
Method Article
Massenspektrometrie-basierte Proteomics Analysen mithilfe der OpenProt-Datenbank zu enthüllen neue Proteine übersetzt aus dem nicht-kanonischen Open Reading Frames
In diesem Artikel
Zusammenfassung
OpenProt ist eine frei zugängliche Datenbank, die eine Polycistronic Modell der eukaryotic Genome erzwingt. Hier präsentieren wir ein Protokoll für die Verwendung von OpenProt Datenbanken bei der Massenspektrometrie Datasets zu verhören. Mit OpenProt kann Datenbank zur Auswertung von Proteomic Experimenten für Entdeckung von Roman und bisher nicht nachweisbar Proteinen.
Zusammenfassung
Genom-Anmerkung steht im Mittelpunkt der heutigen Proteomic Forschung wie die Umrisse der Proteomik Landschaft zieht. Traditionelle Modelle open Frame (ORF) Anmerkung lesen zwei willkürliche Kriterien auferlegen: eine Mindestlänge von 100 Codons und einem einzigen ORF pro Protokoll. Eine wachsende Zahl von Studien berichten jedoch Expression von Proteinen aus angeblich nicht-kodierenden Regionen, die Genauigkeit der aktuellen Genom Anmerkungen eine Herausforderung. Dieser Roman Proteine gefunden wurden kodiert entweder in nicht-kodierende RNAs, 5' oder 3' untranslatierten Regionen (wo) der mRNAs oder überlappende einer bekannten kodierenden Sequenz (CDS) in Alternative ORF. OpenProt ist die erste Datenbank, die eine Polycistronic Modell für eukaryotische Genome, erzwingt Annotation von mehrere ORFs pro Protokoll ermöglicht. OpenProt ist frei zugänglich und bietet benutzerdefinierte Downloads von Proteinsequenzen über 10 Arten. Mit OpenProt Datenbank für Proteomik Experimente ermöglicht neuartige Proteine Entdeckung und betont den Polycistronic Charakter von eukaryotischen Genen. Die Größe der OpenProt-Datenbank (alle vorausgesagt Proteine) ist beträchtlich und Konto für die Analyse getroffen werden muss. Allerdings erhalten Benutzer mit entsprechenden false Discovery Rate (FDR) Einstellungen oder den Einsatz einer eingeschränkten OpenProt Datenbank, einen realistischeren Blick auf die Proteomik-Landschaft. OpenProt ist eine frei verfügbare Tool, die Proteomic Entdeckungen fördern wird.
Einleitung
In den letzten Jahrzehnten geworden Massenspektrometrie (MS)-basierte Proteomics die goldenen Technik Proteome von eukaryotischen Zellen1,2,3,4,5zu entschlüsseln. Diese Methode beruht auf aktuellen Genom Anmerkungen Sequenz Referenzdatenbank Protein zu generieren, die den Umfang der Möglichkeiten6,7,8beschreibt. Allerdings halten Genom Anmerkungen willkürliche Kriterien für ORF-Annotation, z. B. einer Mindestlänge von 100 Codons und einem einzigen ORF pro Protokoll9,10. Eine wachsende Zahl von Studien fordern Sie das aktuelle Modell der Annotation und Entdeckungen der Genomsequenz funktionale ORFs in eukaryotischen Genomen8,11,12,13zu melden, 14. Diese neuartige Proteine codiert in angeblich nicht-kodierende RNAs befinden, in der 5' oder 3' unübersetzt Regionen (UTR) mRNAs oder Überschneidungen der kanonischen kodierenden Sequenz (cCDS) in einem alternativen Rahmen. Obwohl die meisten dieser Entdeckungen glückliche gewesen sind, zeigen sie die Vorbehalte der aktuellen Genom-Anmerkungen und die Polycistronic Art der eukaryotischen Genen8.
Hier heben wir die Verwendung von OpenProt Datenbanken für MS-basierte Proteomics. OpenProt ist die erste Datenbank eine Polycistronic Annotation Modell für eukaryotische Transkriptom festzuhalten. Es ist frei verfügbar bei www.openprot.org15. Ein Teil davon vorhergesagt wäre ORFs zufällig und nicht-funktionale, weshalb OpenProt experimentelle und funktionalen Beweise Vertrauen stärken kumulieren. Experimentelle Beweise sind Protein-Expression (von MS) und Übersetzung Beweis (durch Ribosom Profilierung)15. Funktionelle Beweis einschließen Protein Orthopädie (mit einer In-Paranoid wie Ansatz) und funktionale Domäne Vorhersage15.
OpenProt bietet die Möglichkeit, mehrere Datenbanken, herunterladen, die nur gut unterstützte Proteine auf maßgeschneiderte Datenbanken enthalten. Hier präsentieren wir eine Pipeline für die Verwendung von OpenProt Datenbanken und bieten Einblicke in die Datenbank zu wählen, wenn man bedenkt das experimentelle Ziel. Die Proteomik Analyse Pipeline hier vorgestellten ist von Galaxy Framework unterstützt, da es Open Access und einfach zu bedienende, aber die Datenbanken können mit jedem Workflow16,17,18arbeiten. Wir präsentieren auch wie mithilfe die OpenProt-Website für weitere Informationen über neuartige Proteine erkannt durch MS. Using OpenProt Datenbanken sammeln eine umfassendere Sicht der Proteomik Landschaft bieten wird und die Proteomik und Biomarker Entdeckungen in fördert systematischer als bisherige Methoden.
Dieses Protokoll wird die Verwendung von OpenProt Datenbanken15 hervorgehoben, wenn MS Datasets zu befragen; Es prüft nicht das Design des Experiments überprüft selbst, die gründlich wurde an anderer Stelle20,21,22. In dem Bemühen um vollständig Open Source bleiben ist das Protokoll frei verfügbar (Ergänzende Material S1–S4). Zur besseren Lesbarkeit sind alle Begriffe, die in OpenProt und hiermit in diesem Protokoll in Tabelle 1festgelegt.
Protokoll
1. OpenProt Datenbank herunterladen
Hinweis: Benutzerdefinierte Datenbanken basierend auf RNA-Seq-Daten zum Beispiel auch erzielt werden und das Verfahren ist ausführlich im zweiten Abschnitt dieses Protokolls. Wenn eine benutzerdefinierte Datenbank benötigt wird, überspringen Sie bitte mit dem nächsten Abschnitt.
- Besuchen Sie die OpenProt Website: www.openprot.org und öffnen der Download-Seite mit dem Link im Menü der Startseite.
- Klicken Sie auf die Arten von Interesse, die auf Basis der analysierten experimentelle Daten.
- Klicken Sie auf die gewünschte Proteintyp.
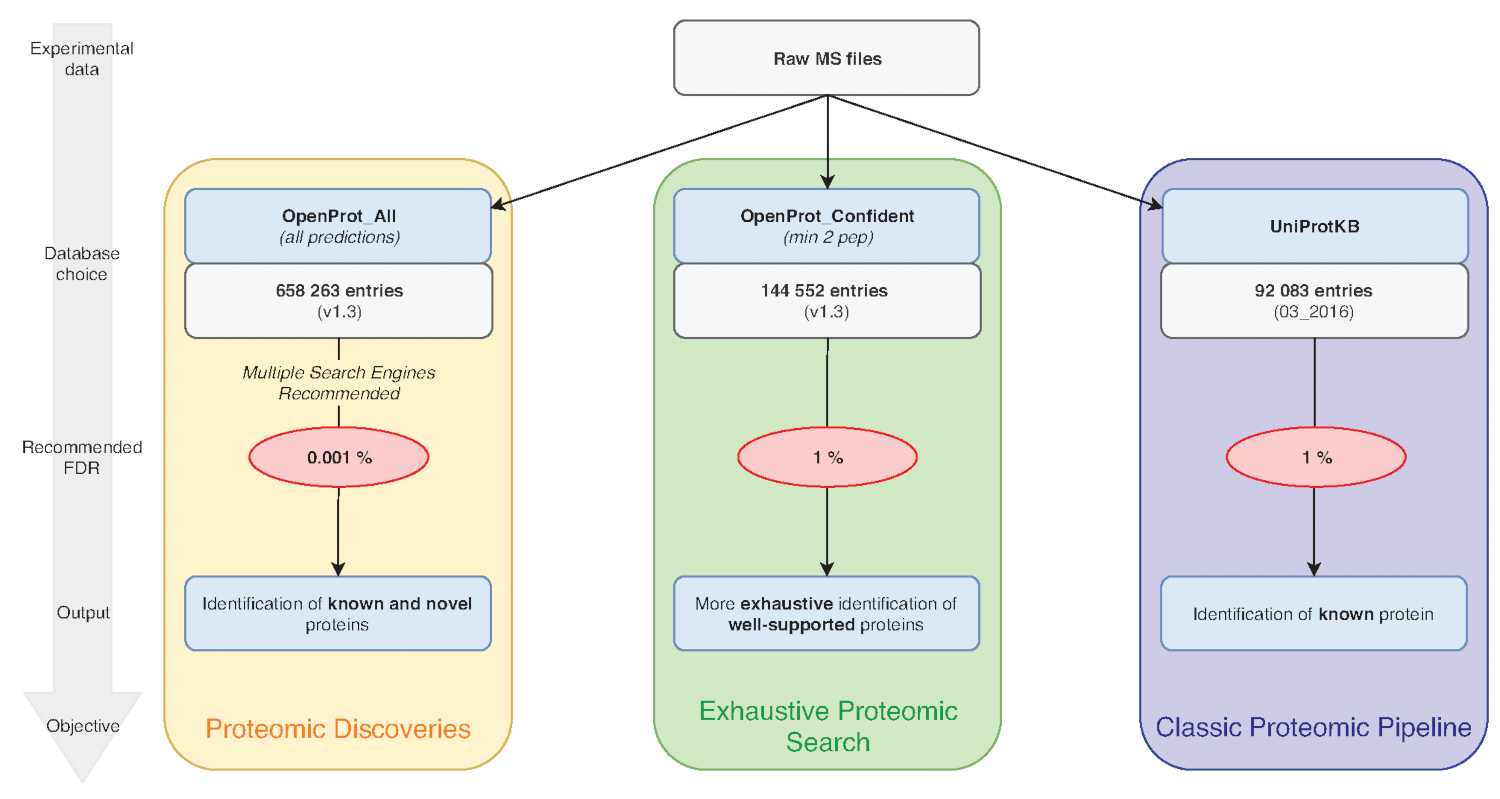
Hinweis: OpenProt bietet drei Klassifizierungen: RefProt, Isoformen und AltProt. Wie in Abbildung 1dargestellt, wird dieser Parameter auf das Forschungsziel variieren.- Klicken Sie auf RefProt allein , nur bekannte Proteine enthalten Dateien erzeugen.
- Klicken Sie auf AltProt und Isoformen Dateien enthalten nur neue Proteine - entweder neuartige Isoformen von bekannten Proteinen (Isoformen) oder durch eine Alternative ORF (AltProts) kodiert zu generieren. Bitte beachten Sie, dass OpenProt eine ORF-Mindestlänge von 30 Codons15erzwingt.
- Klicken Sie auf AltProts, Isoformen und RefProts Dateien, die alle Protein-Arten in der OpenProt Datenbank - bekannte und neuartige Proteine zu generieren.
- Falls verfügbar, klicken Sie auf die Anmerkung von welches, die Protein Sequenzen erstellt werden.
Hinweis: OpenProt bietet eine umfassendere Proteomic Landschaft durch die Kombination mehrere Anmerkungen. Transkriptom Anmerkungen haben eine minimale Überlappung; so kann die ausgewählten Anmerkung der visualisierten Proteomic Profil15,23wesentlich beeinflussen. - Klicken Sie auf das Niveau der sonstige Nachweise zur Protein-Prüfung. Wie in Abbildung 1dargestellt, wird dieser Parameter auf das Forschungsziel variieren.
- Klicken Sie auf mindestens zwei einzigartige Peptide erkannt , Dateien, die nur die zuversichtlichsten Proteine zu generieren.
Hinweis: Ein Kriterium von zwei einzigartigen Peptiden gilt heute als Goldstandard in der Proteomik für Protein-Expression. Wenn experimentelle Ziel ist es, bekannte und gut unterstützte Proteine zu erkennen, empfiehlt sich die Verwendung dieses Parameters. - Klicken Sie auf mindestens eine einzigartige Peptide erkannt erzeugen Dateien mit Proteinen, die bereits mindestens einmal unter die Massenspektrometrie Experimente neu analysiert, indem OpenProt gesehen haben.
Hinweis: Dies ermöglicht eine Berücksichtigung der kürzeren Länge des AltProts und die Wahrscheinlichkeit, dass einige von ihnen nur eine einzigartige tryptic Peptid8,11enthalten kann. - Klicken Sie auf alle vorausgesagt , um Dateien mit allen OpenProt Vorhersagen zu generieren.
Hinweis: Diese Einstellung wird nur empfohlen wenn experimentelle zielt darauf ab, neuartige Proteine (Abbildung 1) zu entdecken. Den nachfolgenden deutlichen Anstieg der Suche Raum fordert eine angepasste Analyse Pipeline wie unter7,15besprochen.
- Klicken Sie auf mindestens zwei einzigartige Peptide erkannt , Dateien, die nur die zuversichtlichsten Proteine zu generieren.
- Klicken Sie auf das gewünschte Dateiformat herunterladen. Proteomic Analysen wählen Sie in der Fasta (Protein)-Datei. Die Readme-Datei enthält alle notwendigen Informationen über das Dateiformat.
2. benutzerdefinierte OpenProt Datenbank herunterladen
Hinweis: Dieser Abschnitt beschreibt, wie eine benutzerdefinierte Datenbank zu erhalten. Wenn keine benutzerdefinierte Datenbank benötigt wird, mit dem nächsten Abschnitt fortfahren.
- Besuchen Sie die Website OpenProt (www.openprot.org) und öffnen Sie die Suchseite, die über den Link im Menü der Startseite.
- Klicken Sie auf die Arten von Interesse, die basierend auf den experimentellen Daten analysiert.
- Geben Sie eine Liste von Genen oder Abschriften von Interesse.
- Wenn Sie eine Liste von Genen zu verwenden, geben Sie ihn im Abfragefeld gen .
- Wenn Sie eine Liste der Protokolle verwenden, geben Sie ihn im Feld Protokoll -Abfrage.
- Kästchen Sie, die für die gewünschte Datenbank gilt.
- Klicken Sie nicht auf einem beliebigen Feld zu einer Tabelle mit allen Arten von Proteinen, die von OpenProt unterstützt: RefProt, Isoformen und AltProts.
- Klicken Sie auf nur Proteine mit experimentellen Beweis zeigen , erhalten eine Tabelle mit allen Arten von Proteinen (Isoformen, RefProts und AltProts), die mindestens einmal durch MS entdeckt wurden bzw. für welche Übersetzung Beweis vom Ribosom gesammelt worden Profilerstellungsdaten.
- In ähnlicher Weise, klicken Sie auf zeigen nur Proteine von MS erkannt oder zeigen nur Proteine vom Ribosom Profilierung erkannt , erhalten Sie eine Tabelle mit allen Arten von Proteinen, die mindestens einmal durch MS oder Ribosom Profilierung bzw. entdeckt wurden.
- Klicken Sie auf nur AltProts zu zeigen oder zeigen nur Isoformen , um eine Tabelle mit nur AltProts oder nur Isoformen bzw. zu erhalten.
- Klicken Sie auf beide zeigen nur AltProts und zeigen nur Isoformen , eine Tabelle mit beiden Arten von Proteinen zu erhalten.
Hinweis: Alle Filter-Kombinationen sind möglich.
- Sobald alle gewünschten Parameter festgelegt sind, klicken Sie auf suchen. Die Tabellenausgabe erscheint unterhalb der Suchfelder Abfrage.
- Klicken Sie auf die Schaltfläche " Download Fasta " in der rechten oberen Ecke der Ausgabetabelle. Dies generiert eine Fasta-Datei mit allen Proteinen infolge der abgefragten Liste von Genen oder Abschriften.
- Bitte beachten Sie, dass aus rechnerischen Gründen OpenProt hält maximal 2.000 Elemente werden abgefragt (Gene oder Abschriften) zu einem Zeitpunkt. Im Falle einer Liste oberhalb dieser Grenze kann mehrere Fasta erzeugt und dann verkettet (wie unten aufgeführt); oder laden Sie die ganze OpenProt-Datenbank und Filtern Sie die erhaltene Datei wie gewünscht.
- Bin die ganze Liste von Genen oder Abschriften in Sub-Listen von bis zu 2.000 Einträge. Für jede Teilliste Herunterladen einer Fasta-Datei wie oben (Schritt 3.3 bis 3.6) beschrieben.
- Melden Sie sich bei der Europäischen Galaxy-Instanz (oder einer anderen Instanz wo Proteomics Werkzeuge zur Verfügung stehen), https://usegalaxy.eu/.
- Erstellen Sie eine neue Geschichte, und importieren Sie alle heruntergeladenen OpenProt Datenbanken (eine pro Teilliste von Genen oder Abschriften) durch Klicken auf das Upload-Logo am linken oberen Rand des Bildschirms.
- Verwenden Sie das Fasta verschmelzen Dateien und Filter einzigartige Sequenzen Tool, entwickelt von den Entwicklern von GalaxyP (https://github.com/galaxyproteomics/). Wählen Sie die Option alle Fasta zusammenführen und geben Sie alle importierten OpenProt Datenbanken.
Hinweis: Jedes Werkzeug kann durchsucht werden, indem mit der Abfrage-Box auf der linken Seite des Bildschirms - Wählen Sie die Option nur Beitritt bewerten Sequenz Unicity und kopieren Sie die OpenProt-ID Parse-Regel (>(.*) \ |), dann klicken Sie auf Ausführen.
- Beachten Sie, dass alle Dateien in einer einzigen Fasta-Datei ohne Redundanz verkettet worden haben, der nun im Bedienfeld "Verlauf" auf der rechten Seite des Bildschirms angezeigt wird. Dies stellt die Datenbank.
(3) Handhabung von Datenbanken
Hinweis: ab sofort wird die Galaxy-Plattform verwendet werden, aber die gleichen Grundsätze können auch auf andere Proteomik-Software.
- Melden Sie sich bei der Europäischen Galaxy-Instanz (oder einer anderen Instanz wo Proteomics Werkzeuge zur Verfügung stehen), https://usegalaxy.eu/.
- Erstellen Sie eine neue Geschichte, und importieren Sie die heruntergeladenen OpenProt Datenbank durch Klicken auf das Upload-Logo am linken oberen Rand des Bildschirms.
- Gehen Sie zur Seite Workflow und Importieren des Datenbank Handhabung Workflows (Ergänzende Material S1) durch Klick auf das Upload-Logo links oben in der mittleren Spalte.
- Klicken Sie auf den Workflow ausführen und wählen Sie die importierten OpenProt-Datenbank als Eingabe.
Hinweis: Dieser Workflow OpenProt Fasta CRAPome Repository hinzufügen und generieren Lockvogel Sequenzen (umgekehrte Sequenzen)24. Wenn eine Shuffle-Köder-Liste gewünscht wird, kann es durch Änderung dieses Parameters auf dem DecoyDatabase-Werkzeug erfolgen. - Benennen Sie die erhaltenen Fasta-Datei etwas Sinnvolles. Die Datenbank ist bereit, für Proteomics Analysen verwendet werden.
4. Massenspektrometrie Dateivorbereitung
Hinweis: Die meisten Proteomics Werkzeuge auf Galaxy Instanzen verwenden Sie das MzML-Format und Peptid-Suchmaschinen bevorzugen Daten im Schwerpunkt-Modus.
- Öffnen Sie das frei verfügbare MSConvert-Tool aus der ProteoWizard Suite und laden Sie die Datendatei analysierten25sein.
- Wählen Sie das Verzeichnis für die Ausgabe und das gewünschte Dateiformat zu MzML.
- Ein Peak picking Filter mit der Wavelet-basierten Algorithmus (CWT) auf MS1 und MS2 Ebenen festgelegt, und starten Sie die Konvertierung26.
(5) Peptid und Protein Identifikation/Quantifizierung
Hinweis: Dieser Teil der Pipeline nutzt Werkzeuge aus der OpenMS Suite, ein vielseitiges und einfach zu bedienenden Rahmen18.
- Melden Sie sich bei der Europäischen Galaxy-Instanz (oder einer anderen Instanz wo Proteomics Werkzeuge zur Verfügung stehen), https://usegalaxy.eu/.
- Erstellen Sie eine neue Geschichte und übertragen Sie die zuvor erstellte Datenbank (Schritt 3.5) auf diese neue Geschichte mit Drag-and-Drop.
- Importieren Sie die transformierte MzML Datendatei (Schritt 4.3) durch Klicken auf das hochladen Logo am linken oberen Rand des Bildschirms.
- Gehen Sie auf die Workflow-Seite und importieren Sie den gewünschten Workflow durch Klick auf das Upload-Logo links oben in der mittleren Spalte.
Hinweis: MS Experimente sind unterschiedlich je nach der gewünschten Endausgabe ausgelegt. Workflows dienen hier zwei häufige Designs: Proteinidentifizierung und Protein Quantifizierung anhand stabiler Isotope Kennzeichnung (SIL). Die Galaxy-Instanz enthält jedoch viele andere Werkzeuge, die andere Arten von Proteomic Analysen27,28unterstützen werden.- Importieren Sie für ein Protein-Identifikation-Design den Workflow in Ergänzenden Material S2zur Verfügung gestellt. Bei der Verwendung dieser Workflow, benutzen Sie bitte nicht die Zlip Kompression beim Konvertieren von Dateien (Schritt 4.2)
- Importieren Sie für ein Protein Quantifizierung anhand stabiler Isotope Labelling Designs den Workflow in Ergänzenden Material S3zur Verfügung gestellt.
- Wählen Sie den Workflow ausführen und überprüfen Sie die verschiedenen Parameter.
- Wählen Sie die importierten MzML Datendatei als Eingang und die zuvor erstellte Datenbank (Schritt 3.5) wie die Datenbankdatei Fasta.
- Da der Workflow die X verwendet! Tandem Suche Motor, das X zu importieren! Tandem Standard Konfiguration Datei (sofern in Ergänzenden Material S4)29 durch Klicken auf das Upload-Logo am linken oberen Rand des Bildschirms.
- Der Workflow verwendet mehrere Suchmaschinen (MS-GF + und X! (Tandem). Fügen Sie andere Suchmaschinen oder wählen Sie einen einzigen einfach durch hinzufügen oder entfernen die Tools aus dem Workflow30,31.
Hinweis: Mit mehreren Suchmaschinen wird empfohlen, da es Sensibilität und Empfindlichkeit der Analyse32erhöht. - Um die erhebliche Zunahme der Größe entfallen, wenn die gesamte OpenProt-Datenbank zu verwenden, verwenden Sie eine strenge FDR-15. Die bereitgestellten Workflow ist standardmäßig für eine 0,001 % FDR, ausreichend für die Nutzung der gesamten OpenProt-Datenbank. Für andere Datenbanken kann dies auf einen gewünschten Wert bearbeitet werden.
Hinweis: Achten Sie darauf, die Parameter für die verschiedenen Instrumente je nach dem Massenspektrometer verwendet und das experimentelle Protokoll (Vorläufer Ionen und Fragment Fehler behoben und Variable Änderungen verwendete Enzym usw.) anzupassen.
- Optional, download Ausgabe für jeden Schritt des Workflows für Lagerung oder Qualitätskontrolle Analyse durch einen Klick auf den gewählten Schritt vom Bedienfeld "Verlauf", dann klicken Sie auf das Speichern -Logo, das unten angezeigt wird.
6. Qualitätskontrolle
Hinweis: Da MS-basierte Proteomics das Ergebnis eines komplexen Prozesses wo jeder Schritt muss ist optimiert werden, um reproduzierbare Ergebnisse zu erzielen, ist die Qualitätskontrolle ein notwendiges Verfahren in der Workflow-33.
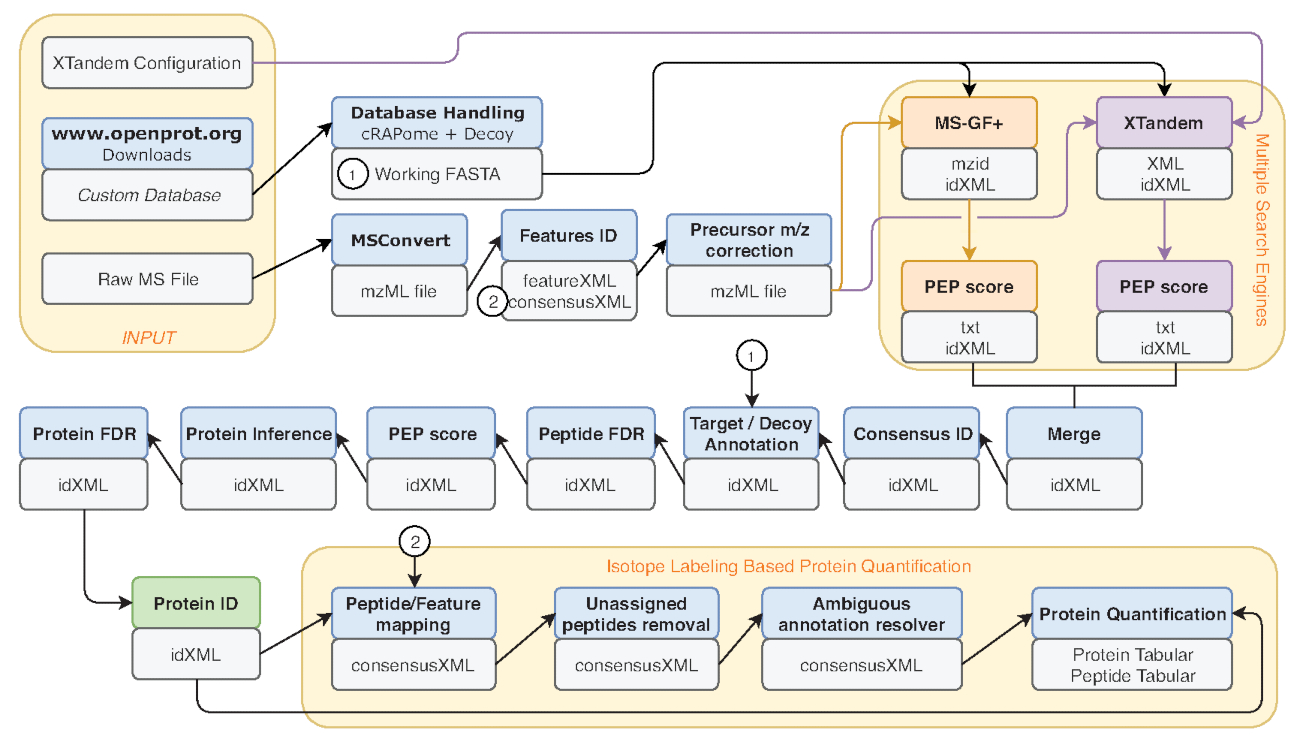
- Mehreren Metriken sind gemeinsame Maßstab für Leistung, wie z. B. die Anzahl der Peptid-Spektrum Übereinstimmungen (PSM), die Anzahl der identifizierten Peptide und Proteine. Führen Sie das Tool Dateiinfo auf der IDFilter-Ausgang (grün in Abbildung 2dargestellt), um solche Metriken zu bieten.
- Zwar gilt nicht für jede Identifikation, vor allem bei großen Datasets sollten Berichte über neue Proteine immer sorgfältig geprüft werden. Inspektion von der Protein-Partitur, die Sequenz-Abdeckung und die Spektren unterstützt die Feststellung ist von entscheidender Bedeutung. Verwenden Sie das TOPPview-Tool aus dem OpenMS-Framework, um dies zu tun; Es ist frei verfügbar und gut dokumentiert18,34,35.
7. OpenProt Datenbank Bergbau
Hinweis: Nach erfolgter eine zuversichtliche Identifizierung ein neuartiges Protein vorhergesagt durch OpenProt (Beitritt Nummern beginnend mit IP_ für AltProts und II_ für neuartige Isoformen) kann weitere biologische Informationen aus der OpenProt-Website-15gesammelt werden.
- Besuchen Sie die OpenProt Website: www.openprot.org und öffnen Sie die Suchseite, die über den Link im Menü Top-Seite.
- Klicken Sie auf die Arten von Interesse (identisch in dem das Protein identifiziert wurde) und geben Sie die Protein-Zbl-Nummer im Abfragefeld Protein .
- Klicken Sie auf suchen, und eine Tabelle mit Basisinformationen über die abgefragten Protein erscheint. Die Tabellenfunktionen: die Protein-Länge (in Aminosäure), Molekulargewicht (kDa) und isoelektrischen Punkt, experimentelle Nachweise von MS oder Ribosom profiling (Übersetzung Beweise, TE) und funktionelle Vorhersagen wie vorhergesagt, Domains und Protein Orthopädie (über der 10 Arten von OpenProt, unterstützt v1. 3). Die Tabelle enthält auch Informationen über das zugehörige gen und Transkript und die Lokalisation des Proteins innerhalb der Niederschrift.
- Klicken Sie auf den Link Details , um weitere Informationen zu sammeln. Die neu eröffnete Seite enthält einen Genom-Browser, die auf die abgefragten Protein und Informationen wie die genomische und transkriptomischen Koordinaten und das Vorhandensein eines Kozak oder Hocheffizienz Übersetzung Einleitung Seite (TIS) Motiv36zentriert ist, 37.
- Klicken Sie auf die Proteine oder DNA- Links von der Registerkarte "Info", um Proteine oder DNA-Sequenzen bzw. zu erhalten.
- Suchen Sie detaillierte Informationen über MS Erkenntnisse zu Ribosom Profilerstellung Erkennung, Erhaltung und identifizierten Protein Domains durch Anklicken der oberen Registerkarten15.
Ergebnisse
Der oben beschriebene Workflow wurde auf eine MS-Dataset auf der PRIDE-Repository38,39angewendet. Die ursprüngliche Studie entwickelt eine Methode (iMixPro), mit stabilen Isotopen Kennzeichnung der Aminosäuren in der Zellkultur (SILAC), um Fehlalarme aus Affinitätsreinigung MS zu beseitigen (AP-MS) Experimente38. Kurz gesagt, besteht darin, ein AP-MS-Experiment Perlen-gebundenen Antikörper, um ein Protein des Interesses (Köder) und seine Interaktoren (Beute) zu holen. Die gesammelten Proteine werden dann verdaut und für MS vorbereitet. Die Sample-Vorbereitung-Methode und die Geräteeinstellungen werden in der ursprünglichen Studie und auf das stolz-Repository (PXD004246) beschrieben. Eine Herausforderung in solchen Experimenten ist die Fülle von false Positives, vor allem aus Proteine binden an die Perlen aber nicht den Köder. Hier wir SILAC verwendet, um unterschiedliche Isotopenverhältnisse zwischen wahren Beute und Fehlalarme zu generieren: 3 Kontrollproben (ohne Köder) kultiviert in Lichtmedium, 1 Probe mit dem Ausdruck des Köders in Lichtmedium kultiviert und 1 Probe mit dem Ausdruck des Köders in schweren Medium kultiviert werden mit Perlen und weiteren Massenspektrometrie Analyse verarbeitet. Mit solch Design haben unspezifische Proteine binden an die Perlen eine schwer-leicht-Verhältnis von 1:4; Wann werden echte Beute haben ein Verhältnis von 1:1-38.
Wir analysiert erneut ihre AP-MS-Daten mithilfe der OpenProt-Datenbank; die Köder enthalten drei körpereigene Proteine (PTPN14, JIP3 und IQGAP1) und zwei ausgedrückt über Proteine (RAF1 und RNF41). Da die Experimente SILAC verwendet, diente der Galaxy-Workflow für Protein Quantifizierung (Ergänzende Material S3, Abbildung 2). Der Workflow wurde mit der ganzen OpenProt (OpenProt_all) oder eingeschränkte OpenProt Datenbank (OpenProt_2pep, einschließlich nur Proteine, die zuvor mit einem Minimum von zwei einzigartigen Peptiden erkannt) ausgeführt.
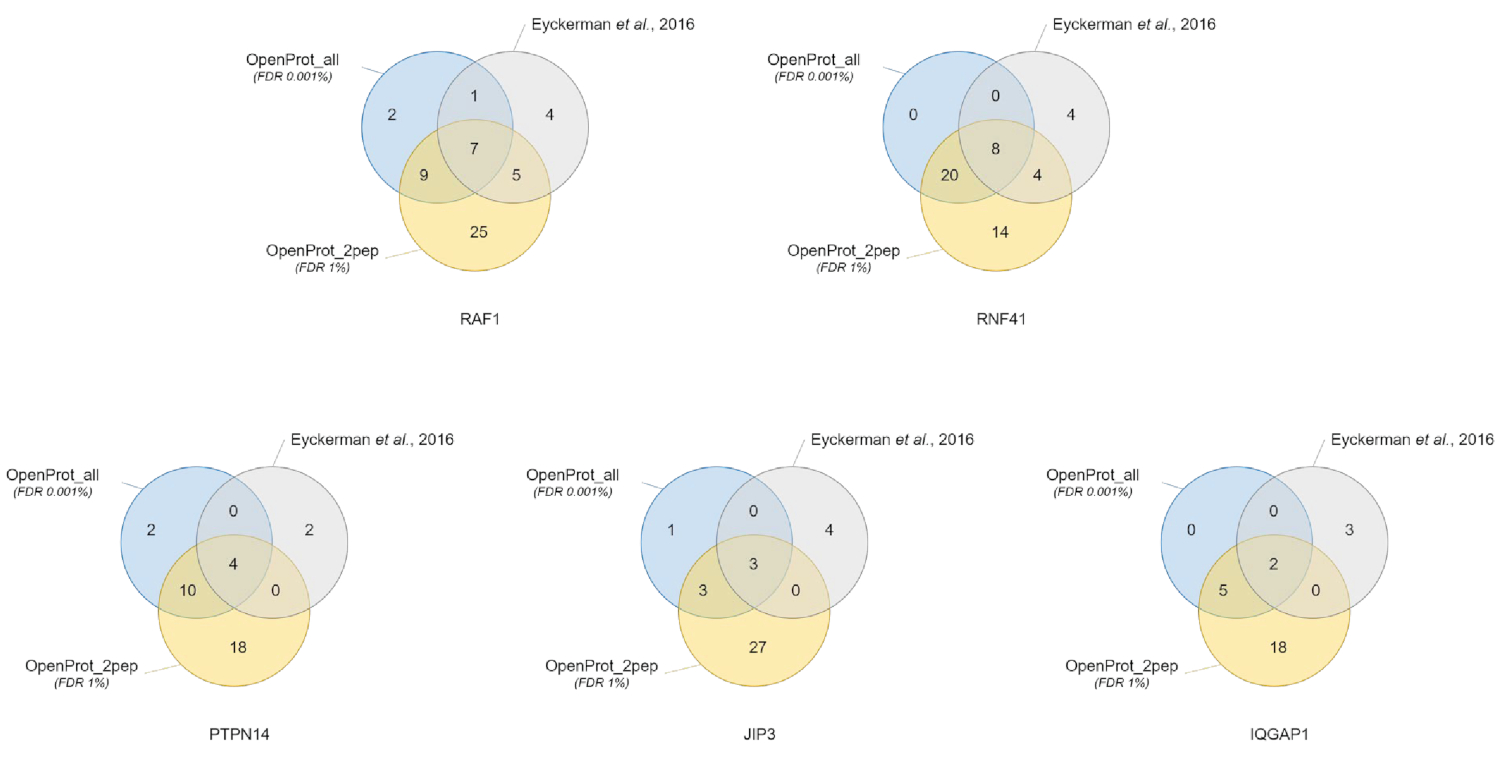
Proteinidentifizierung und Quantifizierung waren gut und reproduzierbar auf die unterschiedlichen verwendeten Datenbanken. Wie in Abbildung 3dargestellt, wurden die meisten Proteine identifiziert in der Originalpublikation auch identifiziert mit Hilfe der OpenProt_2pep oder OpenProt_all Datenbank (eine ausführliche Liste ist verfügbar in Ergänzenden Material S5). Dieses Ergebnis zeigt, dass die hier beschriebenen Pipeline und die OpenProt Datenbanken herstellen Proteinidentifizierung und Quantifizierung des aktuellen Verfahren basierend auf der UniProtKB Datenbanken40vergleichbar sind. Die Verwendung von OpenProt Datenbanken hat jedoch den einzigartigen Vorteil Erkennung neuer und bisher nicht nachweisbar Proteine, wie in diesem Fall zeigt studieren.
11 untermauerten Proteine (1 Isoform und 10 AltProts), aber derzeit nicht kommentierte in Datenbanken wurden über alle Datensätze mit zuversichtlich Peptide, Verwendung der OpenProt_2pep-Datenbank (alle Protein Beitritte, sowie die Anzahl der unterstützen identifiziert. Peptide, gibt es in Ergänzenden Material S5). Diese Datenbank ermöglicht die Verwendung einer traditionellen 1 % FDR als die Erhöhung der Suche Raum bleibt moderat. Diese 11 Proteine wurden nicht in der ursprünglichen Studie identifiziert, als sie abwesend waren aus der Datenbank.
29 neue Proteine (16 Isoformen und 13 AltProts) entdeckte man über alle Datensätze mit zuversichtlich Peptide, Verwendung der OpenProt_all-Datenbank (alle Protein Beitritte, zusammen mit der Anzahl entsprechender Peptide sind erhältlich in ergänzende Material S6 ). Wie in Abbildung 3dargestellt, beeinflusste die empfohlenen strengen FDR die zuversichtlichsten Protein Identifikationen, nicht obwohl es die Gesamtzahl der identifizierten Proteine zu verringern. Vergleichsweise kann mit der OpenProt_2pep-Datenbank, eine höhere Anzahl von neuen Proteine selbstbewusst identifiziert werden. Alle diese neuartige Proteine fehlen aus der OpenProt_2pep-Datenbank. Dies unterstreicht die entscheidende Rolle der ausgewählten Datenbank für MS-basierte Proteomics.
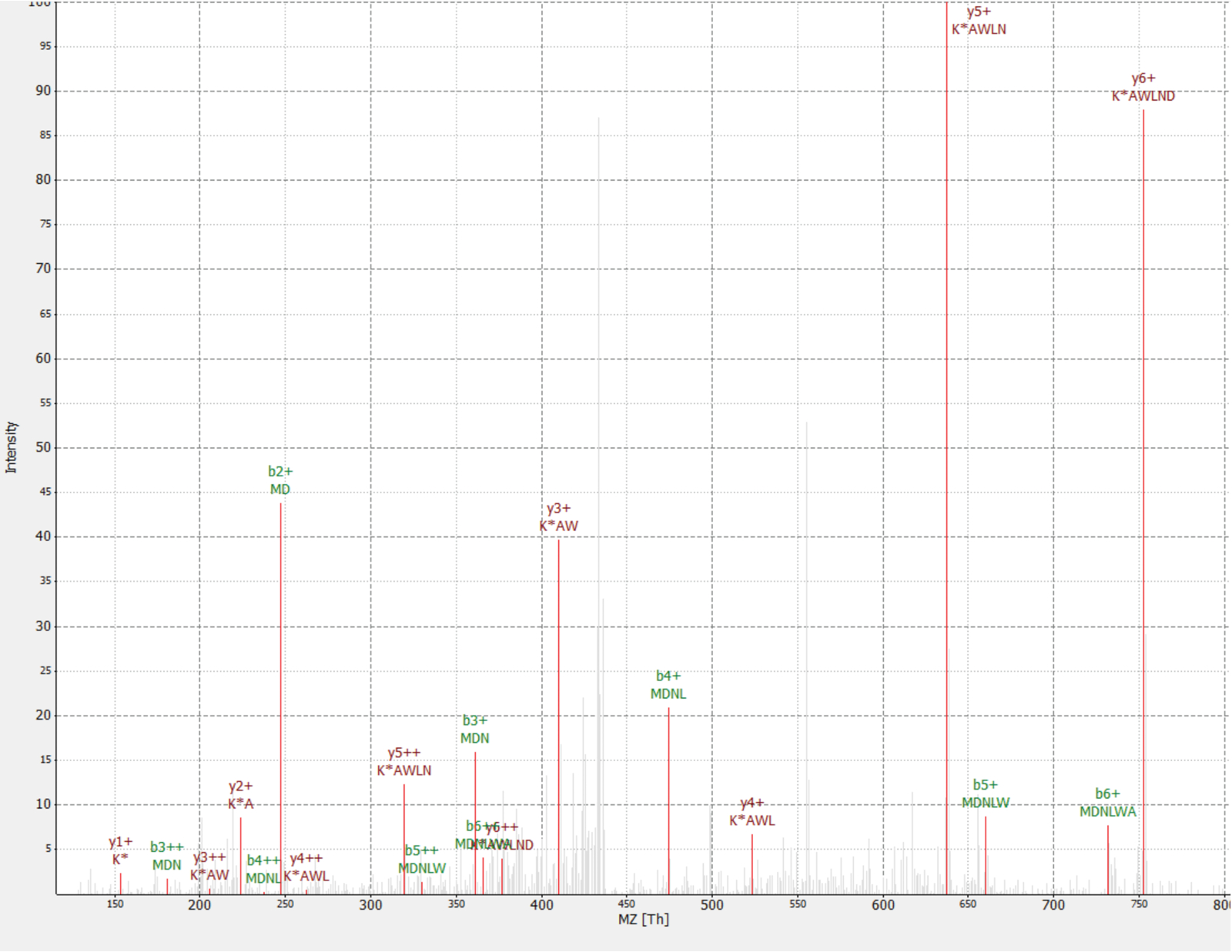
Ein neuartiges Protein wurde als ein Interaktor des RAF1 Proteins (IP_637643) entdeckt. Nutzung der Internetseite OpenProt kann man dieses Protein hatte nicht von MS noch Ribosom Profilerstellung bis jetzt erkannt wurden (OpenProt v1. 3). Das Protein ist 46 Aminosäuren lang und kann nur zwei einzigartige Peptide auf tryptic Verdauung geben. Das Peptid entdeckt in RAF1 AP-MS Dataset (Teil 18) hatte ein gutes Spektrum, wie in Abbildung 4dargestellt, und ein schwer-leicht-Verhältnis von 1,09 angezeigt. Das Protein wird im NANOGNBP1 -Gen kodiert ein Pseudogene NANOGNBist. Das Transkript (ENST00000448444), derzeit als nicht-kodierenden, kommentiert wurde über mehrere Gewebe nach der GTEx Portal40erkannt. Das Protein enthält eine vorhergesagte funktionale Domäne DNA-Bindung (Gene Ontology GO: 0003677)41zugeordnet.

Abbildung 1 : Datenbank-Wahl für Proteomics Analysen Diagramm. Analysen von MS Daten, insbesondere die Datenbank Wahl, hängen die Forschungsziele. Drei gemeinsame Ziele werden in blau (klassische Proteomic Pipeline), grün (erschöpfende Proteomic Suche) und Orange (Proteomic Entdeckung) beschrieben. Jedes Ziel richtet sich nach einer entsprechenden Datenbank und Pipeline. Eine einheitliche Kennzeichnung Werkzeug verwendet werden, für eine erschöpfende und klassischen Proteomics Rohrleitungen. Für die Proteomik-Entdeckung-Pipeline empfehlen wir mehrere Identifikation-Engines. Empfohlene FDRs werden in rot angezeigt, und Protein-Datenbank-Größen werden im grauen Kästchen angezeigt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2 : Grafische Darstellung des Galaxy Workflows verwendet. Schrittweise Darstellung des Proteomic Analysen Workflows für Re-Analyse von Eyckerman Et Al. Daten38verwendet. Eingabedateien, Suche Peptid und Protein Quantifizierung sind durch orange Box angezeigt. Blauen Kästen entsprechen die eingesetzten Werkzeuge und graue Kästchen entsprechen die Ausgabedateien erzeugt. Verschiedene Suchmaschinen (MS-GF + und X! Tandem) werden durch verschiedene Farben (bzw. rot und violett) als auch die Pfeile, die die notwendigen ein- und Ausgänge angezeigt. Das grüne Feld zeigt das Tool eine Liste von Protein Identifikationen erzeugen. Wenn mehrere Ausgaben generiert werden, für die nachgelagerten Stufen angegeben am nächsten auf den Pfeil. Dieser Workflow ist in Ergänzenden Material S2frei verfügbar. X! Tandem Standardkonfigurationsdatei Parameter gibt es in Ergänzenden Material S4. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3 : Vergleich der interactor Identifikation pro Köder mit unterschiedlichen Datenbanken. Venn-Diagramme von Protein Identifikationen mit die zuversichtlichsten OpenProt-Datenbank (in Orange, Nachweise von mindestens 2 einzigartige Peptide, OpenProt_2pep) mit einem 1 % FDR oder die ganze OpenProt Datenbank (in blau, OpenProt_all) mit einer 0,001 % FDR, oder wie berichtet in der ursprünglichen Papier (in grau)38. Jedes Diagramm entspricht identifizierten Interaktoren für die erwähnten Köder: RAF1, RNF41, PTPN14, JIP3 und IQGAP1. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4 : MS/MS-Spektrum der ermittelten MDNLWAK(13 6) Peptid aus neuartiges Protein IP_637643. Intensität ist relativ (0 bis 100 %). Ausgewählten Spitzen sind in rot, y-Ionen Anmerkungen in dunklem Rot und b Ionen Anmerkungen in grün sind angegeben. Auszug aus der TOPPview-Software-34. Vorläufer-Fehler = 2,70 ppm, PEP-Score = 0,12. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Begriff | Definition | Referenz |
| Alternative ORF (AltORF) | nicht-kanonische ORF derzeit nicht im Genom Anmerkungen kommentiert, sondern kommentiert in OpenProt. | 15 |
| Referenz ORF (RefORF) | kanonische ORF kommentierte in Genom Anmerkungen und OpenProt. | 15 |
| Alternative Protein (AltProt) | neuartiges Protein kodiert durch eine AltORF, mit keine deutliche Ähnlichkeit mit einem RefProt. Beitritt-Präfix: IP_. | 15 |
| Referenzproteins (RefProt) | Protein derzeit kommentiert in Protein Sequenzdatenbanken wie UniProtKB, Ensembl oder NCBI RefSeq, und auch in OpenProt. | 15 |
| Neuartige Isoform | neuartiges Protein kodiert durch eine AltORF, mit eine deutliche Ähnlichkeit mit einem RefProt. Beitritt-Präfix: II_. | 15 |
| OpenProt_2pep Datenbank | enthält die Abfolge aller RefProts und neuartige Proteine vorhergesagt durch OpenProt, mit einem Minimum von 2 einzigartige Peptide bereits erkannt. | 15 |
| OpenProt_1pep Datenbank | enthält die Abfolge aller RefProts und neuartige Proteine vorhergesagt durch OpenProt, mit einem Minimum von 1 einzigartige Peptid bereits erkannt. | 15 |
| OpenProt_all Datenbank | enthält die Abfolge aller RefProts und neuartige Proteine von OpenProt vorhergesagt. | 15 |
Tabelle 1: Definition der Begriffe, die in OpenProt und in das Protokoll
Ergänzende Material S1: Galaxy-Workflow für die Handhabung von Datenbanken. Dies wird die CRAPome und Köder-Sequenzen (rückwärts) mit der Eingabe-Datenbank anhängen. Ausgabe ist einer Fasta-Datei. Bitte klicken Sie hier, um download.
Ergänzende Material S2: Galaxy-Workflow für Proteinidentifizierung. Dies erkennt Proteine aus einer Massenspektrometrie-Datendatei mit zwei Suchmaschinen (MS-GF + und X! (Tandem). Jeder Parameter kann abgestimmt werden bevor Sie den Workflow ausführen wie gewünscht. Bitte klicken Sie hier, um download.
Ergänzende Material S3: Galaxy-Workflow für Protein Quantifizierung mit stabilen Isotopen Kennzeichnung (SIL). Dies wird identifizieren und quantifizieren Proteine aus einer Massenspektrometrie-Datendatei mit zwei Suchmaschinen (MS-GF + und X! (Tandem). Jeder Parameter kann abgestimmt werden bevor Sie den Workflow ausführen wie gewünscht. Bitte klicken Sie hier, um download.
Ergänzende Material S4: X! Tandem Parameter Standardkonfigurationsdatei. Dieser XML-Datei ist zum Ausführen von X erforderlich! TandemAdapter-Tool auf der Galaxy-Plattform. Bitte klicken Sie hier, um download.
Ergänzende Material S5: quantifiziert Proteine aus iMixPro Datasets. Datendateien von Eyckerman Et Al. 201638 wurden mit OpenProt Datenbanken verarbeitet und quantifizierte Proteine sind für jede Bedingung aufgeführt. Köder sind PTPN14, JIP3, IQGAP1, RAF1 und RNF41. Gen Namen angegeben in grün entsprechen Proteine auch in der ursprünglichen Papier38identifiziert. Gen Namen angegeben in Orange entsprechen bekannten Interaktoren nach BioGrid, die nicht in der Originalpublikation gemeldet wurden. Gen Namen angegeben in hellblau entsprechen neuartige Proteine identifiziert als Interacter (die entsprechende Protein-Zbl-Nummer ist in Klammern angegeben). Gen Namen angegeben in hellem Grau und kursiv entsprechen wahrscheinlich Verunreinigungen (Keratin Proteine). Bitte klicken Sie hier, um download.
Ergänzende Material S6: neuartige Proteine aus iMixPro Datasets identifiziert. Datendateien von Eyckerman Et Al. 201638 wurden mit OpenProt Datenbanken verarbeitet und neue identifizierte Proteine sind für jede Bedingung aufgeführt. Köder sind PTPN14, JIP3, IQGAP1, RAF1 und RNF41. Protein-Beitritt-Nummern sind aufgeführt, beginnend mit II_ für neuartige Isoformen eines bekannten Proteins und mit IP_ für neue Proteine aus einer alternativen ORF (AltProt). Die Anzahl der Unterstützung Peptide sind in Klammern angegeben. Bitte klicken Sie hier, um download.
Diskussion
Bei der Analyse der Daten von Massenspektrometern die Qualität der Proteinidentifizierung stützt sich teilweise auf die Genauigkeit der verwendeten Datenbank6,20. Aktuelle Ansätze verwenden traditionell UniProtKB Datenbanken, noch diese unterstützt das Genom Anmerkung Modell von einem einzigen ORF pro Abschrift und einer Mindestlänge von 100 Codons (mit Ausnahme der zuvor aufgezeigten Beispiele)40. Mehrere Studien beziehen sich die Mängel solcher Datenbanken mit der Entdeckung der funktionalen ORFs aus angeblich nicht-kodierenden Regionen8,11,12,13. Nun, erlaubt OpenProt für umfassendere Proteinidentifikation wie es mehrere Transkriptom Anmerkungen Proteinsequenzen entlockt. OpenProt ruft NCBI RefSeq (GRCh38.p7) und Ensembl (GRCh38.83) Transkriptom und UniProtKB Anmerkungen (UniProtKB-derjenigen, 2017-09-27)40,42,43. Wie aktuelle Anmerkungen wenig Überlappung präsentieren, zeigt OpenProt somit einen umfassenderen Blick auf die potenziellen Proteomic Landschaft als wenn beschränkt auf eine Anmerkung15.
Darüber hinaus als OpenProt eine Polycistronic Modell erzwingt, ermöglicht es mehrere Protein Anmerkungen pro Protokoll. Statistische und numerische Gründen hält OpenProt noch eine Mindestlänge Schwelle von 30 Codons15. Doch sagt es Tausende von neuartigen Proteinsequenzen, dadurch Erweiterung des Anwendungsbereichs der Möglichkeiten für Proteinidentifizierung voraus. Mit diesem Ansatz unterstützt OpenProt Proteomic Entdeckungen in systematischer Weise.
Die Qualität der Proteinidentifikation kann auch durch die Parameter beeinflusst werden, die verwendet werden. MS-basierte Proteomics Analysen halten in der Regel eine 1 % Protein FDR. Die gesamte OpenProt-Datenbank enthält jedoch ca. 6-Mal mehr Einträge (Abbildung 1). Um diesen erheblichen Anstieg des Suchraums berücksichtigen, empfehlen wir eine strengere FDR von 0,001 %. Dieser Parameter wurde mit Benchmark-Studien und manuelle Auswertung von nach dem Zufallsprinzip ausgewählte Spektren15optimiert. Fehlalarm immer noch eine Möglichkeit, und wir ermutigen gründliche Inspektion und Überprüfung der Belege für ein neuartiges Protein. Ein empfohlener Standard könnte die Identifizierung eines Proteins aus zwei verschiedenen MS-Läufen, sein, wie Datasets15Hintergrunddaten und Fehlalarmen unterscheiden.
Die Pipeline hier bereitgestellt und verwendet für die Fallstudie kann so gerne passen die Versuchsplanung und Parameter geändert werden. Wir würden empfehlen, mit mehreren Suchmaschinen zunehmender Empfindsamkeit und Empfindlichkeit des Peptids Identifikation32. Darüber hinaus fördern wir mit Hilfe der Datenbank entspricht am besten dem experimentellen Ziel (Abbildung 1). Als mit der ganzen OpenProt Datenbank mit einem strengen FDR kommt, können wahre Identifikationen verloren gehen. So sollten die gesamte Datenbank für Entdeckung der neuen Proteine bestimmt, während klassische Proteomics Profilierung der kleineren OpenProt-Datenbanken (z. B. OpenProt_2pep verwendet in der Fallstudie oben) verwendet werden sollte.
OpenProt prognostiziert derzeit Sequenzen beginnend mit ATG-Codon, während mehrere Studien Übersetzung Einleitung bei anderen Codons44,45hervorgehoben. Wenn ein neues Protein durch eine oder mehrere einzigartige Peptide identifiziert wird, ist es möglich die wahre Einleitung Codon nicht die vermuteten ATG ist. Benutzer können nach Übersetzung beweisen auf der Website OpenProt sehen. OpenProt meldet derzeit nur Übersetzung Ereignisse, wenn sie die gesamten prognostizierten Protein Sequenz (100 % Überlappung)15betreffen. So hieße fehlen Übersetzung Beweise nicht, dass das Protein nicht übersetzt wird, aber, dass das Start-Codon möglicherweise nicht die angeblichen ATG.
Trotz ihrer aktuellen Grenzen bietet OpenProt einen umfassenderen Blick auf eukaryotische Genome Codierung Potenzial. OpenProt Datenbanken Proteomic Entdeckungen und das Verständnis der Proteomik Funktionen und Interaktionen zu fördern. Zukünftige Entwicklungen der OpenProt Datenbank werden Anmerkung anderer Arten, Übersetzung Beweis von nicht-ATG beginnen Sie Codon und Entwicklung einer Pipeline, neue Proteine im gesamten Genom und Exome Sequenzierung Studien aufzunehmen.
Offenlegungen
Die Autoren erklären keinen Interessenskonflikt.
Danksagungen
Wir danken Vivian Delcourt für seine Hilfe, Diskussionen und Beratung über diese Arbeit. X.R. ist Mitglied des Fonds de Recherche du Québec Santé FRQS unterstützt Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. Diese Forschung wurde durch eine Canada Research Chair in funktionelle Proteomik und Entdeckung von Roman Proteine, X.R. und CIHR Zuschuss MOP-137056 unterstützt. Wir danken dem Team bei Calcul Québec und Compute Kanada für die Unterstützung bei der Nutzung der Supercomputer mp2 von Université de Sherbrooke. Betrieb der mp2-Supercomputer wird finanziert durch Kanada Foundation of Innovation (CFI), le Ministère de l'Économie, De La Science et de französischen du Québec (MESI) und Les Fonds de Recherche du Québec - Nature et Technologien (FRQ-NT). Die Galaxy-Server, der für einige Proteomics-Berechnungen verwendet wurde ist teilweise finanziert Collaborative Research Center 992 medizinische Epigenetik (DFG Stipendium SFB 992/1 2012) und Bundesministerium für Bildung und Forschung (BMBF gewährt 031 RBC A538A/A538C, 031L0101B /031L0101C de. NBI-Epi, 031L 0106 de. Treppe (de.) NBI)).
Materialien
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
Referenzen
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten