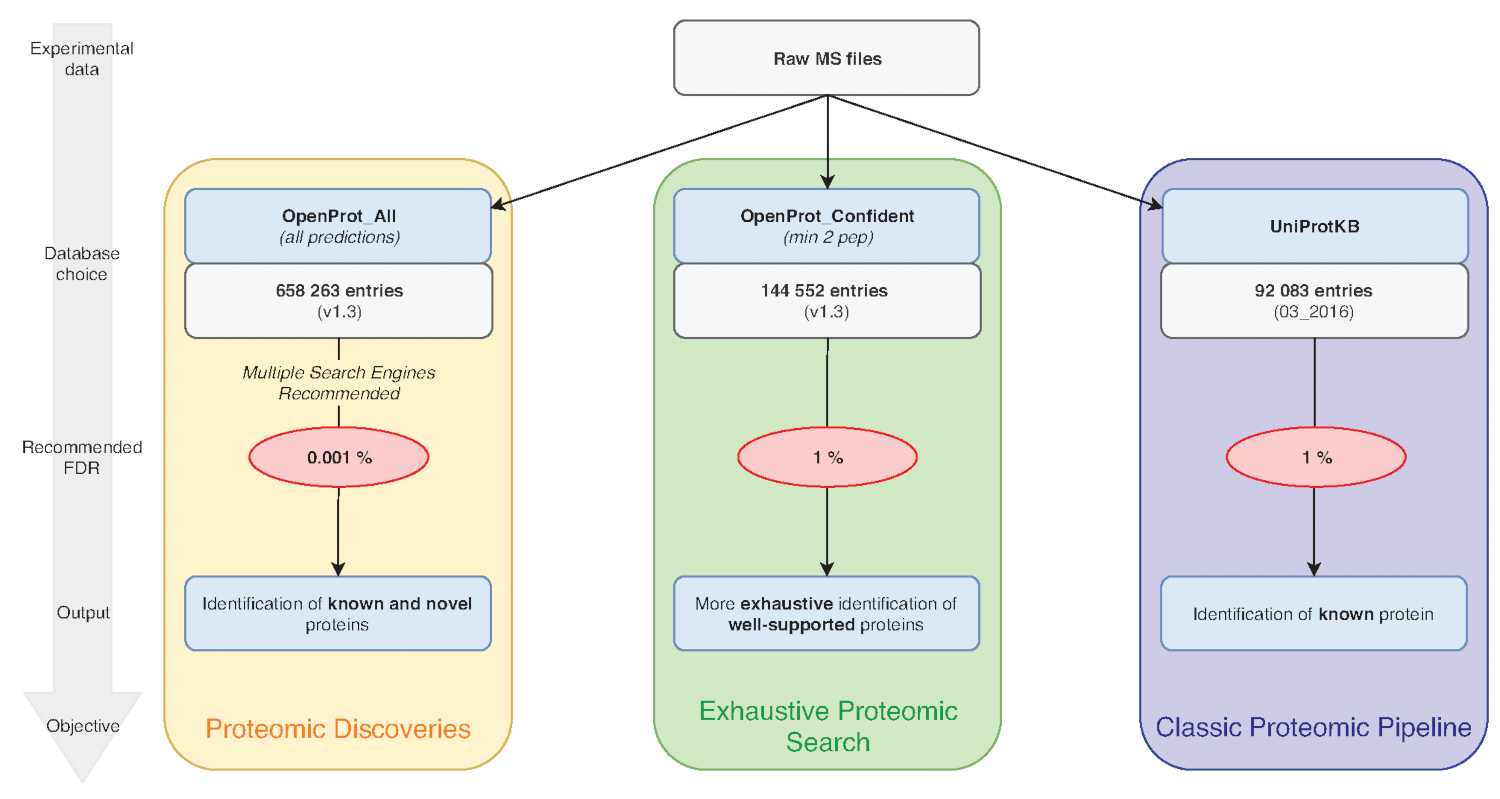
Nota: OpenProt ofrece tres clasificaciones: RefProt, isoformas y AltProt. Como se muestra en la figura 1, este parámetro variará según el objetivo de la investigación.- Haga clic en RefProt solo para generar archivos que contienen sólo las proteínas conocidas.
- Haga clic en AltProt e isoformas para generar archivos que contienen sólo nuevas proteínas - o nuevas isoformas de las proteínas conocidas (isoformas) o codificadas por una alternativa ORF (AltProts). Tenga en cuenta que la OpenProt impone una longitud mínima de la ORF de 30 codones15.
- Haga clic en AltProts, isoformas y RefProts para generar archivos que contienen todos los tipos de proteínas presentes en la base de datos de OpenProt - proteínas conocidas y novedosas.
- Si está disponible, haga clic en la anotación de que proteína se dibujan secuencias.
Nota: OpenProt ofrece un paisaje de proteómica más exhaustivo combinando múltiples anotaciones. Transcriptoma anotaciones tienen un traslapo mínimo; así, la anotación seleccionada puede afectar sustancialmente la proteómica visualizado perfil15,23. - Haga clic en el nivel de evidencias necesarias para la consideración de la proteína. Como se muestra en la figura 1, este parámetro variará según el objetivo de la investigación.
- Haga clic en mínimo de dos péptidos únicos detectados para generar archivos que contienen sólo las proteínas más seguros.
Nota: Un criterio de dos péptidos únicos es considerado un estándar de oro en la proteómica de expresión de la proteína. Si el objetivo experimental es detectar proteínas conocidas y bien soportadas, se recomienda el uso de este parámetro. - Haga clic en mínimo de un único péptidos detectados para generar archivos que contienen las proteínas que ya se han visto al menos una vez entre los experimentos de espectrometría de masas re-analizados por OpenProt.
Nota: Esto permite a la consideración de la longitud más corta del AltProts y la probabilidad de que algunos de ellos pueden contener solamente un único péptido tríptico8,11. - Haga clic en todos predijeron para generar archivos que contienen todas las predicciones de OpenProt.
Nota: Esta configuración sólo se recomienda si el objetivo experimental es descubrir nuevas proteínas (figura 1). El consiguiente aumento considerable en las llamadas a buscar espacio para una tubería de análisis adaptado según lo discutido debajo de7,15.
- Haga clic en mínimo de dos péptidos únicos detectados para generar archivos que contienen sólo las proteínas más seguros.
- Haga clic en el formato de archivo para descargar. Para los análisis proteómicos, seleccionar el archivo Fasta (proteína). El archivo Léame contiene toda la información necesaria en el formato de archivo.
2. encargo descargar base de datos de OpenProt
Nota: Esta sección detalla cómo obtener una base de datos personalizada. Si no hay base de datos personalizada es necesario, vaya a la sección siguiente.
- Visite el sitio web de OpenProt (www.openprot.org) y abra la página de búsqueda mediante el enlace en el menú superior de la página.
- Haga clic en las especies de interés basándose en los datos experimentales analizados.
- Introduzca una lista de genes o transcripciones de interés.
- Cuando se utiliza una lista de genes, introdúzcalo en el cuadro de consulta de Gene .
- Cuando se utiliza una lista de las transcripciones, escríbalo en el cuadro de consulta de la transcripción .
- Marque cualquier casilla que aplique a la base de datos deseada.
- No haga clic en cualquier cuadro para obtener una tabla que contenga todos los tipos de proteína apoyado por OpenProt: RefProt, isoformas y AltProts.
- Haga clic en Mostrar sólo proteínas con evidencia experimental para obtener una tabla que contenga todos los tipos de proteínas (RefProts, isoformas y AltProts) que se han detectado al menos una vez por MS y para cuya traducción se ha recogido evidencia de ribosoma perfiles de datos.
- Del mismo modo, o haga clic en Mostrar sólo las proteínas detectadas por MS Mostrar sólo las proteínas detectadas por ribosome profiling para obtener una tabla que contenga todos los tipos de proteínas que han sido detectados al menos una vez por MS o por ribosome profiling respectivamente.
- O haga clic en Mostrar sólo AltProts Mostrar sólo isoformas para obtener una tabla que contiene sólo AltProts o sólo isoformas respectivamente.
- Haga clic en Mostrar sólo AltProts y Mostrar sólo isoformas para obtener una tabla que contenga ambos tipos de proteínas.
Nota: Todas las combinaciones de filtros son posibles.
- Una vez que se establecen todos los parámetros deseados, haga clic en buscar. La salida de la tabla aparecerá debajo de los campos de la consulta de búsqueda.
- Haga clic en el botón Descargar Fasta en la esquina superior derecha de la tabla de salida. Esto generará un archivo Fasta que contiene todas las proteínas resultantes de la lista consultada de genes o transcritos.
- Tenga en cuenta que por razones computacionales, OpenProt tiene un máximo de 2.000 elementos que preguntó (genes o transcritos) en un momento. En caso de una lista por encima de ese límite, fasta varios puede ser generado y luego concatenar (como se detalla a continuación); o simplemente descargar la base de datos entera de OpenProt y filtrar el archivo obtenido como se desee.
- Bin la lista completa de los genes o transcritos en sublistas de 2.000 entradas o menos. Para cada sublista, descargar un archivo Fasta como se describió anteriormente (paso 3.3 a 3.6).
- Inicie sesión la instancia Europea Galaxy (o cualquier otra instancia que disponen de herramientas de Proteómica), https://usegalaxy.eu/.
- Crear una nueva historia e importar todo descargado OpenProt bases de datos (uno por cada sublista de genes o transcripciones) haciendo clic en el logo de carga en la parte superior izquierda de la pantalla.
- Utilice la herramienta Fasta combinar archivos y secuencias únicas filtro desarrollada por los desarrolladores de GalaxyP (https://github.com/galaxyproteomics/). Seleccione la opción combinar Fasta todos y todas las bases de datos importadas de OpenProt de entrada.
Nota: Cada herramienta puede ser buscado mediante el cuadro de consulta en el lado izquierdo de la pantalla - Seleccione la opción de adhesión sólo para evaluar la unicidad de la secuencia y copia la regla de análisis OpenProt identificador (>(.*) \ |), luego haga clic en Ejecutar.
- Tenga en cuenta que todos los archivos han sido concatenados en un único archivo Fasta con ninguna redundancia que ahora aparece en el panel de historial en el lado derecho de la pantalla. Esto constituye la base de datos de trabajo.
3. manejo de base de datos
Nota: de ahora en adelante, se utilizará la plataforma de la galaxia, pero los mismos principios pueden aplicarse a otros software de proteómica.
- Inicie sesión la instancia Europea Galaxy (o cualquier otra instancia que disponen de herramientas de Proteómica), https://usegalaxy.eu/.
- Crear una nueva historia e importar la base de datos descargado de OpenProt haciendo clic en el logo de carga en la parte superior izquierda de la pantalla.
- Ir a la página de flujo de trabajo y el flujo de trabajo de manejo de base de datos (S1 Material complementario) de importación haciendo clic en el logo de carga en la parte superior izquierda del panel central.
- Haga clic en ejecutar el flujo de trabajo y seleccione la base de datos importada de OpenProt como entrada.
Nota: Este flujo de trabajo anexar el repositorio de CRAPome a la OpenProt fasta y generar señuelo secuencias (secuencias inversas)24. Si desea una lista de señuelo de shuffle, se puede hacer cambiando este parámetro de la herramienta DecoyDatabase. - Cambie el nombre el archivo Fasta obtuvo algo significativo. La base de datos está listo para usarse para análisis de proteómica.
4. preparación de archivo espectrometría de masas
Nota: La mayoría de las herramientas de la proteómica en instancias de galaxia utiliza el formato mzML, y motores de búsqueda de péptido prefieren datos en modo de centroide.
- Abra la herramienta de MSConvert libremente disponible de la suite de ProteoWizard y subir el archivo de datos para ser analizados25.
- Elegir el directorio de la salida y el formato de archivo deseado para mzML.
- Ajustar un pico recogiendo filtro usando el algoritmo basado en wavelets (CWT) MS1 y MS2 y comenzar la conversión26.
5. péptido y proteína identificación/cuantificación
Nota: Esta parte de la tubería utiliza herramientas de la suite de OpenMS, un marco versátil y fácil de usar18.
- Inicie sesión la instancia Europea Galaxy (o cualquier otra instancia que disponen de herramientas de Proteómica), https://usegalaxy.eu/.
- Crear una nueva historia y transferir la base de datos creado anteriormente (paso 3.5) a esta nueva historia con un drag-and-drop.
- Importar el archivo de datos de transformadas mzML (paso 4.3) haciendo clic en el logo de subir en la parte superior izquierda de la pantalla.
- Ir a la página de flujo de trabajo e importar el flujo de trabajo deseado haciendo clic en el logo de carga en la parte superior izquierda del panel central.
Nota: MS experimentos son diferentemente diseñados basado en el resultado final deseado. Flujos de trabajo se incluyen dos diseños frecuentes: identificación de proteínas y cuantificación de la proteína basan en isótopos estables (SIL) de etiquetado. Sin embargo, la instancia de la galaxia contiene muchas otras herramientas que se apoyan otros tipos de análisis de proteómica27,28.- Para un diseño de identificación de proteínas, importar el flujo de trabajo en S2 Material complementario. Cuando utilice este flujo de trabajo, por favor no utilice la compresión zlip al convertir los archivos (paso 4.2)
- Para una cuantificación de la proteína basada en diseño etiquetado del isótopo estable, importar el flujo de trabajo en S3 Material complementario.
- Seleccione ejecutar el flujo de trabajo y revisar los diferentes parámetros.
- Seleccione el archivo de datos importado mzML como entrada y la base de datos creado anteriormente (paso 3.5) como el archivo de base de datos Fasta.
- Puesto que el flujo de trabajo utiliza la X! Búsqueda de tándem motor, importar la X! TANDEM por defecto configuración archivo (proporcionado en S4 de Material suplementario)29 haciendo clic en el logo de carga en la parte superior izquierda de la pantalla.
- El flujo de trabajo utiliza múltiples motores de búsqueda (MS-GF + y X! Tándem). Añadir otros motores de búsqueda o elegir uno solo simplemente por adición o eliminación de las herramientas de flujo de trabajo30,31.
Nota: Utilizando múltiples motores de búsqueda se recomienda ya que aumenta la sensibilidad y la sensibilidad del análisis32. - Para explicar el incremento sustancial en el tamaño cuando se usa la base de datos entera de OpenProt, utilizar un riguroso FDR15. De forma predeterminada, se establece el flujo de trabajo proporcionada para un 0.001% FDR, adecuada para el uso de la base de datos entera de OpenProt. Para otras bases de datos, esto se puede editar cualquier valor deseado.
Nota: Asegúrese de adaptar los parámetros de las diferentes herramientas según el espectrómetro de masas utilizados y el protocolo experimental (precursor ion y fragmento error, fijadas y variable modificaciones, enzima usada, etcetera).
- Descargar opcionalmente, salida para cada paso del flujo de trabajo para el almacenamiento o análisis de control de calidad haciendo clic en el paso solicitado desde el panel de historia, y luego haciendo clic en el logotipo de Save que aparecerá debajo.
6. control de calidad
Nota: Porque proteómica basada en MS es el resultado de un proceso complejo donde cada paso tiene que ser optimizado para producir resultados reproducibles, control de calidad es un procedimiento necesario en el flujo de trabajo33.
- Varias métricas son común punto de referencia de rendimiento, tales como el número de péptido-espectro (PSM), el número de identificación de péptidos y proteínas. Ejecute la herramienta de Información de archivo en la salida de IDFilter (indicada en verde en la figura 2) para proporcionar tales métricas.
- Aunque no se aplica a cada identificación, especialmente con grandes conjuntos de datos, informes de nuevas proteínas siempre deben ser cuidadosamente evaluados. Verificación de la puntuación de la proteína, la cobertura de la secuencia y los espectros apoyando el hallazgo es de vital importancia. Utilice la herramienta TOPPview desde el marco de OpenMS para hacer esto; está libremente disponible y bien documentado18,34,35.
7. OpenProt minería de base de datos
Nota: Una vez que se ha hecho una identificación segura de una nueva proteína predicha por OpenProt (números comenzando con IP_ para AltProts y II_ para novela isoformas), más información biológica se desprende de la Página Web de OpenProt15.
- Ir al sitio web de OpenProt: www.openprot.org y abrir la página de búsqueda mediante el enlace en el menú superior de la página.
- Haga clic en las especies de interés (igual que el uno en el que se identificó la proteína) y escriba el número de proteínas en el cuadro de consulta de proteína .
- Haga clic en buscar y aparecerá una tabla que contiene información básica sobre la proteína consultada. Las características de la mesa: la longitud de la proteína (en aminoácidos), su peso molecular (kDa) y el punto isoeléctrico, evidencias experimentales por MS o ribosome profiling (pruebas de traducción, TE) y predicciones funcionales tales como predijeron dominios y proteína homología (entre las 10 especies apoyadas por OpenProt, v1.3). La tabla también contiene información acerca de los genes relacionados y transcripción y la localización de la proteína dentro de la transcripción.
- Haga clic en el enlace de datos para recabar más información. La recién inaugurada página contiene un browser del genoma que se centra en la proteína consultada y como los genómicos y transcriptómicos coordenadas la presencia de un Kozak o alta eficiencia traducción iniciación sitio (TIS) motivo36, 37.
- Haga clic en la proteína o ADN enlaces de la ficha de información, para obtener proteínas o secuencias de ADN respectivamente.
- Buscar toda la información sobre MS pruebas, perfiles de detección, conservación y dominios de la proteína identificada haciendo clic en las lengüetas superiores15del ribosoma.
Resultados
El flujo de trabajo descrito se aplicó a un conjunto de datos de MS disponible en el repositorio de orgullo38,39. El estudio original desarrolló un método (iMixPro), uso de isótopos estables de etiquetado de aminoácidos en cultura de célula (SILAC), para eliminar los falsos positivos de afinidad-purificación MS (AP-MS) experimentos de38. En Resumen, un experimento AP-MS consiste en utilizar anticuerpos enlazado a los granos para obtener una proteína de interés (cebo) y sus interactianos (presas). Las proteínas recogidas luego son digeridas y preparadas para MS. El método de preparación de la muestra y las opciones están descritas en el estudio original y en el repositorio de orgullo (PXD004246). Un desafío en tales experimentos es la abundancia de falsos positivos, en particular de proteínas vinculantes para los granos pero no el anzuelo. Aquí, utilizamos SILAC generar cocientes del isótopo diferente entre presas verdaderos y falsos positivos: 3 muestras de control (sin cebo) cultivadas en medio ligero, 1 muestra expresando el cebo cultivado en medio luz y 1 muestra expresando el cebo cultivado en medio pesado procesado con las cuentas y posterior análisis de espectrometría de masas. Con tal diseño, proteínas no específicas de enlace a las cuentas tendrá una proporción de pesados a ligeros de 1:4; Cuando verdaderas presas tendrá una relación de 1:138.
Nuevamente analizamos sus datos MS AP usando la base de datos de OpenProt; los cebos incluyeron tres proteínas endógenas (PTPN14, JIP3 y IQGAP1), y dos sobre-expresan proteínas (RAF1 RNF41). Puesto que los experimentos utilizan estándar, se utilizó el flujo de trabajo del Galaxy para la cuantificación de la proteína (Complementario de Material de S3, figura 2). El flujo de trabajo se ejecute usando la base de datos entera de OpenProt (OpenProt_all) o una base de datos restringida de OpenProt (OpenProt_2pep, incluyendo sólo las proteínas detectadas previamente con un mínimo de dos péptidos únicos).
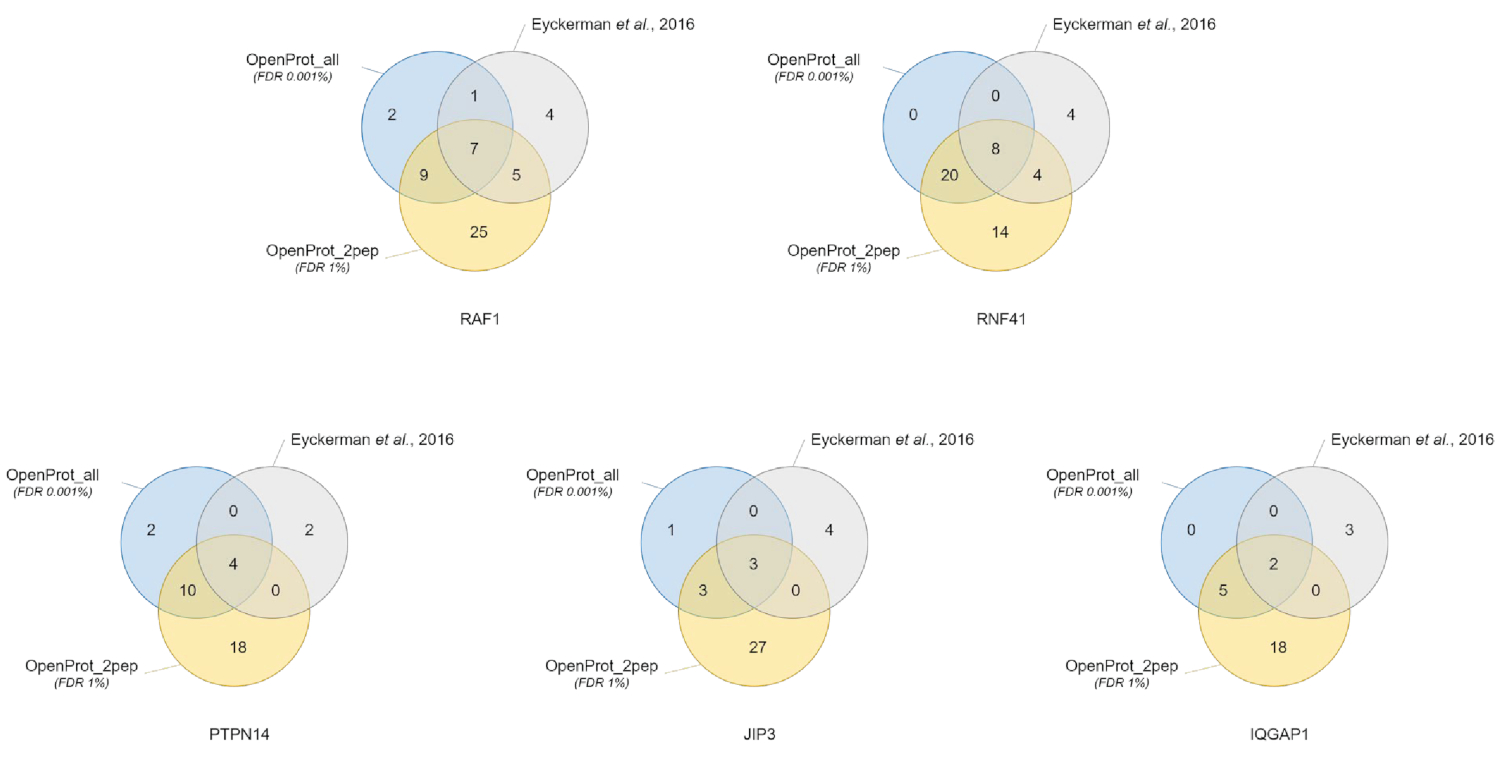
Cuantificación e identificación de proteínas fueron buenos y reproducible a través de las diferentes bases de datos utilizados. Como se muestra en la figura 3, la mayoría proteínas identificadas en el documento original también se identificaron utilizando base de datos OpenProt_2pep o OpenProt_all (una lista detallada está disponible en S5 de Material complementario). Este resultado muestra que la tubería aquí descritos y lo OpenProt son capaces de producir identificación de proteínas y cuantificación comparable a la de los procedimientos actuales basados en el de bases de datos de UniProtKB40bases de datos. Sin embargo, el uso de bases de datos de OpenProt tiene la ventaja de que permiten la detección de novela y proteínas previamente indetectables, como se demuestra en este caso de estudio.
11 bien soportadas proteínas (1 isoforma 10 AltProts), sin embargo, actualmente no está anotados en las bases de datos, se identificaron en todo bases de datos, con péptidos de confianza, utilizando la base de datos de OpenProt_2pep (todos adhesiones de proteína, junto con el número de apoyo péptidos, están disponibles en S5 de Material complementario). Esta base de datos permite el uso del tradicional 1% FDR como el aumento del espacio de búsqueda sigue siendo moderada. Estas 11 proteínas no fueron identificadas en el estudio original como estaban ausentes de la base de datos.
29 nuevas proteínas (16 isoformas y 13 AltProts) fueron descubiertas a través de los conjuntos de datos, con péptidos de confianza, utilizando la base de datos de OpenProt_all (todos adhesiones de proteína, junto con el número de péptidos de apoyo, son S6 de Material suplementario disponible en ). Como se muestra en la figura 3, el FDR estricta recomendada no afectó a las identificaciones más seguros de la proteína, aunque él disminuir el número de proteínas identificadas. Comparativamente a la base de datos de OpenProt_2pep, un mayor número de nuevas proteínas puede ser identificado con toda confianza. Todas estas nuevas proteínas están ausentes de la base de datos de OpenProt_2pep. Esto pone de relieve el papel crucial de la base de datos elegido para proteómica basada en la MS.
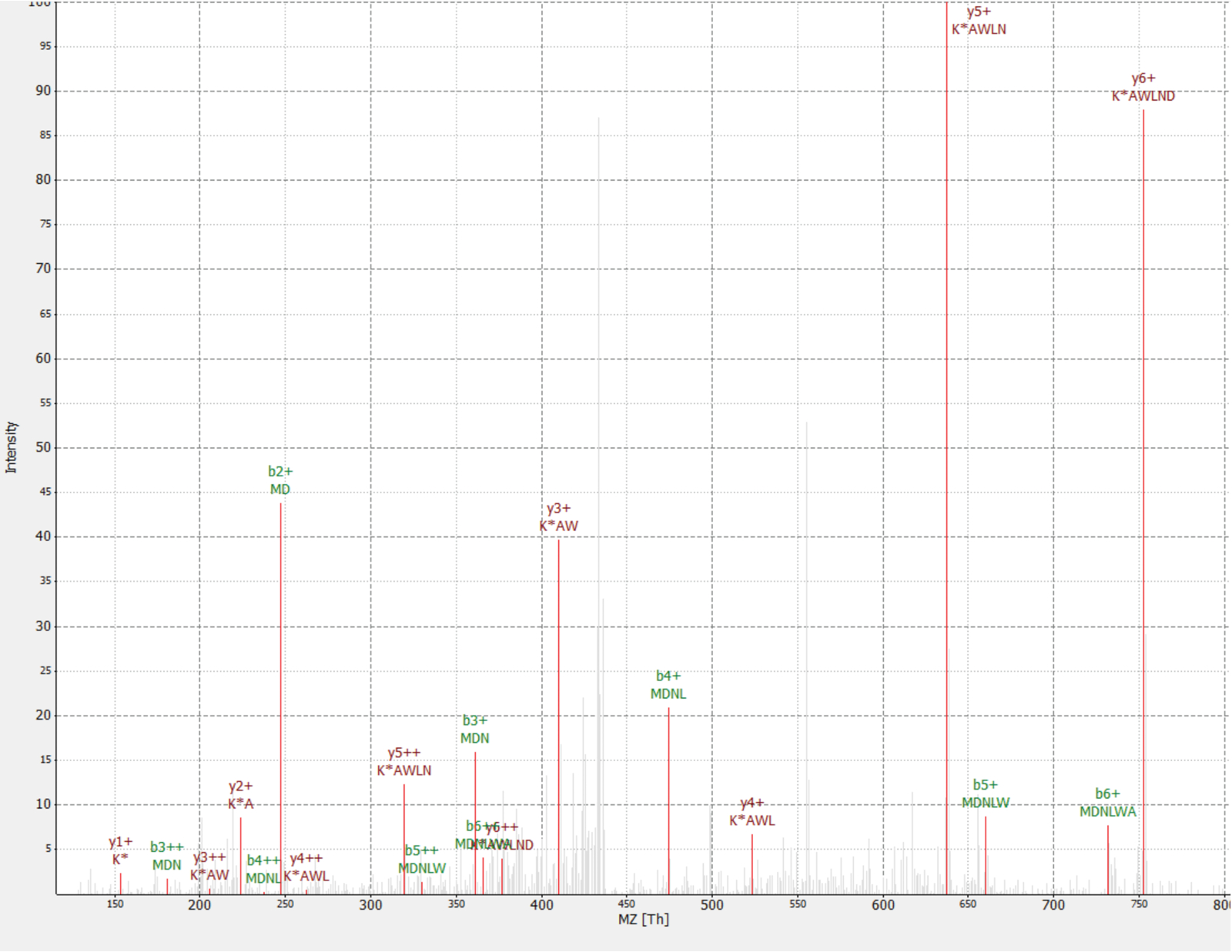
Una nueva proteína fue descubierta como un interactor de la proteína de RAF1 (IP_637643). Utilizar el sitio de OpenProt, uno puede ver esta proteína no había sido detectada por MS ni por ribosoma perfilado hasta ahora (OpenProt v1.3). La proteína es de 46 aminoácidos de largo y sólo puede dar dos péptidos únicos sobre la digestión tríptica. El péptido detectado en RAF1 AP-MS dataset (fracción 18) tenía un espectro de buena calidad, como se muestra en la figura 4y muestran una proporción de pesados a ligeros de 1.09. La proteína está codificada en el gen NANOGNBP1 , que es un pseudogene de NANOGNB. La transcripción (ENST00000448444), actualmente anotada como no-codificación, fue detectada en varios tejidos según el portal GTEx40. La proteína contiene un dominio funcional previsto asociado con ADN vinculante (Gene Ontology GO: 0003677)41.

Figura 1 : Elección de tabla de análisis de Proteómica de la base de datos. Análisis de datos de MS, en particular la elección de la base de datos, dependen de los objetivos de la investigación. Tres objetivos comunes están señaladas en azul (proteómico clásico pipeline), verde (búsqueda exhaustiva de Proteómica) y naranja (descubrimiento de Proteómica). Cada objetivo depende de una base de datos adecuado y tubería. Una herramienta de identificación solo puede usarse para un proteómica clásica y exhaustiva las tuberías. Para la tubería de descubrimiento proteómicos, recomendamos encarecidamente utilizar múltiples motores de identificación. FDRs recomendadas se indican en rojo, y tamaños de base de datos de proteínas están indicados en cuadros de gris. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

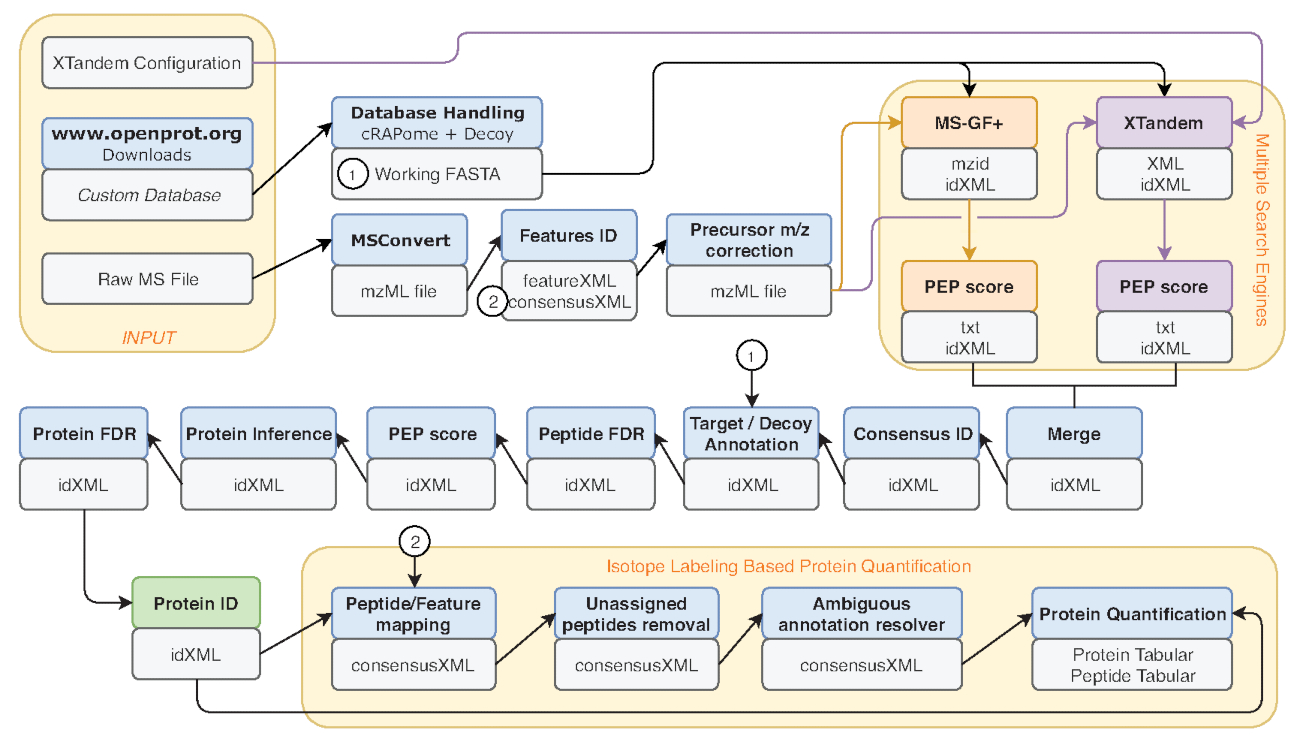
Figura 2 : Representación gráfica del flujo de trabajo galaxia usada. Representación paso a paso del flujo de trabajo de análisis proteómicos utilizado para re-análisis de datos de Eyckerman et al38. Archivos de entrada, péptido búsqueda y cuantificación de proteínas están indicados por las cajas de naranja. Cajas azules corresponden a las herramientas utilizadas y cajas grises corresponden a los archivos de salida generados. Los motores de búsqueda (MS-GF + y X! Tándem) se indican mediante colores diferentes (rojos y morados respectivamente) así como las flechas que indican sus necesarias entradas y salidas. La caja verde destaca la herramienta de generación de una lista de identificaciones de la proteína. Cuando se generan varias salidas, la que se utiliza para los pasos posteriores se indica como la más cercana a la flecha. Este flujo de trabajo está disponible en S2 Material complementario. ¡La X! Archivo de configuración de parámetros de tándem predeterminado está disponible en S4 de Material complementario. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 : Comparación de identificación interactor por cebo utilizando diferentes bases de datos. Diagramas de Venn de identificación de proteínas utilizando la OpenProt más seguro de base de datos (en naranja, evidencias de 2 péptidos únicos mínimos, OpenProt_2pep) con un 1% FDR o el OpenProt toda la base de datos (en azul, OpenProt_all) con un 0,001% FDR, o según en el original del documento (en gris)38. Cada diagrama corresponde a interactianos identificados para el cebo mencionado: RAF1, RNF41, JIP3 y PTPN14, IQGAP1. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4 : Espectro MS/MS de identificado MDNLWAK(6 de 13) péptido de proteína novel IP_637643. La intensidad es relativa (0 a 100%). Las cumbres están indicados en rojo, y anotaciones de los iones están en oscuro rojo y b iones las anotaciones en color verde. Extraído del software de TOPPview34. Precursor Error = 2,70 ppm, cuenta PEP = 0,12. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Plazo | Definición | Referencia |
| Alternativa ORF (AltORF) | ORF no canónico actualmente no está anotado en las anotaciones del genoma, pero anotado en OpenProt. | 15 |
| Referencia ORF (RefORF) | ORF canónico anotado en las anotaciones del genoma y OpenProt. | 15 |
| Proteínas alternativas (AltProt) | nueva proteína codificada por un AltORF, con ninguna semejanza significativa con un RefProt. Prefijo de número de adhesión: IP_. | 15 |
| Proteína de referencia (RefProt) | proteína anotada actualmente en bases de datos de secuencia proteína como UniProtKB, Ensembl o NCBI RefSeq y también en OpenProt. | 15 |
| Isoforma novela | nueva proteína codificada por un AltORF, con una similitud importante con un RefProt. Prefijo de número de adhesión: II_. | 15 |
| OpenProt_2pep la base de datos | contiene la secuencia de todos los RefProts y nuevas proteínas predichas por OpenProt, ya detectado con un mínimo de 2 péptidos únicos. | 15 |
| OpenProt_1pep la base de datos | contiene la secuencia de todos los RefProts y nuevas proteínas predichas por OpenProt, ya detectado con un mínimo de 1 único péptido. | 15 |
| OpenProt_all la base de datos | contiene la secuencia de todas las nuevas proteínas predichas por OpenProt y RefProts. | 15 |
Tabla 1: Definición de términos utilizados en OpenProt y en el protocolo
S1 de Material complementario: flujo de trabajo de galaxia para manejo de base de datos. Esto agregará las secuencias CRAPome y señuelo (inversas) a la base de datos de entrada. Salida es un archivo Fasta. Haga clic aquí para descargar.
S2 Material complementario: flujo de trabajo de galaxia para identificación de proteínas. Esto permitirá identificar proteínas a partir de un fichero de datos de espectrometría de masas con dos motores de búsqueda (MS-GF + y X! Tándem). Cada parámetro se puede ajustar como desee antes de ejecutar el flujo de trabajo. Haga clic aquí para descargar.
S3 Material complementario: flujo de trabajo de galaxia para cuantificación de proteína utilizando isótopos estables etiquetado (SIL). Esto será identificar y cuantificar proteínas a partir de un fichero de datos de espectrometría de masas con dos motores de búsqueda (MS-GF + y X! Tándem). Cada parámetro se puede ajustar como desee antes de ejecutar el flujo de trabajo. Haga clic aquí para descargar.
S4 Material complementario : X! Archivo de configuración de parámetros de defecto de tándem. Este archivo es necesario para el funcionamiento de la X! TandemAdapter herramienta en la plataforma de la galaxia. Haga clic aquí para descargar.
S5 de Material complementario: cuantifican proteínas a partir de conjuntos de datos de iMixPro. Archivos de datos de Eyckerman et al 201638 se procesaron utilizando bases de datos de OpenProt y proteínas cuantificadas se enumeran para cada condición. Los cebos son PTPN14, JIP3, IQGAP1, RAF1 y RNF41. Nombres de gen indicados en verde corresponden a proteínas identificadas también en el papel original38. Nombres de gene indicados en color naranja corresponden a interactianos conocidos según BioGrid que no fueron reportados en el documento original. Nombres de gene indicados en azul claro corresponden a nuevas proteínas identificadas como interactianos (el número correspondiente de la adhesión de proteínas se indica entre corchetes). Nombres de genes indicaron en gris claro y cursiva corresponde a probables contaminantes (proteínas de la queratina). Haga clic aquí para descargar.
S6 de Material complementario: identificar nuevas proteínas de conjuntos de datos de iMixPro. Archivos de datos de Eyckerman et al 201638 se procesaron utilizando bases de datos de OpenProt y nuevas proteínas identificadas se indican para cada condición. Los cebos son PTPN14, JIP3, IQGAP1, RAF1 y RNF41. Proteína de adhesión números aparecen, a partir de II_ para nuevas isoformas de una proteína conocida y con IP_ para nuevas proteínas de un ORF alternativo (AltProt). El número de péptidos de apoyo se indican entre corchetes. Haga clic aquí para descargar.
Discusión
Al analizar los datos de los espectrómetros de masa, la calidad de la identificación de proteínas se basa en parte en la exactitud de la base de datos usado6,20. Enfoques actuales utilizan tradicionalmente UniProtKB las bases de datos, sin embargo, éstos apoyan el modelo de anotación del genoma de una sola ORF por transcripción y una longitud mínima de 100 codones (a excepción de ejemplos previamente demostrados)40. Múltiples estudios relacionan las deficiencias de estas bases de datos con el descubrimiento de ORFs funcionales de supuestamente no codificantes regiones8,11,12,13. Ahora, OpenProt permite más exhaustiva identificación de proteínas como extrae proteínas múltiples anotaciones de transcriptoma. OpenProt recupera NCBI RefSeq (GRCh38.p7) y transcriptomas de Ensembl (GRCh38.83) y anotaciones de UniProtKB (UniProtKB-SwissProt, 2017-09-27)40,42,43. Como anotaciones actuales presentan poco traslapo, OpenProt muestra así una visión más exhaustiva del paisaje proteómicos potencial que cuando limitada a una anotación15.
Además, como OpenProt impone un modelo policistrónico, permite múltiples anotaciones de proteína por transcripción. Por motivos estadísticos y computacionales, OpenProt todavía tiene un umbral mínimo de 30 codones15. Sin embargo, augura miles de secuencias de la nueva proteína, ampliando así el alcance de posibilidades para la identificación de proteínas. Con este enfoque, OpenProt apoya proteómicos descubrimientos de una manera más sistemática.
La calidad de la identificación de proteínas también puede verse afectada por los parámetros que se utilizan. Análisis de proteómica basada en MS tienen típicamente un 1% de proteína FDR. Sin embargo, la base de datos de OpenProt entero contiene aproximadamente 6 veces más entradas (figura 1). Para tener en cuenta este aumento sustancial en el espacio de búsqueda, le recomendamos usar un FDR más estricta de 0.001%. Este parámetro se optimizó mediante estudios de benchmark y manual evaluación de espectros seleccionados al azar15. Falsos positivos siguen siendo una posibilidad, sin embargo, y animamos a inspección y validación de evidencias de una nueva proteína. Un estándar recomendado podría ser la identificación de una proteína de dos pruebas diferentes de EM, como datos de base y falsos positivos varían entre los conjuntos de datos15.
La tubería aquí y utilizado para el estudio de caso puede ser modificada como placer para adaptarse a los parámetros y diseño experimental. Le recomendamos utilizar múltiples motores de búsqueda como aumenta la sensibilidad y la sensibilidad del péptido identificación32. Además, animamos a la base de datos correspondiente mejor para el objetivo experimental (figura 1). Como usar el OpenProt toda la base de datos viene con un FDR estricta, verdaderas identificaciones pueden perderse. Así, la base de datos toda debe ser destinado a descubrimiento de nuevas proteínas, mientras que perfiles de proteómica clásica deberían utilizar las bases de datos de OpenProt más pequeños (como el OpenProt_2pep utilizado en el estudio de caso anterior).
OpenProt en la actualidad predice secuencias a partir de un codón ATG, mientras que varios estudios destacan la iniciación de la traducción en otros codones44,45. Cuando se identifica una nueva proteína por uno o varios péptidos únicos, es posible que el codón de iniciación verdadera no es el presunto ATG. Los usuarios pueden buscar evidencia de traducción en la página web OpenProt. Actualmente, OpenProt informa sólo eventos de traducción si se trata de la proteína prevista toda secuencia (100% de solapamiento)15. Por lo tanto, ausencia de pruebas de traducción no significaría la proteína no está traducida, pero que el codón de inicio no puede ser el supuesta ATG.
A pesar de sus limitaciones actuales, OpenProt ofrece una visión más exhaustiva del potencial de codificación de eukaryotic genomas. Bases de datos de OpenProt fomentan la proteómica descubrimientos y la comprensión de interacciones y funciones de la proteómica. Evolución futura de la base de datos de OpenProt incluye anotaciones de otras especies, pruebas de traducción de ATG no comienzo codon y el desarrollo de un gasoducto para incluir nuevas proteínas en todo el genoma y estudios de secuenciación del exoma.
Divulgaciones
Los autores no declaran conflicto de intereses.
Agradecimientos
Agradecemos a Vivian Delcourt por su ayuda, debates y asesoramiento en obra. X.R. es miembro del Fonds de Recherche du Québec Santé FRQS apoyado Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. Esta investigación fue apoyada por una Cátedra de investigación de Canadá en proteómica funcional y descubrimiento de las proteínas de la novela a la subvención de CIHR, X.R. 137056 fregona. Agradecemos al equipo Calcul Québec y cálculo de Canadá por su apoyo con el uso de la supercomputadora mp2 por la Université de Sheerbrooke. Funcionamiento del superordenador mp2 está financiado por la Fundación Canadá de la innovación (CFI), le ministère de l'Économie, de la science et de l ' Innovation du Québec (MESI) y les Fonds de Recherche du Québec - naturaleza et tecnologías (FRQ-NT). El servidor galaxia que fue utilizado para algunos cálculos de proteómica está financiado en parte por colaboración investigación 992 centro médico epigenética (grant DFG SFB 2012 992/1) y el Ministerio Federal alemán de educación e investigación (BMBF otorga 031 RBC A538A/A538C, 031L0101B De /031L0101C. NBI-epi, 031L 0106 de. ESCALERA (de. NBI)).
Materiales
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
Referencias
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados