
Method Article
Basati sulla spettrometria di massa proteomica analisi utilizzando il Database di OpenProt a svelare nuove proteine tradotte da Non-Canonical Open Reading Frames
In questo articolo
Riepilogo
OpenProt è un database liberamente accessibile che applica un modello di polycistronic dei genomi eucariotici. Qui, presentiamo un protocollo per l'utilizzo di database OpenProt quando interrogare i set di dati di spettrometria di massa. Utilizzo di OpenProt database per l'analisi di esperimenti di proteomica consente per la scoperta del romanzo e proteine precedentemente non rilevabili.
Abstract
Annotazione del genoma è centrale per ricerca proteomica di oggi come disegna i contorni del paesaggio proteomica. I modelli tradizionali di open lettura annotazione frame (ORF) imporre due criteri arbitrari: una lunghezza minima di 100 codoni e un singolo ORF per trascrizione. Tuttavia, un numero crescente di studi segnala espressione delle proteine da presunto non codificante regioni, sfidando l'esattezza delle annotazioni correnti del genoma. Queste proteine sono state trovate di romanzo codificato all'interno di non-codificazione RNAs, 5' o 3' regioni non tradotte (UTR) del mRNA, o la sovrapposizione di una sequenza di codici nota (CDS) in alternativa ORF. OpenProt è il primo database che applica un modello polycistronic per genomi eucariotici, permettendo di annotazione di ORFs multiple per trascrizione. OpenProt è liberamente accessibile e offre il download personalizzato di sequenze proteiche attraverso 10 specie. Utilizzando OpenProt database per esperimenti di proteomica consente l'individuazione di nuove proteine ed evidenzia la natura polycistronic di geni eucariotici. La dimensione del database di OpenProt (tutti predetti proteine) è sostanza e devono essere prese in considerazione per l'analisi. Tuttavia, con appropriato false discovery rate (FDR) impostazioni o l'utilizzo di un database di OpenProt riservato, gli utenti otterrà una visione più realistica del paesaggio proteomica. Nel complesso, OpenProt è uno strumento disponibile gratuitamente che favorirà la proteomica scoperte.
Introduzione
Negli ultimi decenni, spettrometria di massa (MS-) basata proteomica è diventata la tecnica d'oro a decifrare i proteomi di cellule eucariotiche1,2,3,4,5. Questo metodo si basa su annotazioni correnti del genoma per generare un database di sequenza della proteina di riferimento che delinea l'ambito di possibilità6,7,8. Tuttavia, le annotazioni del genoma contenere criteri arbitrari per l'annotazione di ORF, ad esempio una lunghezza minima di 100 codoni e un singolo ORF ogni trascrizione9,10. Un numero crescente di studi sfida l'attuale modello di annotazione e segnala le scoperte di ORFs annotate funzionale in genomi eucarioti8,11,12,13, 14. Queste nuove proteine si trovano codificati in presunto non codificanti, in 5' o 3' non tradotta regioni (UTR) del mRNA, o la sequenza di codificazione canonica (cCDS) di sovrapposizione in un telaio sostitutivo. Sebbene la maggior parte di queste scoperte sono state serendipitous, essi dimostrano i caveat di annotazioni correnti del genoma e la natura polycistronic di geni eucariotici8.
Qui, si evidenzia l'utilizzo di database OpenProt per proteomica basata su MS. OpenProt è il primo database per contenere un modello di annotazione di polycistronic per trascrittomi eucariotiche. È disponibile gratuitamente presso www.openprot.org15. Una parte di questi predetto che ORFS sarebbe casuale e non-funzionali, ed è per questo OpenProt accumula prove sperimentali e funzionale ad accrescere la fiducia. Evidenze sperimentali sono l'espressione della proteina (da MS) e traduzione in evidenza (mediante profilatura, ribosoma)15. Prove funzionali includono proteine ortologhe (con un In-Paranoid come approccio) e dominio funzionale Pronostico15.
OpenProt offre la possibilità di scaricare diversi database, da contenenti solo ben supportati proteine ai database su misura. Qui, presentiamo una pipeline per l'utilizzo di database di OpenProt e offrirà approfondimenti quale database scegliere considerando lo scopo sperimentale. La pipeline di analisi proteomica ha presentata qui è supportata dal framework Galaxy come è accessibile e facile da usare, ma i database possono funzionare con qualsiasi flusso di lavoro16,17,18. Saremo presenti anche come utilizzare il sito Web di OpenProt per raccogliere maggiori informazioni sulle nuove proteine rilevate dai database MS. Using OpenProt fornirà una visione più esaustiva del paesaggio proteomica e favorirà la proteomica e biomarcatori scoperte in modo più sistematico rispetto ai metodi attuali.
Questo protocollo evidenzia l'uso di OpenProt database15 quando interrogare MS DataSet; non procederà a rivedere la progettazione dell'esperimento stesso, che è stato accuratamente Recensito altrove20,21,22. Nel tentativo di rimanere completamente open source, il protocollo è liberamente disponibile (Complementare materiale S1–S4). Per facilitare la lettura, tutti i termini utilizzati in OpenProt e dichiara nel presente protocollo sono definiti nella tabella 1.
Protocollo
1. OpenProt database scaricare
Nota: Database personalizzati sulla base dei dati RNA-seq per esempio possono anche essere ottenuti e la procedura è dettagliata nella seconda sezione del presente protocollo. Se è necessario un database personalizzato, passare alla sezione successiva.
- Vai al sito OpenProt: www.openprot.org e Apri la pagina di download utilizzando il link dal menu di inizio pagina.
- Fare clic su specie di interesse sulla base dei dati sperimentali analizzati.
- Fare clic sul tipo di proteina desiderato.
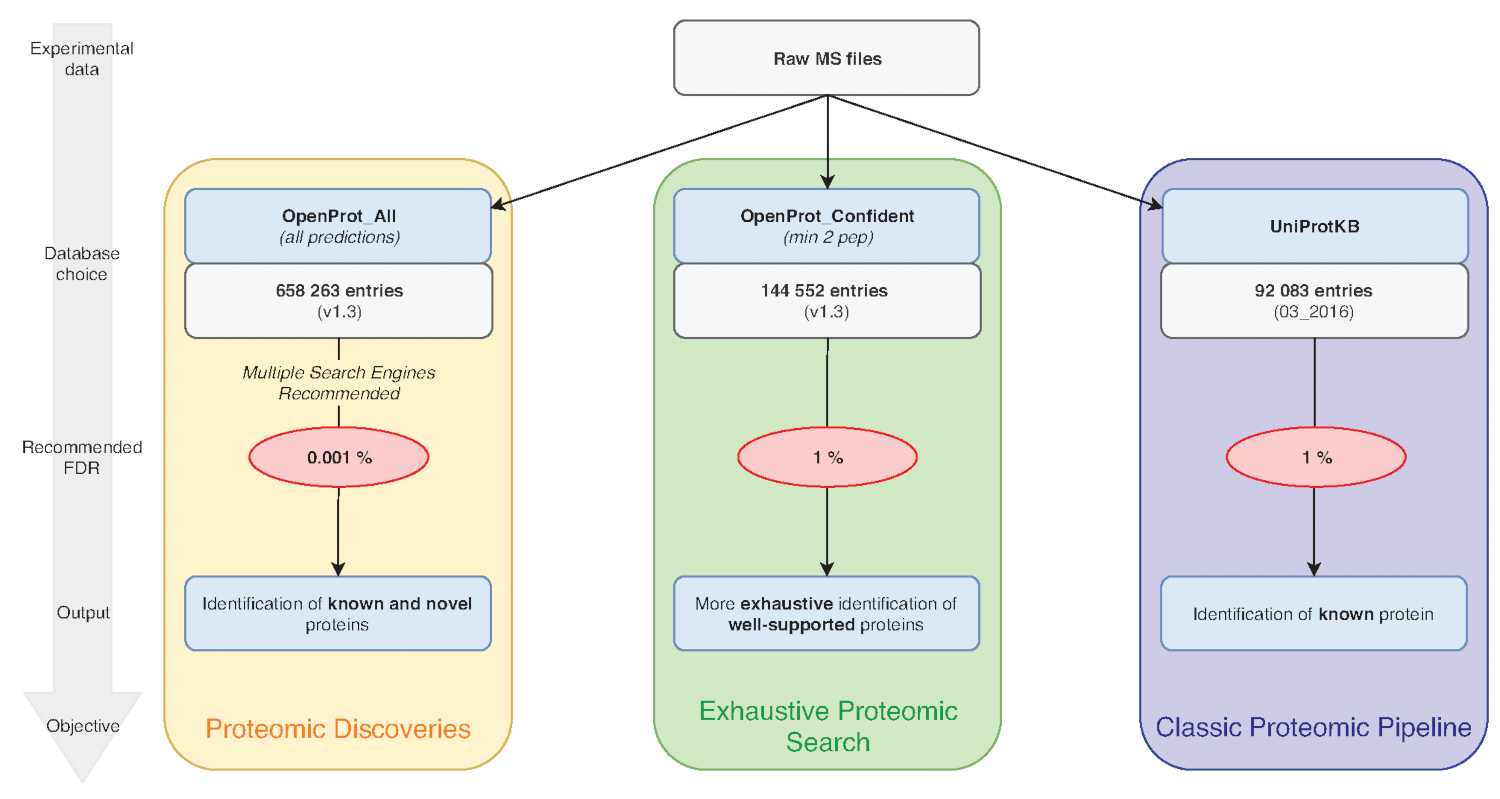
Nota: OpenProt offre tre classificazioni: RefProt, isoforme e AltProt. Come illustrato nella Figura 1, questo parametro varia in base all'obiettivo di ricerca.- Fare clic su RefProt solo per generare i file che contiene solo proteine note.
- Fare clic su AltProt e isoforme per generare i file contenenti solo nuove proteine - entrambi romanzo isoforme di proteine note (isoforme) o codificati da un'alternativa ORF (AltProts). Tieni presente che OpenProt applica una lunghezza minima di ORF di 30 codoni15.
- Clicca su AltProts, isoforme e RefProts per generare i file contenenti tutti i tipi di proteine presenti nel database OpenProt - proteine noti e romanzo.
- Se disponibile, fare clic sull'annotazione dalla quale proteina sequenze vengono disegnate.
Nota: OpenProt offre un paesaggio più esaustivo di proteomica, combinando annotazioni multiple. Le annotazioni del trascrittoma hanno una sovrapposizione minima; così, l'annotazione selezionata possa influenzare sostanzialmente la proteomica visualizzati profilo15,23. - Fare clic sul livello di elementi di prova necessaria per l'esame della proteina. Come illustrato nella Figura 1, questo parametro varia in base all'obiettivo di ricerca.
- Fare clic su un minimo di due peptidi unici rilevati per generare i file contenenti solo le proteine più fiduciosa.
Nota: Un criterio di due peptidi unici è attualmente considerato un gold standard in proteomica di espressione della proteina. Se lo scopo sperimentale è quello di rilevare proteine note e ben supportate, è consigliato l'uso di questo parametro. - Fare clic su minima di un unico peptidi rilevato per generare i file contenenti le proteine che sono già stati visti almeno una volta tra gli esperimenti di spettrometria di massa ri-analizzati da OpenProt.
Nota: In questo modo per la considerazione della lunghezza più corta del AltProts e la probabilità che alcuni di essi possono contenere solo un unico peptide triptico8,11. - Fare clic su tutti predetto per generare i file che contengono tutte le previsioni di OpenProt.
Nota: Questa impostazione è consigliata solo se lo scopo sperimentale è quello di scoprire nuove proteine (Figura 1). L'aumento sostanza successiva chiamate di ricerca di spazio per una pipeline di analisi adattato come discusso di seguito7,15.
- Fare clic su un minimo di due peptidi unici rilevati per generare i file contenenti solo le proteine più fiduciosa.
- Fare clic sul formato di file desiderato per il download. Per analisi proteomica, scegliere il file Fasta (proteina). Il file readme contiene tutte le informazioni necessarie sul formato del file.
2. personalizzato OpenProt database scaricare
Nota: Questa sezione viene illustrato come ottenere un database personalizzato. Se non è necessario nessun database personalizzato, passare alla sezione successiva.
- Visitare il sito Web OpenProt (www.openprot.org) e aprire la pagina di ricerca utilizzando il link dal menu di inizio pagina.
- Fare clic su specie di interesse sulla base dei dati sperimentali analizzati.
- Inserire una lista di geni o trascrizioni di interesse.
- Quando si utilizza una lista di geni, immetterlo nella finestra di query Gene .
- Quando si utilizza un elenco di trascrizioni, immetterlo nella casella di query di trascrizione .
- Seleziona qualsiasi casella che si applica al database desiderato.
- Non fare clic su qualsiasi casella per ottenere una tabella contenente tutti i tipi di proteine supportato da OpenProt: RefProt, isoforme e AltProts.
- Fare clic su Visualizza solo proteine con prove sperimentali per ottenere una tabella contenente tutti i tipi di proteine (RefProts, isoforme e AltProts) che sono stati rilevati almeno una volta da MS e/o per quale traduzione prova è stati raccolti dal ribosoma i dati di profilatura.
- Allo stesso modo, fare clic su Show solo proteine rilevate da MS o Show solo proteine rilevati dal ribosoma profilatura per ottenere una tabella contenente tutti i tipi di proteine che sono stati rilevati almeno una volta da MS o da ribosoma profilatura rispettivamente.
- Fare clic su Visualizza solo AltProts o Show solo isoforme per ottenere una tabella contenente solo AltProts o solo isoforme rispettivamente.
- Fare clic su due Show solo AltProts e mostrare solo isoforme per ottenere una tabella che contiene entrambi i tipi di proteine.
Nota: Sono possibili tutte le combinazioni di filtri.
- Una volta impostati tutti i parametri desiderati, fare clic su Cerca. Output della tabella apparirà sotto i campi di query di ricerca.
- Fare clic sul pulsante Scarica Fasta nell'angolo in alto a destra della tabella di output. Questo genererà un file Fasta che contiene tutte le proteine derivanti dall'elenco richiesto dei geni o trascrizioni.
- Siete pregati di notare che per motivi di calcolo, OpenProt contiene un massimo di 2.000 elementi per essere interrogato (geni o trascrizioni) alla volta. In caso di un elenco sopra tale limite, fasta diversi può essere generata e quindi concatenato (come dettagliato di seguito); o semplicemente scaricare l'intero database di OpenProt e filtrare il file ottenuto come desiderato.
- Bin tutto l'elenco dei geni o trascrizioni in sub-elenchi di 2.000 voci o meno. Per ogni sub-elenco, è necessario scaricare un file Fasta come descritto in precedenza (punto 3.3-3.6).
- Login per l'istanza di Galaxy europeo (o qualsiasi altra istanza dove sono disponibili strumenti di proteomica), https://usegalaxy.eu/.
- Creare una nuova storia e importare tutti i database di OpenProt scaricati (uno per ogni sub-elenco dei geni o trascrizioni) cliccando sul logo caricamento in alto a sinistra dello schermo.
- Utilizzare lo strumento Fasta unire file e sequenze uniche filtro sviluppato dagli sviluppatori GalaxyP (https://github.com/galaxyproteomics/). Selezionare l'opzione Unisci tutte le Fasta e tutti i database di OpenProt importati in ingresso.
Nota: Ogni strumento possa essere cercato utilizzando la casella query sul lato sinistro dello schermo - Selezionare l'opzione di adesione solo per valutare la sequenza unicity e copiare la regola di OpenProt identificatore parse (>(.*) \ |), quindi fare clic su Execute.
- Si noti che tutti i file sono stati concatenati in un unico file Fasta senza ridondanza che ora viene visualizzata nel pannello cronologia sul lato destro dello schermo. Questo costituisce il database di lavoro.
3. la gestione del database
Nota: da questo momento in poi, verrà utilizzata la piattaforma di Galaxy, ma gli stessi principi possono essere applicati ad altri software di proteomica.
- Login per l'istanza di Galaxy europeo (o qualsiasi altra istanza dove sono disponibili strumenti di proteomica), https://usegalaxy.eu/.
- Creare una nuova storia e importare il database di OpenProt scaricato cliccando sul logo caricamento in alto a sinistra dello schermo.
- Vai alla pagina del flusso di lavoro e importare il Database Gestione flusso di lavoro (Complementare materiale S1) cliccando sul logo in alto a sinistra sul pannello centrale upload.
- Fare clic su eseguire il flusso di lavoro e selezionare il database di OpenProt importato come input.
Nota: Questo flusso di lavoro verrà aggiungere il repository di CRAPome alla fasta OpenProt e generare esca sequenze (sequenze inversa)24. Se si desidera un elenco di decoy shuffle, può essere fatto modificando questo parametro sullo strumento DecoyDatabase. - Rinominare il file Fasta ottenuto a qualcosa di significativo. Il database è pronto per essere utilizzato per le analisi di proteomica.
4. preparazione dei file spettrometria di massa
Nota: la maggior parte degli strumenti di proteomics disponibili su istanze di Galaxy utilizza il formato di Simone, e peptide motori di ricerca preferiscono i dati in modalità centroide.
- Aprire lo strumento di MSConvert liberamente disponibile dalla suite ProteoWizard e caricare il file di dati per essere analizzati25.
- Scegliere la directory per l'output e il formato di file desiderato per Simone.
- Impostare un picco raccolta filtro utilizzando l'algoritmo basato su wavelet (CWT) sui livelli di MS1 e MS2 e avviare la conversione26.
5. peptidi e proteine Identificazione/quantificazione
Nota: Questa parte della pipeline utilizza strumenti della suite di OpenMS, un quadro versatile e facile da usare18.
- Login per l'istanza di Galaxy europeo (o qualsiasi altra istanza dove sono disponibili strumenti di proteomica), https://usegalaxy.eu/.
- Creare una nuova storia e trasferire il database creato in precedenza (punto 3.5) a questa nuova storia con un drag-and-drop.
- Importare il file di dati di simone trasformato (punto 4.3) cliccando sul logo caricare in alto a sinistra dello schermo.
- Vai alla pagina del flusso di lavoro e importare il flusso di lavoro desiderato cliccando sul logo in alto a sinistra sul pannello centrale upload.
Nota: Gli esperimenti MS sono diversamente progettati in base al risultato finale desiderato. I flussi di lavoro sono forniti qui per due disegni frequenti: proteina identificazione e quantificazione di proteine basato su isotopo stabile etichettatura (SIL). Tuttavia, l'istanza di Galaxy contiene molti altri strumenti che sosterranno altri tipi di analisi proteomica27,28.- Per una progettazione di identificazione di proteine, importare il flusso di lavoro fornito in S2 materiale supplementare. Quando si utilizza questo flusso di lavoro, si prega di non utilizzare la compressione di zlip quando si convertono i file (passo 4.2)
- Per una quantificazione della proteina basata sul design d'etichettatura isotopo stabile, importare il flusso di lavoro fornito in S3 materiale supplementare.
- Selezionare eseguire il flusso di lavoro e rivedere i vari parametri.
- Selezionare il file di dati importati Simone come input e il database creato in precedenza (punto 3.5) del file di database Fasta.
- Poiché il flusso di lavoro utilizza la X! Ricerca di tandem motore, importare la X! Tandem predefinito configurazione file (fornito in Supplementari materiale S4)29 cliccando sul logo caricamento in alto a sinistra dello schermo.
- Il flusso di lavoro utilizza più motori di ricerca (MS-GF + e X! Tandem). Aggiungere altri motori di ricerca o scegliere una sola semplicemente di aggiungere o rimuovere gli strumenti dal flusso di lavoro30,31.
Nota: Utilizzando più motori di ricerca è consigliabile in quanto aumenta la sensibilità e la sensibilità delle analisi32. - Per tenere conto per il notevole aumento delle dimensioni quando si utilizza l'intero database di OpenProt, utilizzare un FDR rigorose15. Per impostazione predefinita, il flusso di lavoro fornito è impostata per un 0,001% FDR, adeguata per l'utilizzo dell'intero database OpenProt. Per altri database, questo può essere modificato a qualsiasi valore desiderato.
Nota: Assicurarsi di adattare i parametri degli strumenti diversi a seconda lo spettrometro di massa utilizzato e il protocollo sperimentale (precursore dello ione e frammento, corretto errore e modifiche variabile, enzima usato, ecc.).
- Facoltativamente, è possibile scaricare output per ogni fase del flusso di lavoro per l'archiviazione o analisi di controllo qualità cliccando sul pulsante passo selezionato dal pannello Cronologia, quindi fare clic su Salva logo che apparirà sotto.
6. controllo di qualità
Nota: Poiché proteomica basata su MS-è il risultato di un processo complesso in cui ogni passo deve essere ottimizzato per produrre risultati riproducibili, controllo di qualità è una procedura necessaria nel flusso di lavoro33.
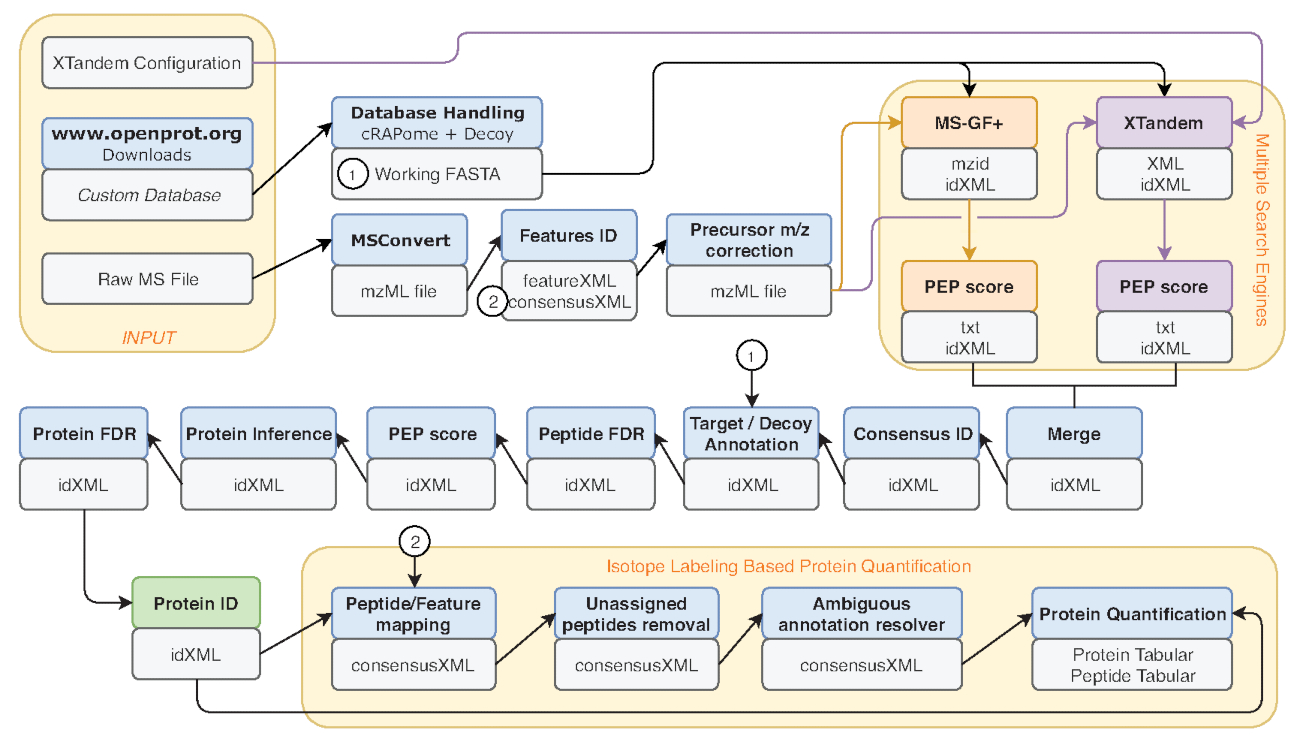
- Diversi criteri di misurazione sono comuni benchmark delle prestazioni, come ad esempio il numero di corrispondenze di peptide-spettro (PSM), il numero di proteine e peptidi identificati. Eseguire lo strumento di File Info sull'uscita IDFilter (indicata in verde nella Figura 2) per fornire tali metriche.
- Anche se non è applicabile a ogni identificazione, soprattutto con grandi set di dati, rapporti di nuove proteine dovrebbero sempre essere valutati con attenzione. Ispezione di punteggio nella proteina, la copertura di sequenza e gli spettri sostenendo l'individuazione è di vitale importanza. Utilizzare lo strumento di TOPPview da quadro OpenMS per farlo; è liberamente disponibile e ben documentato18,34,35.
7. OpenProt data mining di database
Nota: Una volta stabilita un'identificazione fiduciosa di una nuova proteina predetta da OpenProt (numeri di adesione a partire con IP_ per AltProts e II_ per le isoforme di romanzo), più biologiche informazioni possono essere raccolte dal sito Web OpenProt15.
- Vai al sito OpenProt: www.openprot.org e Apri la pagina di ricerca utilizzando il link nel menu di inizio pagina.
- Fare clic sulle specie di interesse (uguale a quello in cui la proteina è stata identificata) e immettere il numero di adesione di proteine nella finestra di query della proteina .
- Fare clic su ricerca e verrà visualizzata una tabella contenente informazioni di base sulla proteina richiesta. Le funzionalità di tabella: la lunghezza della proteina (in amminoacido), suo peso molecolare (kDa) e punto isoelettrico, sostenendo la prova sperimentale di MS o ribosoma profilatura (prove di traduzione, TE) e le previsioni funzionali come predetto domini e proteina ortologhe (tra le 10 specie supportati da OpenProt, v 1.3). La tabella contiene anche informazioni relative al gene e trascrizione e la localizzazione della proteina all'interno della trascrizione.
- Fare clic sul collegamento Dettagli per raccogliere ulteriori informazioni. La pagina appena aperta contiene un browser del genoma che è centrato sulla proteina query e informazioni quali la genomica e trascrittomica coordinate e la presenza di un Kozak o ad alta efficienza traduzione iniziazione sito (TIS) motivo36, 37.
- Fare clic sulla proteina o DNA collegamenti dalla scheda info, per ottenere proteine o sequenze di DNA rispettivamente.
- Sfoglia informazioni dettagliate sulle prove di MS, ribosoma profiling rilevamento, conservazione e domini proteici identificati cliccando sulle linguette superiori15.
Risultati
Il flusso di lavoro descritto in precedenza è stato applicato a un oggetto dataset MS disponibile sui repository orgoglio38,39. Lo studio originale ha sviluppato un metodo (iMixPro), mediante marcatura a isotopi stabili di amminoacidi nella coltura delle cellule (SILAC), per eliminare i falsi positivi da MS di purificazione di affinità (AP-MS) esperimenti38. In breve, un esperimento di AP-MS consiste di usando gli anticorpi associati a perline per recuperare una proteina di interesse (esca) e suoi interattori (prede). Le proteine raccolte sono poi digerite e preparate per MS. Il metodo di preparazione del campione e le impostazioni dello strumento sono descritti nello studio originale e sul repository di orgoglio (PXD004246). Una sfida in tali esperimenti è l'abbondanza di falsi positivi, in particolare dalle proteine leganti le perle ma non l'esca. Qui, abbiamo usato SILAC per generare rapporti isotopici diversi tra prede veri e falsi positivi: sono 3 campioni di controllo (nessun esca) coltivati in chiaro e medio, 1 campione esprimendo l'esca coltivata in medium leggero e 1 campione esprimendo l'esca coltivato nel mezzo pesante elaborato con le perline e ulteriori analisi di spettrometria di massa. Con tale disegno, non specifiche proteine leganti ai talloni avrà un pesante-to-light in rapporto di 1:4; Quando vere prede avrà un rapporto di 1:138.
Abbiamo analizzato nuovamente i dati AP-MS utilizzando il database di OpenProt; le esche incluso tre proteine endogene (PTPN14, JIP3 e IQGAP1), e due sovra-espresse proteine (RAF1 e RNF41). Poiché gli esperimenti utilizzato SILAC, il flusso di lavoro di Galaxy per quantificazione della proteina è stato usato (S3 materiale complementare, Figura 2). Il flusso di lavoro è stato eseguito utilizzando l'intero database di OpenProt (OpenProt_all) o un database riservato di OpenProt (OpenProt_2pep, tra cui solo proteine precedentemente rilevati con un minimo di due peptidi unici).
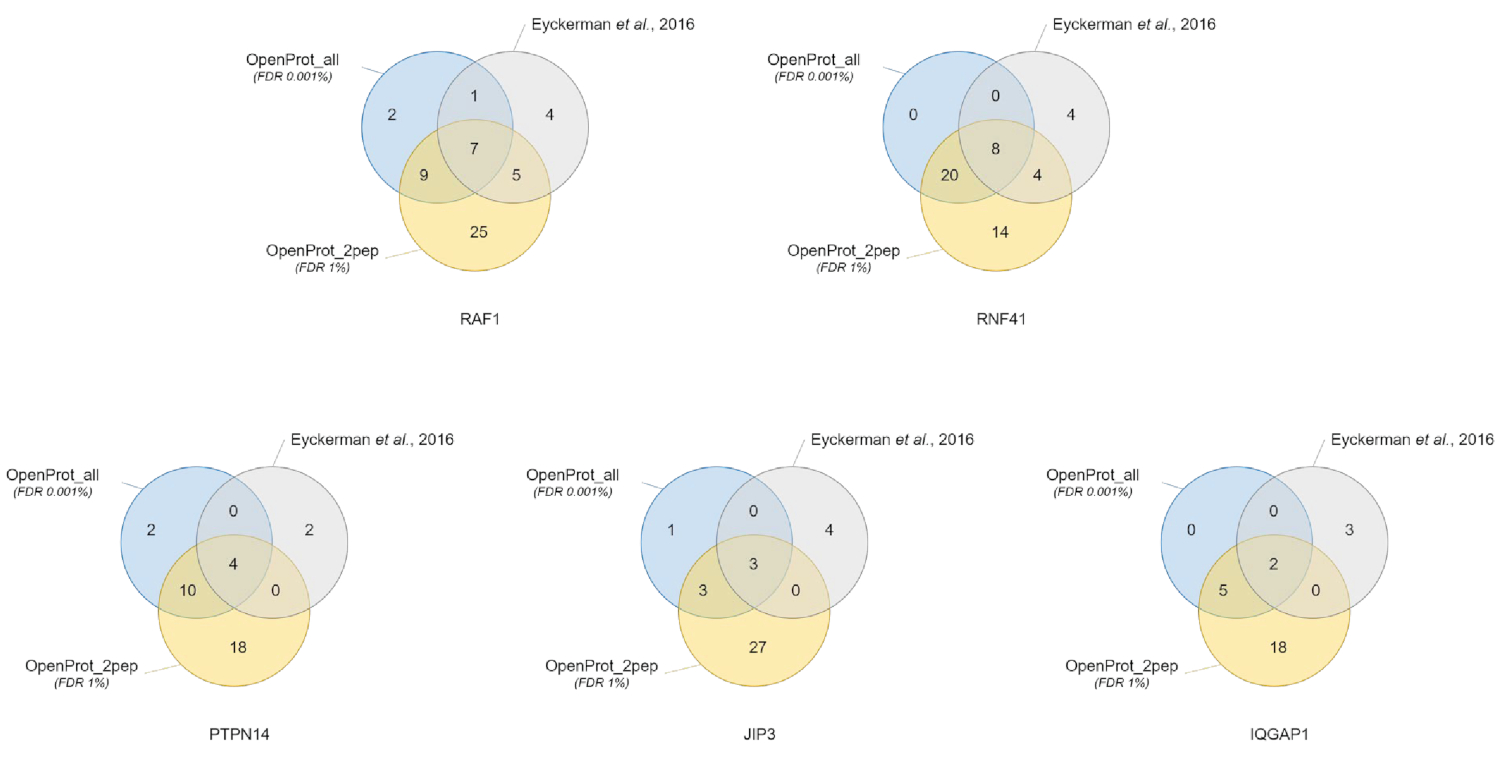
Quantificazione e identificazione delle proteine erano buoni e riproducibili attraverso i diversi database utilizzati. Come mostrato nella Figura 3, la maggior parte delle proteine identificate nel documento originale inoltre sono state identificate usando il OpenProt_2pep o il OpenProt_all database (un elenco dettagliato è disponibile in S5 materiale supplementare). Questo risultato dimostra che la pipeline descritta qui e il OpenProt database sono in grado di produrre la proteina identificazione e quantificazione paragonabile a quella delle attuali procedure basate sul database UniProtKB40. Tuttavia, l'utilizzo di database di OpenProt ha il vantaggio unico di permettendo la rilevazione di romanzo e proteine precedentemente non rilevabili, come dimostrato in questo caso di studio.
11 proteine ben supportati (1 isoforma e 10 AltProts), ma attualmente non annotato nel database, sono stati identificati attraverso tutti i DataSet, con peptidi fiduciosi, utilizzando il database di OpenProt_2pep (tutte le adesioni di proteina, insieme al numero di supporto peptidi, sono disponibili in S5 materiale supplementare). Questo database consente l'utilizzo di un tradizionale 1% FDR come l'aumento di spazio di ricerca rimane moderato. Queste 11 proteine non sono state identificate nello studio originale come erano assenti dal database.
29 nuove proteine (16 isoforme e 13 AltProts) sono stati scoperti attraverso tutti i DataSet, con peptidi fiduciosi, utilizzando il database di OpenProt_all (tutte le adesioni di proteina, insieme al numero di peptidi di supporto, sono disponibili in supplementari materiale S6 ). Come mostrato nella Figura 3, il FDR rigorose consigliati non ha colpito le identificazioni di proteine più fiduciosa, anche se esso ha fatto diminuire il numero totale di proteine identificate. Comparativamente al database OpenProt_2pep, un numero maggiore di nuove proteine possa essere identificato con sicurezza. Tutte queste proteine romanzo sono assenti dal database di OpenProt_2pep. Questo sottolinea il ruolo cruciale del database selezionato per proteomica basata su MS.
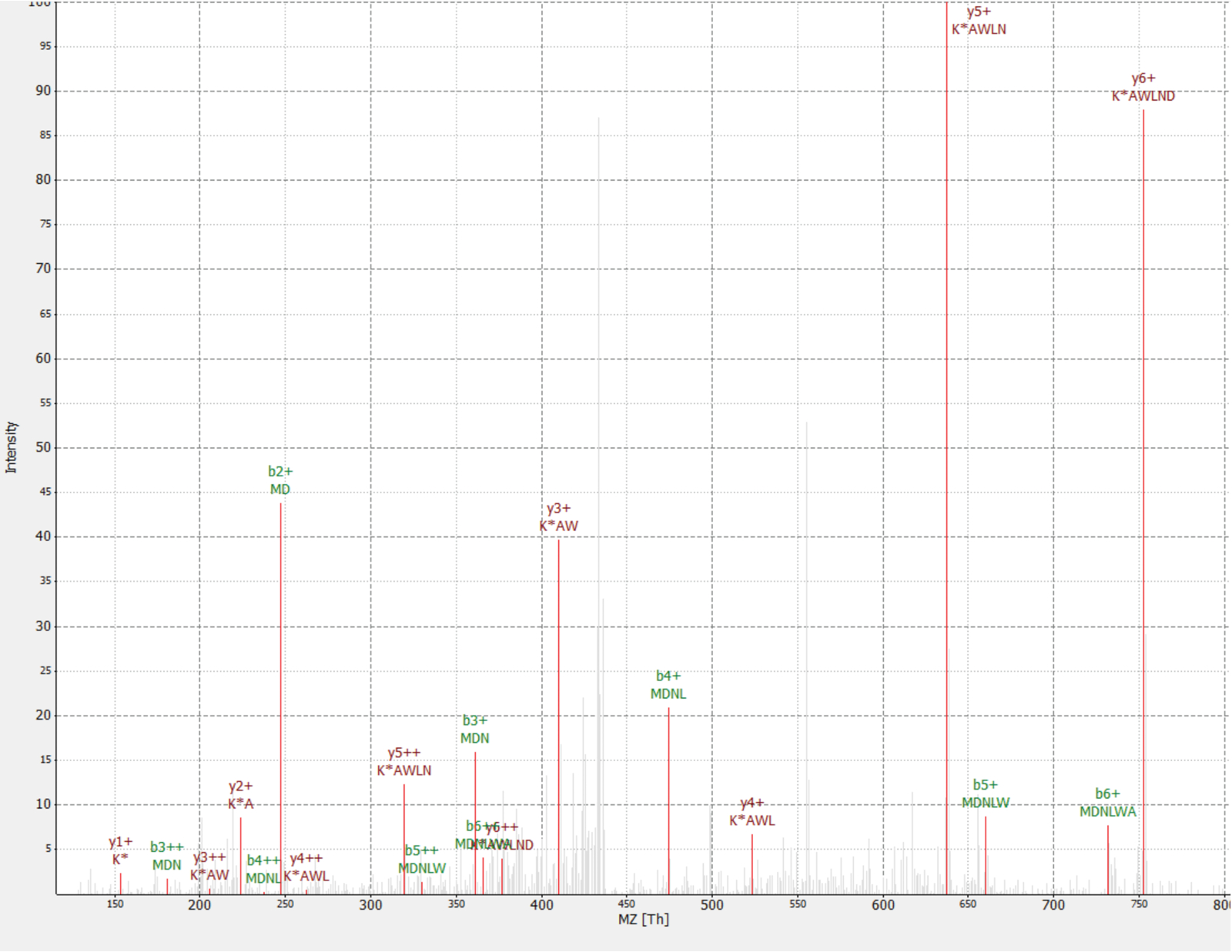
Una nuova proteina è stata scoperta come un'interazione della proteina RAF1 (IP_637643). Usando il sito Web OpenProt, si può vedere questa proteina non è stata rilevata da MS né ribosoma profilatura fino ad ora (OpenProt v. 1.3). La proteina è 46 aminoacidi lunghe e può solo dare due peptidi unici sulla digestione trittica. Il peptide rilevato in RAF1 AP-MS dataset (frazione 18) ha avuto un spettro di buona qualità, come mostrato nella Figura 4e visualizzato un rapporto di pesante-to-light di 1,09. La proteina è codificata nel gene NANOGNBP1 , che è uno pseudogene di NANOGNB. La trascrizione (ENST00000448444), attualmente annotata come non-codificazione, è stata rilevata in parecchi tessuti secondo il sesso portale40. La proteina contiene un dominio funzionale previsto associato con DNA binding (Gene Ontology GO: 0003677)41.

Figura 1 : Database scelta per grafico analisi proteomica. Analisi dei dati di MS, in particolare la scelta del database, dipendono gli obiettivi della ricerca. Tre obiettivi comuni sono delineati in blu (proteomica classica pipeline), verde (ricerca esaustiva proteomica) e arancio (proteomica scoperta). Ogni obiettivo dipende da un database appropriato e pipeline. Uno strumento di identificazione unico può essere utilizzato per una proteomica classica ed esaustivo condotte. Per la pipeline di scoperta di proteomica, si consiglia di utilizzare più motori di identificazione. FDRs consigliati sono indicati in rosso, e dimensioni del database di proteine sono indicati nelle caselle grigie. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2 : Rappresentazione grafica del flusso di lavoro Galaxy usato. Rappresentazione dettagliata del flusso di lavoro di analisi proteomica utilizzato per ri-analisi di dati di Eyckerman et al.38. File di input, la ricerca del peptide e quantificazione della proteina sono indicati da scatole arancioni. Scatole blu corrispondono agli strumenti usati e grigi caselle corrispondono al file di output generati. I motori di ricerca diversi (MS-GF + e X! Tandem) sono indicati da colori differenti (rispettivamente rossi e viola) così come le frecce che indicano loro necessari ingressi e uscite. Casella verde mette in evidenza lo strumento di generazione di un elenco di identificazioni di proteina. Quando uscite multiple vengono generati, quello utilizzato per la procedura a valle è indicato come la più vicina alla freccia. Questo flusso di lavoro è liberamente disponibile in S2 materiale supplementare. La X! File di configurazione dei parametri di impostazione predefinita tandem è disponibile in S4 materiale supplementare. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3 : Confronto di interactor identificazione per esca usando diversi database. Diagrammi di Venn di identificazioni di proteina utilizzando il OpenProt più fiducioso del database (in arancione, elementi di prova di minimi 2 unici peptidi, OpenProt_2pep) con un 1% FDR, o il OpenProt intero database (in blu, OpenProt_all) con un 0,001% FDR, o come riportato in originale carta (in grigio)38. Ogni diagramma corrisponde a interattori identificati per l'esca accennato: RAF1, RNF41, PTPN14, JIP3 e IQGAP1. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4 : Spettro MS/MS di identificato MDNLWAK(13 6) peptide da proteina novella IP_637643. L'intensità è relativa (0-100%). Picchi selezionati sono evidenziati in rosso, le annotazioni di ioni y sono nelle annotazioni di ioni scure rosso e b in verde. Estratti dal software di TOPPview34. Errore di precursore = 2,70 ppm, il Punteggio di PEP = 0.12. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Termine | Definizione | Riferimento |
| ORF alternativi (AltORF) | ORF non canonico attualmente non annotata nelle annotazioni di genoma, ma annotati in OpenProt. | 15 |
| Riferimento ORF (RefORF) | canonico ORF annotato nel genoma annotazioni e OpenProt. | 15 |
| Alternativi della proteina (AltProt) | nuova proteina codificata da un AltORF, con nessuna somiglianza significativa con un RefProt. Prefisso di adesione: IP_. | 15 |
| Proteina di riferimento (RefProt) | proteina attualmente annotata nelle banche dati di sequenza della proteina come UniProtKB, Ensembl o NCBI RefSeq e anche in OpenProt. | 15 |
| Isoforma romanzo | nuova proteina codificata da un AltORF, con una somiglianza significativa con un RefProt. Prefisso di adesione: II_. | 15 |
| OpenProt_2pep database | contiene la sequenza di tutti i RefProts e nuove proteine preveduti di OpenProt, già rilevato con un minimo di 2 peptidi unici. | 15 |
| OpenProt_1pep database | contiene la sequenza di tutte le RefProts e le nuove proteine preveduti di OpenProt, già rilevato con un minimo di 1 peptide unico. | 15 |
| OpenProt_all database | contiene la sequenza di tutti i RefProts e nuove proteine preveduti da OpenProt. | 15 |
Tabella 1: Definizione dei termini utilizzati in OpenProt e in tutto il protocollo
Complementare materiale S1: flusso di lavoro Galaxy per la gestione del database. Questo aggiungerà le sequenze CRAPome e decoy (inversione) al database di input. Output è un file Fasta. Per favore clicca qui per scaricare.
Complementare materiale S2: flusso di lavoro Galaxy per identificazione della proteina. Questo identificherà proteine da un file di dati di spettrometria di massa utilizzando due motori di ricerca (MS-GF + e X! Tandem). Ogni parametro può essere sintonizzato come desiderato prima di eseguire il flusso di lavoro. Per favore clicca qui per scaricare.
Complementare materiale S3: flusso di lavoro Galaxy per quantificazione della proteina usando isotopo stabile etichettatura (SIL). Questo identificare e quantificare le proteine da un file di dati di spettrometria di massa utilizzando due motori di ricerca (MS-GF + e X! Tandem). Ogni parametro può essere sintonizzato come desiderato prima di eseguire il flusso di lavoro. Per favore clicca qui per scaricare.
S4 materiale complementare: X! File di configurazione di parametri di default di tandem. Questo file XML è necessario per l'esecuzione di X! TandemAdapter strumento sulla piattaforma Galaxy. Per favore clicca qui per scaricare.
Complementare materiale S5: quantificato proteine da DataSet iMixPro. File di dati da Eyckerman et al 201638 sono stati elaborati utilizzando i database OpenProt e proteine quantificati sono elencati per ogni condizione. Le esche sono PTPN14, JIP3, IQGAP1, RAF1 e RNF41. Nomi di gene indicati in verde corrispondono alle proteine identificate anche nella carta originale38. Nomi di gene indicati in arancio corrispondono al noti Interactiani secondo BioGrid che non sono stati segnalati nel documento originale. Nomi di gene indicati in blu chiaro corrispondono a nuove proteine identificate come Interactiani (il corrispondente numero di adesione di proteine è indicato tra parentesi). Nomi di gene indicata in grigio chiaro e corsivo corrisponda alla probabile contaminanti (proteine di cheratina). Per favore clicca qui per scaricare.
Complementare materiale S6: identificare nuove proteine da DataSet iMixPro. File di dati da Eyckerman et al 201638 sono stati elaborati utilizzando i database OpenProt e romanzo proteine identificate sono elencati per ogni condizione. Le esche sono PTPN14, JIP3, IQGAP1, RAF1 e RNF41. Numeri di accessione della proteina sono elencati, a partire con II_ per le isoforme di romanzo di una proteina conosciuta e con IP_ per nuove proteine da un'alternativa ORF (AltProt). Il numero di peptidi di supporto sono indicati tra parentesi. Per favore clicca qui per scaricare.
Discussione
Quando si analizzano dati da spettrometri di massa, la qualità di identificazione delle proteine si basa in parte sull'accuratezza del database utilizzato6,20. Attuali approcci tradizionalmente utilizzano database UniProtKB, eppure questi supportano il modello di annotazione del genoma di un singolo ORF per trascrizione e una lunghezza minima di 100 codoni (ad eccezione di esempi precedentemente dimostrati)40. Più studi riguardano le carenze di tali database con la scoperta di ORFs funzionale da presunto non codificante regioni8,11,12,13. Ora, OpenProt consente per l'identificazione di proteine più esauriente quanto richiama sequenze proteiche da annotazioni multiple del trascrittoma. OpenProt recupera NCBI RefSeq (GRCh38.p7) e trascrittomi Ensembl (GRCh38.83) e le annotazioni UniProtKB (UniProtKB-SwissProt, 2017-09-27)40,42,43. Come annotazioni correnti presentano scarsa sovrapposizione, OpenProt così una visualizzazione più esaustivo del paesaggio proteomica potenziali rispetto a quando limitato a un'annotazione15.
Inoltre, come OpenProt applica un modello di polycistronic, consente annotazioni multiple di proteina per trascrizione. Per motivi statistici e computazionali, OpenProt detiene ancora una soglia di lunghezza minima di 30 codoni15. Ancora, esso predice migliaia di sequenze proteiche romanzo, quindi ampliare la sfera di possibilità per l'identificazione della proteina. Con questo approccio, OpenProt supporta proteomica scoperte in maniera più sistematica.
La qualità di identificazione della proteina può anche dipendere dai parametri che vengono utilizzati. Analisi proteomica basata su MS-tengono tipicamente una proteina 1% FDR. Tuttavia, l'intero database OpenProt contiene circa 6 volte più voci (Figura 1). Per tenere conto di questo aumento sostanza nello spazio ricerca, si consiglia di utilizzare un FDR più rigorose di 0.001%. Questo parametro è stato ottimizzato utilizzando studi di benchmark e valutazione manuale di spettri selezionato casualmente15. Falso positivo sono ancora una possibilità, però, e incoraggiamo ispezione accurata e convalida di elementi di prova per una nuova proteina. Un standard consigliato potrebbe essere l'identificazione di una proteina da due diverse esecuzioni di MS, come dati in background e falsi positivi variano tra i set di dati15.
La pipeline fornito qui e utilizzato per lo studio di caso può essere modificata come lieti di inserire il disegno sperimentale e parametri. Lo consigliamo utilizzando più motori di ricerca come aumenta la sensibilità e la sensibilità del peptide identificazione32. Inoltre, incoraggiamo utilizzando il database corrisponde al meglio allo scopo sperimentale (Figura 1). Come usare il OpenProt intero database viene fornito con un FDR rigorose, veri identificazioni potrebbero andare persi. Così, l'intero database dovrebbe essere destinato per la scoperta di nuove proteine, mentre analisi proteomica classica dovrebbe essere utilizzando i database di OpenProt più piccoli (ad esempio OpenProt_2pep utilizzato nel caso di studio qui sopra).
OpenProt predice attualmente sequenze a partire con un codone ATG, mentre diversi studi ha evidenziato l'inizio della traduzione presso altri codoni44,45. Quando una nuova proteina è identificata da uno o più peptidi univoci, è possibile che il codone di inizio allineare non è la presunta ATG. Gli utenti possono cercare prove di traduzione sul sito OpenProt. Attualmente, OpenProt segnala eventi traduzione solo se esse riguardano l'intera proteina preveduta sequenza (100% sovrapposizione)15. Così, assenza di prove di traduzione non significherebbe la proteina non è tradotto, ma che il codone di inizio non può essere presunta ATG.
Nonostante i suoi limiti attuali, OpenProt offre una visione più esaustiva delle potenziale codifica dei genomi eucariotici. OpenProt database favoriscono la proteomica scoperte e la comprensione delle funzioni di proteomica e interazioni. Gli sviluppi futuri del database OpenProt includerà annotazione di altre specie, prove di traduzione da non-ATG avviare codone e lo sviluppo di una pipeline da includere nuove proteine nell'intero genoma e gli studi di sequenziamento dell'esoma.
Divulgazioni
Gli autori non dichiarano alcun conflitto di interessi.
Riconoscimenti
Ringraziamo Vivian Delcourt per suo aiuto, discussioni e consigli su questo lavoro. X.R. è un membro del Fonds de Recherche du Québec Santé FRQS-supportato Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. Questa ricerca è stata sostenuta da un Canada Research Chair in proteomica funzionale e scoperta delle proteine di romanzo a grant X.R. e CIHR MOP-137056. Ringraziamo il team di Calcul Québec e Compute Canada per il loro sostegno con l'uso del mp2 di supercomputer da Université de Sherbrooke. Funzionamento del supercomputer mp2 è finanziato da Fondazione Canada di innovazione (CFI), le ministère de il Économie, de la science et de l'Innovation du Québec (MESI) e les Fonds de Recherche du Québec - natura et technologies (FRQ-NT). Il server Galaxy che è stato utilizzato per alcuni calcoli di proteomica è in parte finanziato dal centro ricerca collaborativo 992 epigenetica medica (grant DFG SFB 992/1 2012) e Ministero federale tedesco dell'istruzione e della ricerca (BMBF concede 031 RBC A538A/A538C, 031L0101B De /031L0101C. NBI-epi, L 031 0106 de. SCALA (de. NBI)).
Materiali
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
Riferimenti
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati