
Method Article
Basé sur la spectrométrie de masse protéomique des Analyses à l’aide de la base de données OpenProt de dévoiler de nouvelles protéines traduit de cadres ouverts de lecture Non canoniques
Dans cet article
Résumé
OpenProt est une base de données librement accessible qui applique un modèle de polycistronique du génome des eucaryotes. Nous présentons ici un protocole pour l’utilisation des bases de données de OpenProt lors de l’interrogation des ensembles de données de spectrométrie de masse. À l’aide de OpenProt base de données pour l’analyse des expériences de la protéomique permet la découverte du roman et protéines indétectables.
Résumé
Annotation du génome est au centre de recherche protéomique d’aujourd'hui qu’il dessine les contours du paysage protéomique. Les modèles traditionnels d’open lecture annotation de cadre (ORF) imposer deux critères arbitraires : une longueur minimale de 100 codons et une seule ORF par transcription. Toutefois, un nombre croissant d’études signalent l’expression de protéines provenant prétendument non codantes régions, contester l’exactitude des annotations de génome actuel. Ces roman protéines trouvées codés au sein non codantes RNAs, 5' ou 3' régions non traduites (RTNs) de l’ARNm, ou chevauchant une séquence codante connue (CD) dans une solution de rechange ORF. OpenProt est la première base de données qui permet d’appliquer un modèle de polycistronique des génomes eucaryotes, permettant l’annotation de multiples ORF par transcription. OpenProt est librement accessible et offre personnalisées téléchargements de séquences protéiques sur 10 espèces. À l’aide de OpenProt base de données pour les expériences de la protéomique permet la découverte de nouvelles protéines et met en évidence le caractère polycistronique des gènes eucaryotes. La taille de base de OpenProt (tous prédit protéines) est importante et doit être prise en compte pour l’analyse. Cependant, avec les paramètres de fréquence (FDR) découverte de faux appropriées ou l’utilisation d’une base de données OpenProt restreinte, utilisateurs gagneront une vision plus réaliste du paysage protéomique. Dans l’ensemble, OpenProt est un outil disponible gratuitement qui favorisera les découvertes de la protéomique.
Introduction
Ces dernières décennies, protéomique de la spectrométrie de masse (MS-) basée est devenue la technique or à déchiffrer les protéomes de cellules eucaryotes1,2,3,4,5. Cette méthode s’appuie sur les annotations de génome actuel pour générer une base de référence protéine séquence qui décrit la portée des possibilités6,7,8. Toutefois, les annotations de génome tenir des critères arbitraires pour l’annotation de l’ORF, comme une longueur minimale de 100 codons et une seule ORF par transcription9,10. Un nombre croissant d’études conteste le modèle actuel d’annotation et de faire rapport des découvertes de non annotées ORF fonctionnelle dans les génomes eucaryotes8,11,12,13, 14. Ces nouvelles protéines se trouvent encodées en ARN prétendument non codantes, dans les 5' ou 3' non traduite régions (UTR) du mRNA, ou chevauchant la séquence codante canonique (cCDS) dans un autre cadre. Bien que la plupart de ces découvertes ont été heureux hasard, ils démontrent les mises en garde des annotations de génome actuel et la nature de polycistronique des gènes eucaryotes8.
Ici, nous mettons en évidence l’utilisation de bases de données OpenProt pour la protéomique axée sur le MS. OpenProt est la première base de données de détenir un modèle annotation polycistronique transcriptomes eucaryotes. Il est disponible gratuitement au www.openprot.org15. Une proportion d'entre eux prédit Qu'orfs serait aléatoire et non fonctionnelles, c’est pourquoi OpenProt cumule les preuves expérimentales et fonctionnelle d’accroître la confiance. Les preuves expérimentales incluent expression de la protéine (en MS) et traduction de preuve (par ribosome profilage)15. Preuves fonctionnelles comprennent le niveau de protéine (avec un In-paranoïaque comme approche) et le domaine fonctionnel prédiction15.
OpenProt offre la possibilité de télécharger plusieurs bases de données, de contenant uniquement des protéines bien soutenus aux bases de données sur mesure. Ici, nous présenterons un pipeline pour l’utilisation des bases de données OpenProt et offrira des aperçus de quelle base de données choisir étant donné le but expérimental. Le pipeline d’analyse protéomique présenté ici est pris en charge par l’infrastructure de la galaxie comme il est accessible et facile à utiliser, mais les bases de données peuvent fonctionner avec n’importe quel workflow16,17,18. Nous présenterons également comment utiliser le site Web OpenProt pour recueillir des informations complémentaires sur nouvelles protéines détectées par MS. Using OpenProt bases de données fournira une vue plus exhaustive du paysage protéomiques et favorisera la protéomique et les biomarqueurs découvertes dans une manière plus systématique que les méthodes actuelles.
Ce protocole met en évidence l’utilisation de bases de données de OpenProt15 lors de l’interrogation de datasets MS ; Elle n’examinera pas la conception de l’expérience elle-même, qui a été complètement revu ailleurs20,21,22. Dans le but de rester entièrement open source, le protocole est librement disponible (S1 de matériel complémentaire–S4). Pour faciliter la lecture, tous les termes utilisés en OpenProt et par les présentes tout au long de ce protocole sont définies au tableau 1.
Protocole
1. téléchargement de base de données de OpenProt
Remarque : les bases de données personnalisées basées sur des données de RNA-seq par exemple peuvent également être obtenues et la procédure est détaillée dans la deuxième partie du présent protocole. Si une base de données personnalisé est nécessaire, veuillez passer à la section suivante.
- Allez sur le site de OpenProt : www.openprot.org et ouvrez la page de téléchargements en utilisant le lien dans le menu haut de page.
- Cliquez sur les espèces d’intérêt basé sur les données expérimentales analysées.
- Cliquez sur le type de protéine désiré.
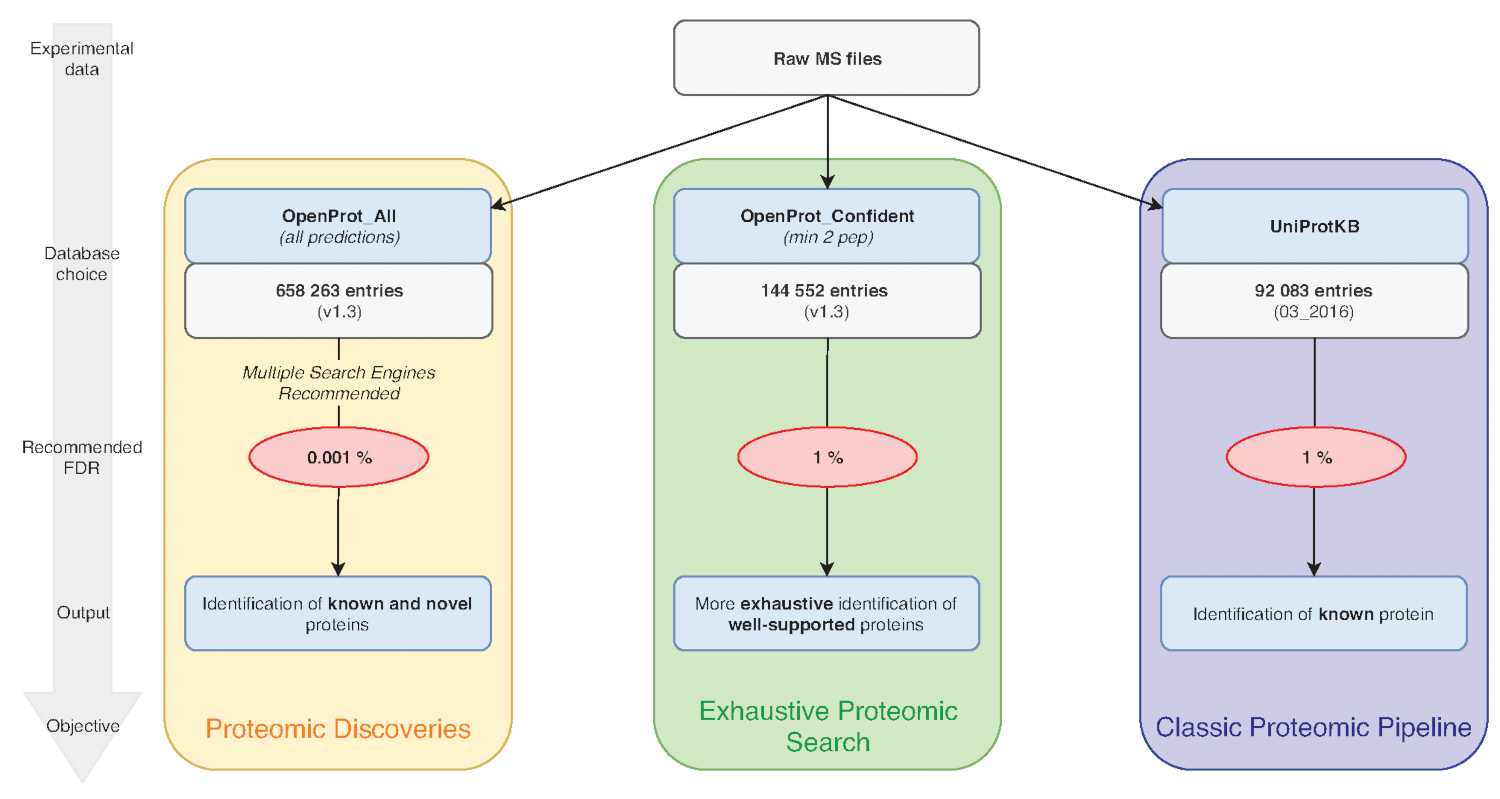
Remarque : OpenProt offre trois classifications : RefProt, isoformes et AltProt. Comme illustré à la Figure 1, ce paramètre varie selon l’objectif de la recherche.- Cliquez sur RefProt uniquement pour générer des fichiers qui contiennent uniquement des protéines connues.
- Cliquez sur AltProt et les isoformes pour générer des fichiers contenant uniquement des protéines nouvelles - deux nouveaux isoformes de protéines connues (isoformes) ou codées par une solution de rechange ORF (AltProts). Notez que OpenProt applique une longueur minimale de ORF de 30 codons15.
- Cliquez sur AltProts, isoformes et RefProts pour générer des fichiers contenant tous les types de protéines présentes dans la base de données OpenProt - protéines connues et nouvelles.
- S’il est disponible, cliquez sur l’annotation de quelles protéines séquences sont tirées.
Remarque : OpenProt offre un paysage plus exhaustif de la protéomique en combinant plusieurs annotations. Les annotations de transcriptome ont un chevauchement minimal ; ainsi, l’annotation sélectionnée peut affecter considérablement la protéomique visualisée profil15,23. - Cliquez sur le niveau de pièces justificatives nécessaires à l’examen de la protéine. Comme illustré à la Figure 1, ce paramètre varie selon l’objectif de la recherche.
- Cliquez sur au minimum deux peptides uniques détectés pour générer des fichiers contenant seulement les protéines plus confiants.
Remarque : Un critère de deux peptides uniques est considéré actuellement comme un étalon-or en protéomique pour l’expression de la protéine. Si expérimental vise à détecter les protéines connues et bien soutenus, l’utilisation de ce paramètre est recommandée. - Cliquez sur au minimum un peptides uniques détecté pour générer des fichiers contenant des protéines qui ont déjà été vus au moins une fois entre les expériences de spectrométrie de masse, ré-analysés par OpenProt.
Remarque : Ceci permet à l’examen de la plus courte longueur de AltProts et la probabilité que certains d'entre eux peuvent contenir qu’un seul peptide trypsique unique8,11. - Cliquez sur tout prédit pour générer des fichiers contenant l’ensemble des prédictions OpenProt.
Remarque : Ce paramètre est recommandé seulement si expérimental vise à découvrir de nouvelles protéines (Figure 1). L’augmentation substantielle subséquente dans les appels de l’espace de recherche pour un pipeline d’analyse adaptés tel que discuté ci-dessous7,15.
- Cliquez sur au minimum deux peptides uniques détectés pour générer des fichiers contenant seulement les protéines plus confiants.
- Cliquez sur le format de fichier souhaité à télécharger. Pour les analyses protéomiques, choisir le fichier Fasta (protéine). Le fichier readme contient toutes les informations nécessaires sur le format de fichier.
2. custom OpenProt téléchargement de base de données
Remarque : Cette section décrit en détail comment obtenir une base de données personnalisée. Si aucune base de données personnalisé n’est nécessaire, passez à la section suivante.
- Allez sur le site de OpenProt (www.openprot.org) et ouvrez la page de recherche en utilisant le lien dans le menu haut de page.
- Cliquez sur les espèces d’intérêt basé sur les données expérimentales analysées.
- Entrez une liste des gènes ou des transcriptions d’intérêt.
- Lorsque vous utilisez une liste de gènes, entrez-le dans la boîte de requête de gène .
- Lorsque vous utilisez une liste de transcriptions, entrez-le dans la boîte de requête de transcription .
- Cocher toute case qui s’applique à la base de données souhaitée.
- Ne cliquez pas sur n’importe quelle boîte pour obtenir un tableau contenant tous les types de protéines pris en charge par OpenProt : RefProt, isoformes et AltProts.
- Cliquez sur Afficher seulement les protéines avec les données expérimentales afin d’obtenir un tableau contenant tous les types de protéines (isoformes, RefProts et AltProts) qui ont été détectés au moins une fois par SM et/ou pour laquelle traduction preuves ont été recueillies du ribosome données de profilage.
- De même, cliquez sur Afficher seulement les protéines détectées par SM ou montrer seulement les protéines détectées par ribosome profilage pour obtenir un tableau contenant tous les types de protéines qui ont été détectés au moins une fois par SM ou par ribosome profilage respectivement.
- Cliquez sur afficher uniquement AltProts ou montrent seulement des isoformes d’obtenir une table contenant uniquement AltProts ou seulement les isoformes respectivement.
- Cliquez sur fois Show seulement AltProts et ne montrer que les isoformes pour obtenir une table qui contient les deux types de protéines.
Remarque : Toutes les combinaisons de filtres sont possibles.
- Une fois que tous les paramètres désirés sont définies, cliquez sur rechercher. La sortie de la table s’affiche en dessous des champs de requête de recherche.
- Cliquez sur le bouton Télécharger Fasta dans le coin supérieur droit de la table de sortie. Cela va générer un fichier de Fasta contenant toutes les protéines résultant de la liste a interrogé des gènes ou des transcriptions.
- Veuillez noter que pour des raisons informatiques, OpenProt peut contenir un maximum de 2 000 éléments d’être interrogé (gènes ou relevés de notes) à la fois. Dans le cas d’une liste au-dessus de cette limite, fasta plusieurs peut être généré et ensuite concaténées (tel que décrit ci-après) ; ou tout simplement télécharger l’ensemble de la base OpenProt et filtrer le fichier obtenu comme vous le souhaitez.
- Bin la liste entière des gènes ou des transcriptions en sous-listes ou inférieure à 2 000 entrées. Pour chaque sous-liste, télécharger un fichier de Fasta tel que décrit ci-dessus (étape 3.3 à 3.6).
- Connectez-vous à l’instance européenne Galaxy (ou toute autre instance où les outils de la protéomique sont disponibles), https://usegalaxy.eu/.
- Créer une nouvelle histoire et toutes les bases de données OpenProt téléchargés (un par sous-liste des gènes ou des transcriptions) importer en cliquant sur le logo de téléchargement en haut gauche de l’écran.
- Utilisez l’outil Fasta Merge Files et filtre des séquences uniques mis au point par les développeurs de GalaxyP (https://github.com/galaxyproteomics/). Sélectionnez l’option fusionner tous Fasta et toutes les bases de données OpenProt importés d’entrée.
Remarque : Chaque outil peut être recherché à l’aide de la boîte de requête sur le côté gauche de l’écran - Sélectionnez l’option d’adhésion seulement pour évaluer l’unicité de la séquence et copier la règle d’analyse identifiant OpenProt (>(.*) \ |), puis cliquez sur Execute.
- Notez que tous les fichiers ont été concaténés dans un unique fichier Fasta avec aucune redondance qui apparaît à présent dans le panneau historique sur le côté droit de l’écran. Ceci constitue la base de données de travail.
3. manipulation des bases de données
NOTE : à l’avenir, la plateforme Galaxy sera utilisée, mais les mêmes principes peuvent servir à d’autres logiciels de la protéomique.
- Connectez-vous à l’instance européenne Galaxy (ou toute autre instance où les outils de la protéomique sont disponibles), https://usegalaxy.eu/.
- Créer une nouvelle histoire et importer la base de données OpenProt téléchargée en cliquant sur le logo de téléchargement en haut gauche de l’écran.
- Accédez à la page de flux de travail et importer le flux de travail de base de données de gestion (S1 de matériel supplémentaire) en cliquant sur le logo de téléchargement en haut gauche du panneau central.
- Cliquez sur exécuter le workflow et sélectionnez la base de données importée de OpenProt comme entrée.
Remarque : Ce flux de travail sera ajouter le référentiel CRAPome à la OpenProt fasta et générer des séquences de leurre (séquences inverse)24. Si vous souhaitez une liste de leurre shuffle, il peut être fait en changeant ce paramètre sur l’outil DecoyDatabase. - Renommez le fichier de Fasta obtenu en quelque chose de significatif. La base de données est prêt à être utilisé pour l’analyse protéomique.
4. préparation de fichier de spectrométrie de masse
Remarque : La plupart des outils protéomique disponibles sur des instances de la galaxie utilise le format de Hatta dit Harber, et moteurs de recherche de peptide préfèrent des données en mode centroïde.
- Ouvrez l’outil MSConvert disponible gratuitement de la suite ProteoWizard et télécharger le fichier de données pour être analysées25.
- Choisissez le répertoire pour la sortie et le format de fichier souhaité pour Hatta dit Harber.
- Mettez un pic cueillette filtre à l’aide de l’algorithme ondelettes basée (CWT) niveau MS1 et MS2 et passez la conversion26.
5. peptide et protéine d’identification et de quantification
Remarque : Cette partie du pipeline utilise des outils de la suite de OpenMS, un cadre polyvalent et facile à utiliser-18.
- Connectez-vous à l’instance européenne Galaxy (ou toute autre instance où les outils de la protéomique sont disponibles), https://usegalaxy.eu/.
- Créer une nouvelle histoire et transférer la base de données créé précédemment (étape 3.5) dans cette nouvelle histoire avec un glisser-déplacer.
- Importez le fichier de données transformées Hatta dit Harber (étape 4.3) en cliquant sur le logo à télécharger en haut gauche de l’écran.
- Accédez à la page de flux de travail et importer le flux de travail désiré en cliquant sur le logo de téléchargement en haut gauche du panneau central.
NOTE : MS expériences sont conçus différemment selon le résultat final souhaité. Flux de travail est fournis ici pour deux motifs fréquents : identification des protéines et la quantification des protéines basé sur les isotopes stables d’étiquetage (SIL). Toutefois, l’instance de galaxie contient beaucoup d’autres outils qui aider les autres types de proteomic analyses27,28.- Pour une conception d’identification de protéine, importer le flux de travail fourni en S2 de matériel supplémentaire. Lorsque vous utilisez ce flux de travail, s’il vous plaît ne pas utiliser la compression zlip lors de la conversion de vos fichiers (étape 4.2)
- Pour une quantification de protéines basée sur le design étiquetage isotopique, importer le flux de travail fourni dans S3 de matériel supplémentaire.
- Sélectionnez exécuter le flux de travail et examiner les différents paramètres.
- Sélectionnez le fichier de données importé Hatta dit Harber comme entrée et la base de données créé précédemment (étape 3.5) tant que le fichier de base de données Fasta.
- Étant donné que le flux de travail utilise le X ! Recherche tandem moteur, importer le X ! En tandem par défaut configuration file (fournie dans S4 de matériel supplémentaire)29 en cliquant sur le logo de téléchargement en haut gauche de l’écran.
- Le flux de travail utilise plusieurs moteurs de recherche (MS-GF + et X ! Tandem). Ajouter d’autres moteurs de recherche ou choisir un seul simplement en ajoutant ou supprimant les outils de workflow30,31.
Remarque : En utilisant plusieurs moteurs de recherche est recommandée car elle augmente la sensibilité et la sensibilité de l’analyse de32. - Afin de tenir compte de l’augmentation substantielle de taille lors de l’utilisation de l’ensemble de la base OpenProt, utilisez un rigoureux FDR15. Par défaut, le flux de travail fourni est définie pour un 0,001 % FDR, adéquate pour l’utilisation de l’ensemble de la base OpenProt. Pour les autres bases de données, cela peut être édité aux valeurs souhaitées.
Remarque : N’oubliez pas d’adapter les paramètres des outils différents selon le spectromètre de masse utilisé et le protocole expérimental (précurseur ion et fragment de correction d’une erreur et modifications variables, enzyme utilisée, etc.).
- Vous pouvez également télécharger sortie pour chaque étape du flux de travail pour le stockage ou l’analyse du contrôle de la qualité en cliquant sur l’étape choisie depuis le panneau historique, puis en cliquant sur le logo de sauver qui apparaîtra sous.
6. contrôle de la qualité
NOTE : Protéomique axée sur le MS étant le résultat d’un processus complexe où chaque étape doit être optimisé pour produire des résultats reproductibles, contrôle de qualité est une procédure nécessaire dans le flux de travail33.
- Plusieurs mesures sont commune benchmark de la performance, comme le nombre de correspondances de peptide-spectrum (PSM), le nombre de protéines et peptides identifiés. Exécutez l’outil de Fichier d’informations sur la sortie de IDFilter (indiquée en vert sur la Figure 2) afin de fournir de telles mesures.
- Bien que non applicable à chaque identification, surtout avec les grands ensembles de données, rapports de nouvelles protéines devraient toujours être soigneusement évaluées. Inspection de la partition de la protéine, la couverture de la séquence et les spectres d’étayer la conclusion est d’une importance vitale. Utiliser l’outil TOPPview dans le cadre de OpenMS du pour faire ; Il est disponible gratuitement et bien documentée18,34,35.
7. extraction de base de données de OpenProt
Remarque : Après avoir procédé à une identification confiante d’une nouvelle protéine prédite par OpenProt (numéros commençant par IP_ pour AltProts et II_ nouvelles isoformes), plus d’informations biologiques peuvent être collectées depuis le site Web de OpenProt15.
- Allez sur le site de OpenProt : www.openprot.org et ouvrez la page de recherche en utilisant le lien dans le menu haut de page.
- Cliquez sur les espèces d’intérêt (le même que celui dans lequel la protéine a été identifiée) et entrez le numéro d’ordre de protéine dans la boîte de requête de protéine .
- Cliquez sur rechercher et une table qui contient des informations de base sur la protéine interrogée apparaîtra. Les caractéristiques de la table : la longueur de protéine (en acides aminés), son poids moléculaire (kDa) et le point isoélectrique, soutenant des preuves expérimentales par MS ou le profilage ribosome (traduction signes, TE) et prédictions fonctionnelles tels que prédit des domaines et des protéines niveau (à travers les 10 espèces prises en charge par OpenProt, v1.3). La table contient également des informations sur le gène connexe et de transcription et de la localisation de la protéine dans la transcription.
- Cliquez sur le lien détails à recueillir des renseignements. La page nouvellement ouverte contient un fureteur du génome qui est centré sur la protéine interrogé et les informations telles que la génomique et transcriptomique coordonnées et la présence d’un Kozak ou haute efficacité translation initiation site (TIS) motif36, 37.
- Cliquez sur les protéines ou l’ADN de liens dans l’onglet info, pour obtenir des séquences d’ADN ou de protéines respectivement.
- Accédez à des informations détaillées quant aux preuves de MS, ribosome, profilage de détection, de conservation et de domaines de protéines identifiées en cliquant sur les onglets de haut de la page15.
Résultats
Le flux de travail décrit ci-dessus a été appliqué à un ensemble de données MS disponible sur la fierté référentiel38,39. L’étude originale a développé une méthode (iMixPro), utilisant des isotopes stables d’étiquetage des acides aminés dans la culture de cellules (SILAC), afin d’éliminer les faux positifs de la milliseconde purification d’affinité (AP-MS) expériences38. En bref, une expérience AP-MS consiste à utiliser des anticorps liés aux perles pour extraire une protéine d’intérêt (appâts) et ses interacteurs (proies). Les protéines collectées sont ensuite digérées et préparés pour les États membres. La méthode de préparation d’échantillon et les paramètres de l’instrument sont décrites dans l’étude originale et sur le référentiel de la fierté (PXD004246). Un défi dans de telles expériences est l’abondance de faux positifs, notamment des protéines liant les perles mais pas l’appât. Ici, nous avons utilisé SILAC pour générer les rapports des isotopes différents entre proies vrais et faux positifs : 3 échantillons de contrôle (pas d’appât) cultivés en clair moyen, 1 échantillon exprimant l’appât cultivée dans un milieu lumineux et 1 échantillon exprimant l’appât cultivée dans un milieu lourd sont traitées avec les perles et l’analyse ultérieure de la spectrométrie de masse. Avec une telle structure, non-spécifique protéines liant aux talons aura un ratio de lourds à la lumière de 1:4 ; Lorsque les proies vrais aura un ratio de 1:1,38.
Nous avons ré-analysé leurs données AP-MS à l’aide de la base de données OpenProt ; les appâts inclus trois protéines endogènes (PTPN14, JIP3 et IQGAP1), et deux surexprimée protéines (RAF1 et RNF41). Étant donné que les expériences utilisé SILAC, le workflow de Galaxy pour la quantification des protéines a été utilisé (S3 de matériel supplémentaire, Figure 2). Le flux de travail a été exécuté à l’aide de l’ensemble de la base OpenProt (OpenProt_all) ou une base de données restreinte de l’OpenProt (OpenProt_2pep, dont seules les protéines précédemment détectés avec un minimum de deux peptides uniques).
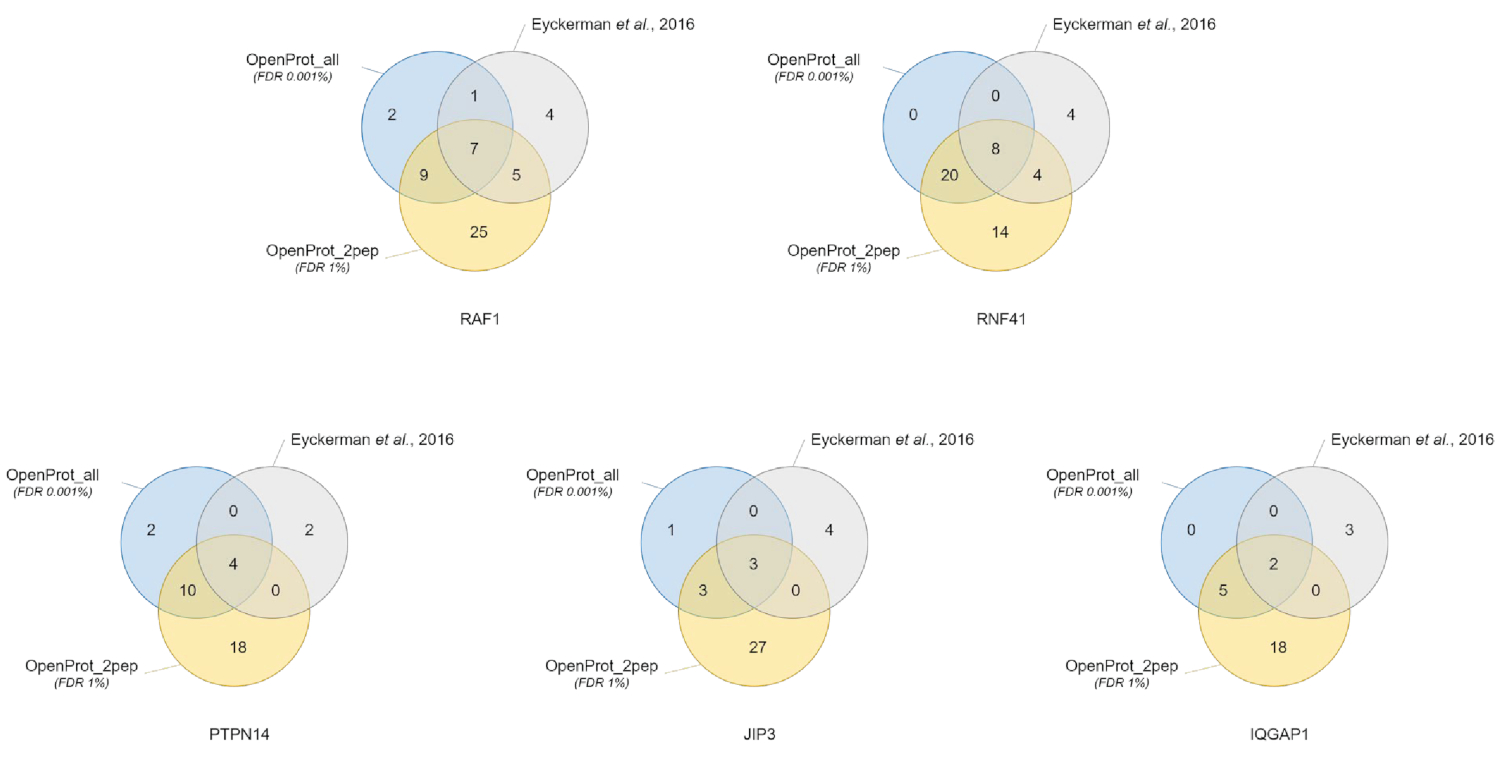
Quantification et identification des protéines étaient bonnes et reproductibles dans les différentes bases de données utilisées. Comme illustré à la Figure 3, la plupart des protéines identifiées dans le document original ont également identifiés à l’aide de la OpenProt_2pep ou la OpenProt_all de base de données (une liste détaillée est disponible dans S5 de matériel supplémentaire). Ce résultat montre que le pipeline décrite ici et le OpenProt bases de données sont capables de produire la protéine identification et la quantification comparable à celle des actuelles procédures fondées sur les bases de données de UniProtKB40. Cependant, l’utilisation de bases de données OpenProt a l’avantage unique de détection de roman et indétectables protéines, tel que démontré dans ce cas étudier.
11 bien supporté de protéines (1 isoforme et 10 AltProts), mais actuellement non annotés en bases de données, ont été identifiés dans l’ensemble de tous les ensembles de données, avec des peptides confiants, à l’aide de la base de données OpenProt_2pep (toutes les accessions de protéines, ainsi que le nombre de supporter peptides, sont disponibles en S5 de matériel supplémentaire). Cette base de données permet l’utilisation d’un traditionnel 1 % FDR que l’augmentation de l’espace de recherche reste modérée. Ces 11 protéines ne figuraient pas dans l’étude originale, tels qu’ils étaient absents de la base de données.
29 nouvelles protéines (16 isoformes et 13 AltProts) ont été découverts dans l’ensemble de tous les ensembles de données, avec des peptides confiants, à l’aide de la base de données OpenProt_all (toutes les accessions de protéines, ainsi que le nombre de supporter des peptides, sont disponibles dans S6 de matériel supplémentaire ). Comme illustré à la Figure 3, le FDR strict recommandé n’affecte pas l’identification de protéines plus confiants, bien qu’il ne diminue pas le nombre total des protéines identifiées. Relativement à la base de données OpenProt_2pep, peut être identifié en toute confiance un plus grand nombre de nouvelles protéines. Toutes ces nouvelles protéines sont absentes de la base de données OpenProt_2pep. Cela met en évidence le rôle crucial de la base de données choisie pour la protéomique axée sur le MS.
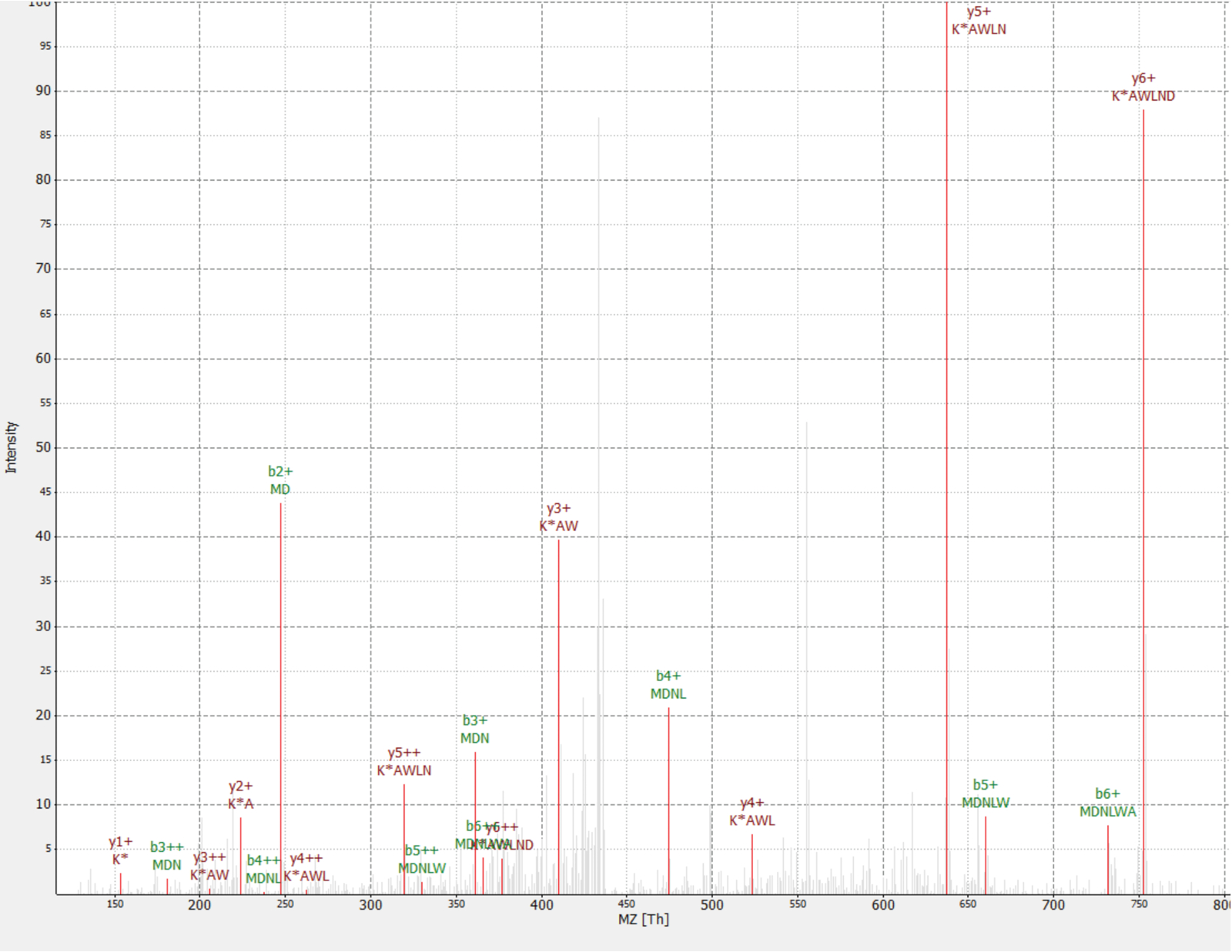
Une nouvelle protéine détectée comme un interacteur de la protéine RAF1 (IP_637643). En utilisant le site de OpenProt, on peut voir cette protéine n’avait pas été détectée par MS, ni ribosomes profilage jusqu'à présent (OpenProt v1.3). La protéine est 46 acides aminés et ne peut donner deux peptides uniques digestion trypsique. Le peptide détecté dans l’AP-MS RAF1 dataset (fraction 18) a un spectre de bonne qualité, comme illustré à la Figure 4et affiche un ratio de lourds à la lumière de 1,09. La protéine est codée dans le gène NANOGNBP1 , qui est un pseudogène de NANOGNB. La transcription (ENST00000448444), actuellement annotée comme non codantes, a été détectée dans plusieurs tissus selon le portail GTEx40. La protéine contient un domaine fonctionnel prévu associé à ADN de liaison (Gene Ontology GO : 0003677)41.

Figure 1 : Choix graphique analyse protéomique des bases de données. Analyses de données de MS, notamment le choix de la base de données, dépendent des objectifs de recherche. Trois objectifs communs figurent en bleu (protéomique classique pipeline), vert (recherche protéomique exhaustive) et orange (protéomique découverte). Chaque objectif repose sur une base de données appropriée et pipeline. Un outil unique d’identification peut être utilisé pour un protéomique exhaustive et classique pipelines. Pour le pipeline de découverte de protéomique, nous recommandons fortement d’utiliser plusieurs moteurs d’identification. FDR recommandées est indiquées en rouge et tailles de base de données de protéine sont indiqués dans les cases grises. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

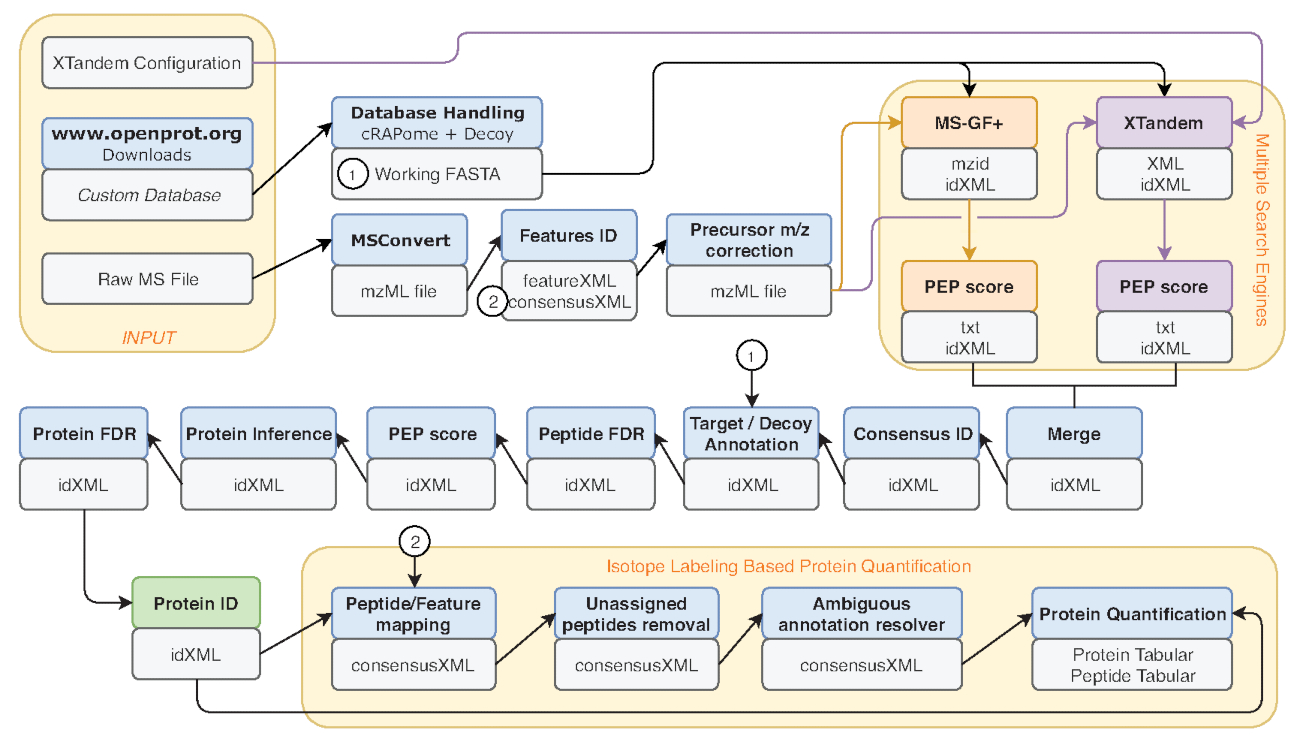
Figure 2 : Représentation graphique du flux de la galaxie utilisé. Représentation par étapes du workflow analyse protéomique utilisé pour ré-analyse de Eyckerman et coll. données38. Fichiers d’entrée, recherche de peptide et quantification des protéines sont indiquées par des boîtes orange. Boîtes bleues correspondent aux outils utilisés et les zones gris correspondent aux fichiers de sortie générés. Les moteurs de recherche différents (MS-GF + et X ! Tandem) sont indiquées par des couleurs différentes (respectivement rouges et violets) ainsi que les flèches indiquant leurs intrants nécessaires et les sorties. La boîte verte met en évidence l’outil générant une liste des identifications de protéine. Lorsque plusieurs sorties sont générés, celle utilisée pour les étapes en aval est indiqué comme le plus proche de la flèche. Ce flux de travail est librement disponible en S2 de matériel supplémentaire. Le X ! Fichier de configuration de paramètres par défaut en tandem est disponible sur S4 de matériel supplémentaire. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : Comparaison d’identification interactor par appât à l’aide de bases de données différentes. Les diagrammes de Venn des identifications de protéine à l’aide de la OpenProt plus confiant des bases de données (en orange, pièces justificatives de minimums 2 peptides uniques, OpenProt_2pep) avec un 1 % rad, ou le OpenProt de toute base de données (en bleu, OpenProt_all) avec un 0,001 % FDR, ou comme indiqué à l’origine de papier (en gris)38. Chaque diagramme correspond aux Interactiens identifiés pour l’appât mentionné : RAF1, RNF41, PTPN14, JIP3 et IQGAP1. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : Spectre MS/MS d’identifié MDNLWAK(13, 6) peptide de la protéine nouvelle IP_637643. L’intensité est relative (0 à 100 %). Les sommets sélectionnés sont indiquées en rouge, les annotations d’ions y sont en noir rouge et b les annotations des ions en vert. Extrait de la TOPPview logiciel34. Précurseur erreur = 2,70 ppm, score de PEP = 0,12. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Terme | Définition | Référence |
| ORF alternatif (AltORF) | ORF non canoniques ne sont actuellement pas annoté dans les annotations du génome, mais annoté dans OpenProt. | 15 |
| Référence ORF (RefORF) | ORF canonique annoté dans les annotations de génome et OpenProt. | 15 |
| Autres protéines (AltProt) | nouvelle protéine codée par un AltORF, avec aucune similitude significative avec un RefProt. Préfixe de l’adhésion : IP_. | 15 |
| Protéine de référence (RefProt) | protéines actuellement annotée dans des bases de données de protéine séquence tels que UniProtKB, Ensembl ou NCBI RefSeq, ainsi que dans OpenProt. | 15 |
| Isoforme roman | nouvelle protéine codée par un AltORF, avec une similitude importante avec un RefProt. Préfixe de l’adhésion : II_. | 15 |
| OpenProt_2pep base de données | contient la séquence de tous les RefProts et les nouvelles protéines prévues par OpenProt, déjà détecté avec un minimum de 2 peptides uniques. | 15 |
| OpenProt_1pep base de données | contient la séquence de tous les RefProts et les nouvelles protéines prévues par OpenProt, déjà détecté avec un minimum de 1 peptide unique. | 15 |
| OpenProt_all base de données | contient la séquence de tous les RefProts et les nouvelles protéines prévues par OpenProt. | 15 |
Tableau 1 : Définition des termes utilisés au OpenProt et dans le protocole
S1 de matériel supplémentaire : "workflow" Galaxy pour la manipulation de la base de données. Cela va ajouter les séquences CRAPome et leurre (inverses) à la base de données d’entrée. Sortie est un fichier de Fasta. S’il vous plaît cliquez ici pour télécharger.
S2 de matériel supplémentaire : "workflow" Galaxy pour l’identification des protéines. Il identifiera des protéines à partir d’un fichier de données de spectrométrie de masse à l’aide de deux moteurs de recherche (MS-GF + et X ! Tandem). Chaque paramètre peut être réglé comme vous le souhaitez avant d’exécuter le flux de travail. S’il vous plaît cliquez ici pour télécharger.
S3 de matériel supplémentaire : "workflow" Galaxy pour la quantification de protéines en utilisant des isotopes stables, étiquetage (SIL). Cela va identifier et quantifier les protéines à partir d’un fichier de données de spectrométrie de masse à l’aide de deux moteurs de recherche (MS-GF + et X ! Tandem). Chaque paramètre peut être réglé comme vous le souhaitez avant d’exécuter le flux de travail. S’il vous plaît cliquez ici pour télécharger.
S4 de matériel supplémentaire : X ! Fichier de configuration des paramètres de défaut en tandem. XML ce fichier est nécessaire pour faire fonctionner le X ! Outil TandemAdapter sur la plate-forme de la galaxie. S’il vous plaît cliquez ici pour télécharger.
S5 de matériel supplémentaire : quantifiée des protéines à partir des ensembles de données iMixPro. Fichiers de données de Eyckerman Al 201638 ont été traitées à l’aide de bases de données OpenProt et protéines chiffrés sont répertoriés pour chaque condition. Les appâts sont PTPN14, JIP3, IQGAP1, RAF1 et RNF41. Gène noms indiqués en vert correspondent aux protéines également identifiés dans l’original papier38. Gène noms indiqués en orange correspondent aux Interactiens connus selon BioGrid qui n’étaient pas indiqués dans le document original. Noms de gène indiqués en bleu correspondent aux nouvelles protéines identifiées comme interacteurs (le numéro d’ordre de protéine correspondante est indiqué entre parenthèses). Gène noms indiqués en gris clair et italique correspondre à des contaminants susceptibles (protéines de kératine). S’il vous plaît cliquez ici pour télécharger.
S6 de matériel supplémentaire : identifié de nouvelles protéines de datasets iMixPro. Fichiers de données de Eyckerman Al 201638 ont été traitées à l’aide de bases de données OpenProt et nouvelles protéines identifiées sont répertoriées pour chaque condition. Les appâts sont PTPN14, JIP3, IQGAP1, RAF1 et RNF41. Protéine adhésion sont listés, commençant par II_ nouvelles isoformes d’une protéine connue et avec IP_ de nouvelles protéines de l’ORF de rechange (AltProt). Le nombre de supporter les peptides sont indiqués entre parenthèses. S’il vous plaît cliquez ici pour télécharger.
Discussion
Lors de l’analyse des données de spectromètres de masse, la qualité de l’identification des protéines s’appuie en partie sur l’exactitude de la base de données utilisée6,20. Les approches actuelles utilisent traditionnellement UniProtKB bases de données, mais ceux-ci soutiennent le modèle d’annotation du génome d’un ORF unique par transcription et une longueur minimale de 100 codons (à l’exception des exemples précédemment démontrées)40. Plusieurs études rapportent les lacunes de ces bases de données avec la découverte de l’ORF fonctionnelle du prétendument non codantes régions8,11,12,13. Maintenant, OpenProt permet l’identification de protéines plus exhaustive qu’il puise les annotations de transcriptome plusieurs séquences protéiques. OpenProt récupère RefSeq NCBI (GRCh38.p7) et les transcriptomes Ensembl (GRCh38.83) et les annotations de UniProtKB (UniProtKB-SwissProt, 2017-09-27)40,42,43. Lorsque les annotations actuelles présentent peu de chevauchement, OpenProt affiche donc une vue plus exhaustive du paysage protéomiques potentiel que lorsque limité à une annotation15.
En outre, comme OpenProt applique un modèle de polycistronique, il permet plusieurs annotations de protéine par transcription. Pour des raisons de statistiques et de calculs, OpenProt détient toujours un seuil de longueur minimale de 30 codons15. Pourtant, il prédit des milliers de séquences de protéines nouvelles, élargissant ainsi le champ des possibilités pour l’identification des protéines. Avec cette approche, OpenProt prend en charge les découvertes de protéomique d’une manière plus systématique.
La qualité de l’identification des protéines peut également être affectée par les paramètres qui sont utilisés. Analyse protéomique axée sur le MS détiennent généralement un 1 % de protéines FDR. Toutefois, la base de données OpenProt entier contient environ 6 fois plus d’entrées (Figure 1). Pour expliquer cette augmentation substantielle dans l’espace de recherche, nous recommandons d’utiliser un FDR plus strict de 0,001 %. Ce paramètre a été optimisé à l’aide d’études comparatives et évaluation manuelle des spectres choisis au hasard15. Faux positif sont toujours une possibilité, bien que, et nous encourageons inspection approfondie et la validation de pièces justificatives pour une nouvelle protéine. Une norme recommandée pourrait être l’identification d’une protéine de deux séries différentes de MS, comme données de base et des faux positifs varient entre les ensembles de données15.
Le pipeline fournis ici et utilisés pour l’étude de cas peut être modifié aussi heureux d’adapter le protocole expérimental et paramètres. Nous recommandons l’utilisation de plusieurs moteurs de recherche car elle augmente la sensibilité et la sensibilité du peptide identification32. En outre, nous encourageons l’utilisation de la base de données correspondant le mieux au but expérimental (Figure 1). Comme à l’aide de la OpenProt toute base de données est livré avec un FDR rigoureux, véritables identifications peuvent être perdues. Ainsi, l’ensemble de la base devrait être destiné à la découverte de nouvelles protéines, tandis que profilage protéomique classique devrait utiliser les petites bases de données OpenProt (tels que les OpenProt_2pep utilisés dans l’étude de cas ci-dessus).
OpenProt prévoit actuellement des séquences commençant par un codon ATG, alors que plusieurs études ont souligné initiation de la traduction à autres codons44,45. Lorsqu’une nouvelle protéine est identifiée par un ou plusieurs peptides uniques, il est possible du que codon d’initiation véritable n’est pas l’ATG présumée. Utilisateurs peuvent chercher des preuves de traduction sur le site OpenProt. Actuellement, OpenProt signale uniquement les événements de traduction si elles concernent l’ensemble des protéines prédites séquence (100 % de chevauchement)15. Ainsi, l’absence de preuve de la traduction ne signifierait pas la protéine n’est pas traduite, mais que le codon de début ne peut pas être l’ATG présumée.
Malgré ses limites actuelles, OpenProt offre une vue plus exhaustive du potentiel de codage des génomes eucaryotes. OpenProt bases de données favorisent les découvertes de la protéomique et la compréhension des fonctions de la protéomique et les interactions. Les développements futurs de la base de données OpenProt comprendra annotation d’autres espèces, preuve de la traduction de non-ATG start codon et le développement d’un pipeline d’inclure des protéines nouvelles dans l’ensemble du génome et des études de séquençage de l’exome.
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts.
Remerciements
Nous remercions Vivian Delcourt pour son aide, des discussions et des conseils sur ce travail. X.R. est membre du Fonds de Recherche du Québec Santé FRQS appuyés par le Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. Cette recherche a été financée par une chaire de recherche du Canada en protéomique fonctionnelle et découverte de la protéine de roman à grant X.R. et IRSC MOP-137056. Nous remercions l’équipe Calcul Québec et calcul Canada pour leur soutien à l’utilisation de la mp2 supercalculateur de l’Université de Sherbrooke. Du supercalculateur mp2 est financée par la Fondation Canada de l’Innovation (FCI), le ministère de l’Économie, de la science et de l’innovation du Québec (MESI) et les Fonds de Recherche du Québec - Nature et technologies (FRQ-NT). Le serveur Galaxy qui a été utilisé pour des calculs de la protéomique est en partie financé par Collaborative Research Centre 992 médical épigénétique (subvention DFG SFB/992/1 2012) et ministère fédéral allemand de l’éducation et la recherche (BMBF accorde 031 RBC A538A/A538C, 031L0101B De /031L0101C. NBI-epi, 031L 0106 de. ESCALIER (de. NBI)).
matériels
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
Références
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.