
Method Article
Espectrometria de massa-baseado Proteomics análises usando banco de dados OpenProt para desvendar novas proteínas traduzidas de Frames de leitura abertos não-canônicos
Neste Artigo
Resumo
OpenProt é um banco de dados livremente acessível que impõe um modelo policistrônico de genomas eucarióticos. Aqui, apresentamos um protocolo para o uso de bancos de dados OpenProt quando interrogando datasets de espectrometria de massa. Usando o OpenProt banco de dados para análise de experimentos de proteomic permite a descoberta do romance e proteínas anteriormente indetectáveis.
Resumo
Anotação do genoma é central para a pesquisa de proteomic de hoje como ele desenha os contornos da paisagem proteomic. Os modelos tradicionais de abrir lendo anotação de quadro (ORF) impor dois critérios arbitrários: um comprimento mínimo de 100 códons e um único ORF por transcrição. No entanto, um número crescente de estudos relatam a expressão de proteínas de supostamente não-codificantes regiões, desafiando a exatidão das anotações atual do genoma. Estas proteínas foram encontradas de romance codificado ou não-codificantes RNAs, 5' ou 3' regiões untranslated (UTRs) de mRNAs, ou sobrepondo uma sequência de codificação conhecida (CDS) em alternativa ORF. OpenProt é o primeiro banco de dados, o que impõe um modelo policistrônico para genomas eucarióticos, permitindo a anotação de ORFs múltiplos por transcrição. OpenProt é livremente acessível e oferece downloads personalizados de sequências de proteínas através de 10 espécies. Usando o OpenProt o banco de dados para experimentos de proteomic permite a descoberta de novas proteínas e destaca a natureza policistrônico de genes eukaryotic. O tamanho do banco de dados OpenProt (tudo previsto proteínas) é substancial e precisa ser levado em conta para a análise. No entanto, com configurações de taxa (FDR) apropriado descoberta falsa ou o uso de um banco de dados OpenProt restrito, os usuários ganharão uma visão mais realista da paisagem proteomic. Em geral, OpenProt é uma ferramenta disponível gratuitamente que fomentará proteomic descobertas.
Introdução
Nas últimas décadas, a espectrometria de massa (MS-) baseada proteomics tornou-se a técnica dourada para decifrar proteomes de células eucarióticas,1,2,3,4,5. Este método baseia-se na atual anotações de genoma para gerar um referência proteína sequência de dados que descreve o escopo de possibilidades6,7,8. No entanto, as anotações de genoma manter critérios arbitrários para anotação de ORF, tais como um comprimento mínimo de 100 códons e um único ORF por transcrição9,10. Um número crescente de estudos desafia o actual modelo de anotação e relatar as descobertas de ORFs funcionais anotadas em genomas eucarióticos8,11,12,13, 14. Estas novas proteínas encontram-se codificados em RNAs supostamente não-codificantes, no 5' ou 3' não traduzidas (UTR) de regiões de mRNAs, ou sobrepondo a sequência de código canônica (cCDS) em um quadro de alternativo. Embora a maioria destas descobertas foram acidentais, eles demonstram que as advertências de anotações atual do genoma e a natureza de policistrônico de genes eukaryotic8.
Aqui, destaca-se o uso de bancos de dados OpenProt para proteômica baseada em MS. OpenProt é o primeiro banco de dados para manter um modelo de anotação de policistrônico para transcriptomes eucarióticas. Está livremente disponível em www.openprot.org15. Uma proporção desses previu que ORFS seria aleatório e não-funcionais, por isso é que OpenProt acumula evidências experimentais e funcional para aumentar a confiança. Evidências experimentais incluem a expressão da proteína (pelo MS) e tradução de provas (pelo Ribossoma perfilação)15. Provas funcionais incluem proteína homologia (com um In-paranoico como abordagem) e previsão de domínio funcional15.
OpenProt oferece a possibilidade de baixar vários bancos de dados, desde que contenham apenas bem suportadas proteínas para bancos de dados sob medidas. Aqui, apresentaremos um pipeline para o uso de bancos de dados OpenProt e irá oferecer insights sobre qual banco de dados para escolher tendo em conta o objectivo experimental. O pipeline de análise proteômica apresentado aqui é suportado pela estrutura Galaxy como é acesso livre e fácil de usar, mas os bancos de dados podem funcionar com qualquer fluxo de trabalho16,17,18. Também apresentaremos como usar o site OpenProt para reunir mais informações sobre novas proteínas detectadas pelos bancos de dados MS. Using OpenProt irá fornecer uma visão mais exaustiva da paisagem proteomic e fomentem descobertas proteomics e biomarcadores em uma maneira mais sistemática do que métodos atuais.
Este protocolo destaca o uso de bancos de dados de OpenProt15 quando interrogando MS datasets; Isso não irá rever o desenho do experimento em si, que tem sido exaustivamente analisado em outro lugar20,21,22. Em um esforço para permanecer totalmente aberto, o protocolo é livremente disponível (S1 de Material complementar–S4). Para facilitar a leitura, todos os termos usados em OpenProt e, por este meio, em todo este protocolo são definidos na tabela 1.
Protocolo
1. OpenProt download de banco de dados
Nota: Bancos de dados personalizados com base em dados de RNA-seq por exemplo também podem ser obtidos e o procedimento é detalhado na segunda seção do presente protocolo. Se houver necessidade de um banco de dados personalizado, por favor, pule para a próxima seção.
- Acesse o site OpenProt: www.openprot.org e abra a página de Downloads usando o link no menu do topo da página.
- Clique sobre as espécies de interesse, com base nos dados experimentais analisados.
- Clique no tipo de proteína desejado.
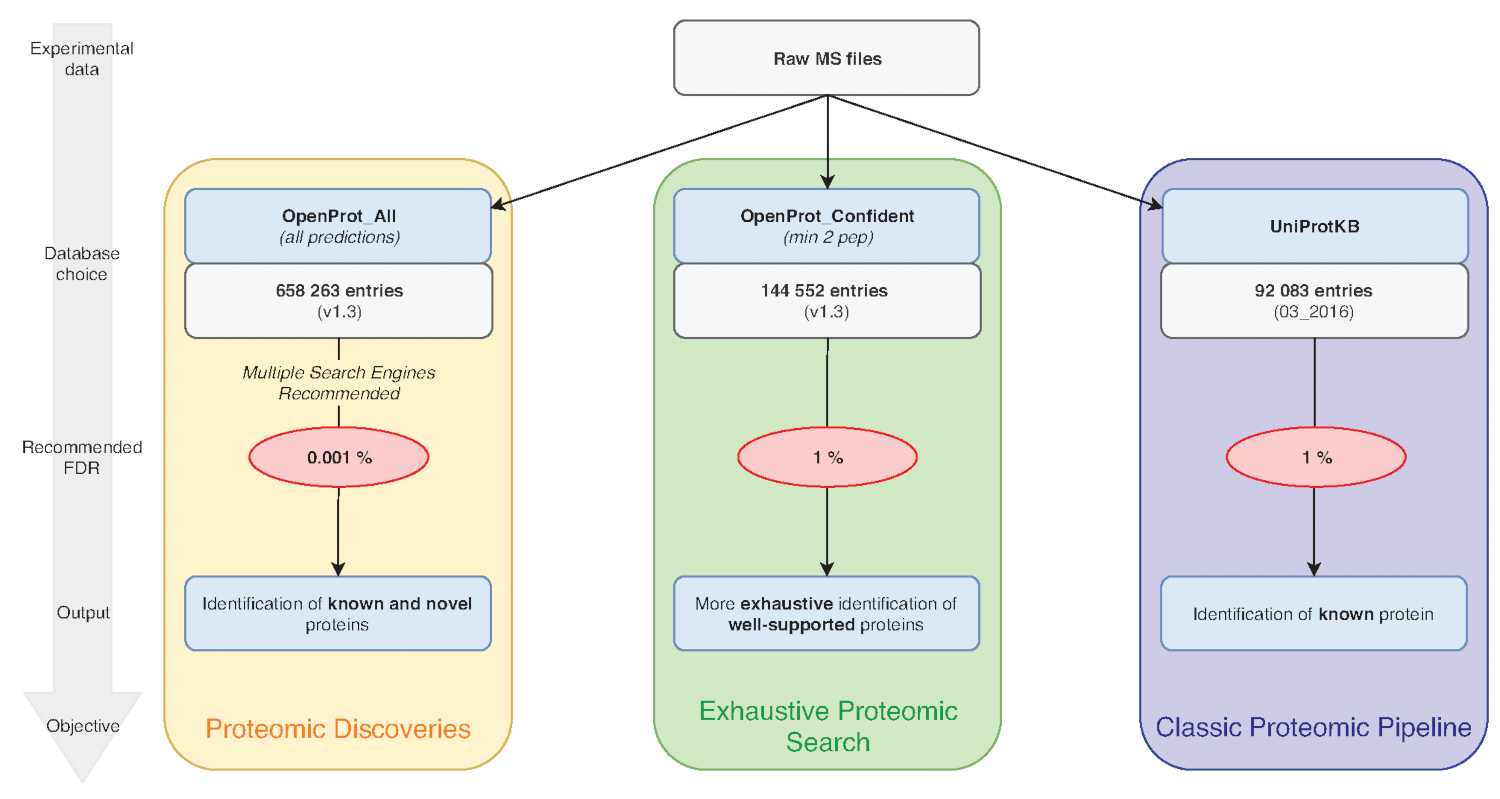
Nota: O OpenProt oferece três classificações: RefProt, isoformas e AltProt. Como mostrado na Figura 1, este parâmetro varia com base no objectivo da investigação.- Clique no RefProt sozinho para gerar arquivos que contém somente proteínas conhecidas.
- Clique em AltProt e isoformas para gerar arquivos contendo apenas novas proteínas - qualquer romance isoformas de proteínas conhecidas (isoformas) ou codificados por uma alternativa ORF (AltProts). Por favor, note que OpenProt impõe um comprimento mínimo de ORF de 30 códons15.
- Clique em AltProts, isoformas e RefProts para gerar arquivos contendo todos os tipos de proteínas presentes no banco de dados OpenProt - proteínas conhecidas e romance.
- Se estiver disponível, clique na anotação de qual proteína sequências são desenhadas.
Nota: OpenProt oferece uma paisagem mais exaustiva de proteomic combinando múltiplas anotações. Anotações de transcriptoma tem uma sobreposição mínima; assim, a anotação selecionada pode afectar substancialmente o perfil de proteomic visualizado15,23. - Clique sobre o nível de provas necessárias para a consideração de proteína. Como mostrado na Figura 1, este parâmetro varia com base no objectivo da investigação.
- Clique no mínimo de dois peptídeos únicos detectados para gerar arquivos contendo somente as proteínas mais confiantes.
Nota: Um critério de dois peptídeos exclusivos atualmente é considerado um padrão-ouro em proteômica na expressão de proteínas. Se o objetivo experimental é detectar proteínas conhecidas e bem suportadas, recomenda-se o uso desse parâmetro. - Clique no mínimo de um único peptídeos detectado para gerar arquivos contendo proteínas que já foram vistas pelo menos uma vez entre os experimentos de espectrometria de massa, re-analisados pelo OpenProt.
Nota: Isto permite a consideração de menor comprimento de AltProts e a probabilidade de que alguns deles podem conter apenas um único peptídeo tryptic8,11. - Clique em tudo o que previu para gerar arquivos contendo todas as previsões OpenProt.
Nota: Essa configuração é recomendada apenas se o objetivo experimental é descobrir novas proteínas (Figura 1). O subsequente aumento substancial nas chamadas busca espaço para um gasoduto de análise adaptados como discutido abaixo de7,15.
- Clique no mínimo de dois peptídeos únicos detectados para gerar arquivos contendo somente as proteínas mais confiantes.
- Clique sobre o formato de arquivo desejado para fazer o download. Para análises de proteomic, escolha o arquivo Fasta (proteína). O arquivo Leiame contém todas as informações necessárias sobre o formato de arquivo.
2. custom download de banco de dados de OpenProt
Nota: Esta seção fornece detalhes sobre como obter um banco de dados personalizado. Não se houver necessidade de nenhum banco de dados personalizado, pule para a próxima seção.
- Acesse o site OpenProt (www.openprot.org) e abra a página de busca usando o link no menu do topo da página.
- Clique sobre as espécies de interesse, com base nos dados experimentais analisados.
- Insira uma lista de genes ou transcrições de interesse.
- Quando usando uma lista de genes, inseri-lo na caixa de consulta de Gene .
- Quando usando uma lista de transcrições, inseri-lo na caixa de consulta de transcrição .
- Marque qualquer caixa que se aplica a banco de dados desejado.
- Não clique em qualquer caixa para obter uma tabela que contém todos os tipos de proteína, apoiado por OpenProt: RefProt, isoformas e AltProts.
- Clique em Mostrar apenas proteínas com evidências experimentais , para obter uma tabela que contém todos os tipos de proteínas (isoformas, RefProts e AltProts) que foram detectados pelo menos uma vez por MS e/ou para qual tradução provas foram coletadas do Ribossoma dados de perfil.
- Da mesma forma, clique em Mostrar apenas proteínas detectadas pelo MS ou em Mostrar apenas proteínas detectadas pelo Ribossoma perfis para obter uma tabela contendo todos os tipos de proteínas que foram detectados pelo menos uma vez por MS ou Ribossoma perfis respectivamente.
- Clique em Mostrar apenas AltProts ou em Mostrar apenas isoformas para obter uma tabela que contém somente AltProts ou apenas isoformas respectivamente.
- Clique em Mostrar apenas AltProts e Mostrar apenas isoformas para obter uma tabela que contém os dois tipos de proteínas.
Nota: Todas as combinações de filtros são possíveis.
- Uma vez que todos os parâmetros desejados estiverem definidos, clique em Pesquisar. A saída de tabela aparecerá abaixo os campos de consulta de pesquisa.
- Clique no botão Download Fasta no canto superior direito da tabela de saída. Isso irá gerar um arquivo Fasta que contém todas as proteínas resultantes da lista consultada de genes ou transcrições.
- Por favor, note que, por razões computacionais, OpenProt detém um máximo de 2.000 elementos para ser consultado (genes ou transcrições) de cada vez. No caso de uma lista acima desse limite, fasta vários pode ser gerados e depois concatenadas (como detalhado abaixo); ou simplesmente baixar o banco de dados inteiro OpenProt e filtrar o arquivo obtido conforme desejado.
- Bin a lista inteira de genes ou transcrições em sub-listas de 2.000 entradas ou menos. Para cada lista de sub, baixe um arquivo Fasta como descrito acima (passo 3.3 a 3.6).
- Acessar a instância Europeia Galaxy (ou qualquer outra instância onde proteômica ferramentas estão disponíveis), https://usegalaxy.eu/.
- Criar uma nova história e importar todos os bancos de dados baixados de OpenProt (um por sublista dos genes ou transcrições) clicando no logotipo no canto superior esquerdo da tela do carregamento.
- Use a ferramenta Fasta mesclar arquivos e sequências únicas de filtro desenvolvida pelos desenvolvedores do GalaxyP (https://github.com/galaxyproteomics/). Selecione a opção de mesclar Fasta todas e todos os importados OpenProt bancos de dados de entrada.
Nota: Cada ferramenta pode ser pesquisada usando a caixa de consulta no lado esquerdo da tela - Selecione a opção de adesão apenas para avaliar a unicidade de sequência e copiar a regra de análise de identificador de OpenProt (>(.*) \ |), em seguida, clique em executar.
- Observe que todos os arquivos tenham sido concatenados em um único arquivo Fasta com nenhuma redundância que agora aparece no painel Histórico do lado direito da tela. Isto constitui o trabalho de banco de dados.
3. manipulação de dados
Nota: de agora em diante, a plataforma da galáxia será usada, mas os mesmos princípios podem ser aplicados a outros softwares de proteômica.
- Acessar a instância Europeia Galaxy (ou qualquer outra instância onde proteômica ferramentas estão disponíveis), https://usegalaxy.eu/.
- Criar uma nova história e importar o banco de dados OpenProt baixado clicando no logotipo no canto superior esquerdo da tela do carregamento.
- Vá para a página de fluxo de trabalho e o fluxo de trabalho de manipulação de banco de dados (S1 de Material complementar) de importação clicando no logotipo do carregamento na parte superior esquerda do painel do meio.
- Clique em executar o fluxo de trabalho e selecione o banco de dados OpenProt importado como entrada.
Nota: Este fluxo de trabalho irá acrescentar o repositório CRAPome a fasta de OpenProt e gerar chamariz sequências (sequências reversa)24. Se uma lista de chamariz shuffle é desejada, pode ser feito alterando este parâmetro na ferramenta DecoyDatabase. - Renomeie o arquivo Fasta obtido para algo significativo. O banco de dados está pronto para ser usado para análise proteômica.
4. preparação de arquivo de espectrometria de massa
Nota: A maioria das ferramentas disponíveis em instâncias de galáxia proteomics usa o formato de mzML, e os motores de busca de peptídeo preferem dados no modo de centroide.
- Abra a ferramenta de MSConvert livremente disponível da suite ProteoWizard e carregar o arquivo de dados para ser analisados25.
- Escolha o diretório para a saída e o formato de arquivo desejado para mzML.
- Definir um pico escolher o filtro usando o algoritmo wavelet baseado (CWT) em níveis MS1 e MS2 e iniciar a conversão de26.
5. identificação/quantificação peptídeos e proteínas
Nota: Esta parte do pipeline utiliza ferramentas da suite OpenMS, uma estrutura versátil e fácil de usar,18.
- Acessar a instância Europeia Galaxy (ou qualquer outra instância onde proteômica ferramentas estão disponíveis), https://usegalaxy.eu/.
- Criar uma nova história e transferir o banco de dados criado anteriormente (passo 3.5) para esta nova história com um arrastar-e-soltar.
- Importe o arquivo de dados mzML transformado (passo 4.3) clicando no logotipo do Upload no canto superior esquerdo da tela.
- Vá para a página de fluxo de trabalho e importar o fluxo de trabalho desejado clicando no logotipo do carregamento na parte superior esquerda do painel do meio.
Nota: MS experimentos diferente são projetados com base na saída final desejada. Fluxos de trabalho são fornecidos aqui para dois projetos frequentes: proteína identificação e quantificação de proteínas com base no isótopo estável rotulagem (SIL). No entanto, a instância de galáxia contém muitas outras ferramentas que oferecerá suporte a outros tipos de análises de proteomic27,28.- Para um projeto de identificação de proteínas, importe o fluxo de trabalho fornecido no S2 de Material complementar. Ao usar este fluxo de trabalho, por favor não use a compressão zlip ao converter seus arquivos (etapa 4.2)
- Para uma quantificação de proteína baseada no projeto de etiquetagem de isótopo estável, importe o fluxo de trabalho fornecido em S3 de Material complementar.
- Selecione executar o fluxo de trabalho e analisar os diferentes parâmetros.
- Selecione o arquivo de dados importados mzML como entrada e o banco de dados criado anteriormente (passo 3.5) como o arquivo Fasta de banco de dados.
- Desde que o fluxo de trabalho usa o X! Pesquisar em tandem do motor, o X de importação! Conjunto padrão configuração arquivo (fornecido em S4 de Material suplementar)29 clicando no logotipo no canto superior esquerdo da tela do carregamento.
- O fluxo de trabalho usa vários mecanismos de busca (MS-GF + e X! Tandem). Acrescentar outros motores de busca ou escolher uma única simplesmente, adicionando ou removendo as ferramentas do fluxo de trabalho de30,31.
Nota: Usar vários motores de busca é recomendado como aumenta a sensibilidade e a sensibilidade da análise32. - Para dar conta do aumento substancial do tamanho quando usando o banco de dados inteiro de OpenProt, use um rigoroso FDR15. Por padrão, o fluxo de trabalho fornecido é definido para um 0.001% FDR, adequada para a utilização da base de dados inteira de OpenProt. Para outros bancos de dados, isso pode ser editado para qualquer valor desejado.
Nota: Certifique-se de adaptar os parâmetros das ferramentas diferentes dependendo do espectrômetro de massa utilizado e o protocolo experimental (precursor do íon e do fragmento, Corrigido erro e modificações variáveis, enzima usada, etc.).
- Opcionalmente, download saída para cada etapa do fluxo de trabalho para armazenamento ou análise de controle de qualidade, clique na etapa do painel Histórico escolhida, clicando no salvar logotipo que irá aparecer por baixo.
6. controle de qualidade
Nota: Porque proteômica baseada em MS é o resultado de um processo complexo, onde cada passo precisa ser otimizado para produzir resultados reprodutíveis, controle de qualidade é um procedimento necessário para o fluxo de trabalho de33.
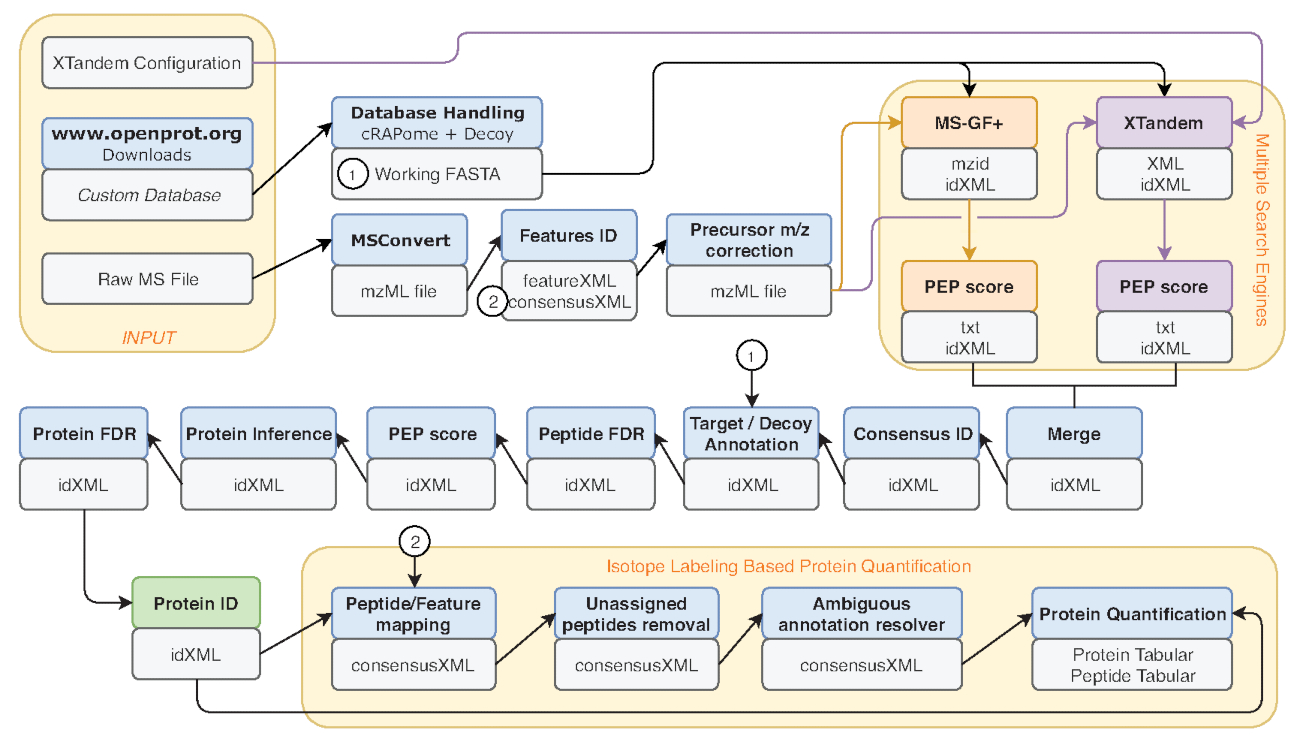
- Várias métricas são comuns benchmark de desempenho, tais como o número de correspondências de peptídeo-espectro (PSM), o número de identificação de peptídeos e proteínas. Execute a ferramenta de Informações do arquivo na saída do IDFilter (indicada em verde na Figura 2) para fornecer tais métricas.
- Embora não aplicáveis para cada identificação, especialmente com grandes conjuntos de dados, relatórios de novas proteínas sempre devem ser cuidadosamente avaliados. Inspeção de Pontuação a proteína, a cobertura de sequência e os espectros apoiando a constatação é de vital importância. Use a ferramenta de TOPPview do quadro de OpenMS para fazer isso; é livremente disponível e bem documentado18,34,35.
7. OpenProt mineira de banco de dados
Nota: Uma vez que foi feita uma identificação confiante de uma proteína romance prevista pela OpenProt (números de adesão começando com IP_ para AltProts e II_ para romance isoformas), mais informações biológicas podem ser obtidas da OpenProt site15.
- Acesse o site OpenProt: www.openprot.org e abra a página de pesquisa usando o link no menu do topo da página.
- Clique sobre as espécies de interesse (mesmo que aquele no qual a proteína foi identificada) e digite o número de adesão de proteína na caixa de consulta de proteína .
- Clique em Pesquisar e aparecerá uma tabela contendo informações básicas sobre a proteína consultada. As características da mesa: o comprimento de proteína (em aminoácidos), seu peso molecular (kDa) e ponto isoelétrico, evidência experimental por MS ou Ribossoma perfis (prova de tradução, TE) e previsões funcionais de apoio tais como previram domínios e proteína homologia (do outro lado as 10 espécies suportadas pelo OpenProt, v 1.3). A tabela também contém informações sobre o gene relacionado e transcrição e a localização da proteína dentro da transcrição.
- Clique no link detalhes para reunir mais informações. A recém-inaugurado página contém um navegador do genoma que é centralizado nos consultado de proteína e informações como a genômica e coordenadas de transcriptomic e a presença de um Kozak ou alta eficiência tradução iniciação local (TIS) motivo36, 37.
- Clique no DNA ou proteína links da guia de informação, para obter proteína ou sequências de DNA, respectivamente.
- Procure informações detalhadas sobre provas de MS, Ribossoma perfilação deteção, conservação e domínios da proteína identificada clicando sobre os guias top15.
Resultados
O fluxo de trabalho descrito acima foi aplicado a um conjunto de dados do MS disponível sobre o repositório de orgulho38,39. O estudo original desenvolveu um método (iMixPro), utilizando o isótopo estável rotulagem de aminoácidos na cultura de pilha (SILAC), para eliminar falsos positivos de afinidade-purificação MS (AP-MS) experimentos38. Em breve, um experimento de AP-MS consiste no uso de anticorpos ligados a grânulos para buscar uma proteína de interesse (isca) e suas interactianos (presas). As proteínas coletadas são então digeridas e preparadas para MS. O método de preparação de amostra e as configurações do instrumento são descritas no estudo original e no repositório do orgulho (PXD004246). Um desafio em tais experiências é a abundância de falsos positivos, nomeadamente de proteínas de ligação para as contas, mas não a isca. Aqui, usamos SILAC para gerar diferentes isótopos entre presas verdadeiras e falsos positivos: são 3 amostras de controle (sem isca) cultivadas em luz médio, 1 amostra expressando a isca cultivada num meio de luz e 1 amostra expressando a isca cultivada em meio pesado processado com os grânulos e posterior análise de espectrometria de massa. Com tal projeto, proteínas não-específica vinculação aos talões terá uma relação de pesados-à-luz de 1:4; Quando verdadeiras presas terá uma proporção de 1:1,38.
Re-analisamos seus AP-MS dados usando o banco de dados OpenProt; as iscas incluíam três proteínas endógenas (PTPN14, JIP3 e IQGAP1), e dois over expressaram proteínas (RAF1 e RNF41). Desde os experimentos usado SILAC, utilizou-se o fluxo de trabalho do Galaxy para quantificação de proteína (S3 de Material complementar, Figura 2). O fluxo de trabalho foi executado usando o inteiro OpenProt banco de dados (OpenProt_all) ou um restrito OpenProt (OpenProt_2pep, incluindo apenas proteínas anteriormente detectadas com um mínimo de dois peptídeos exclusivos).
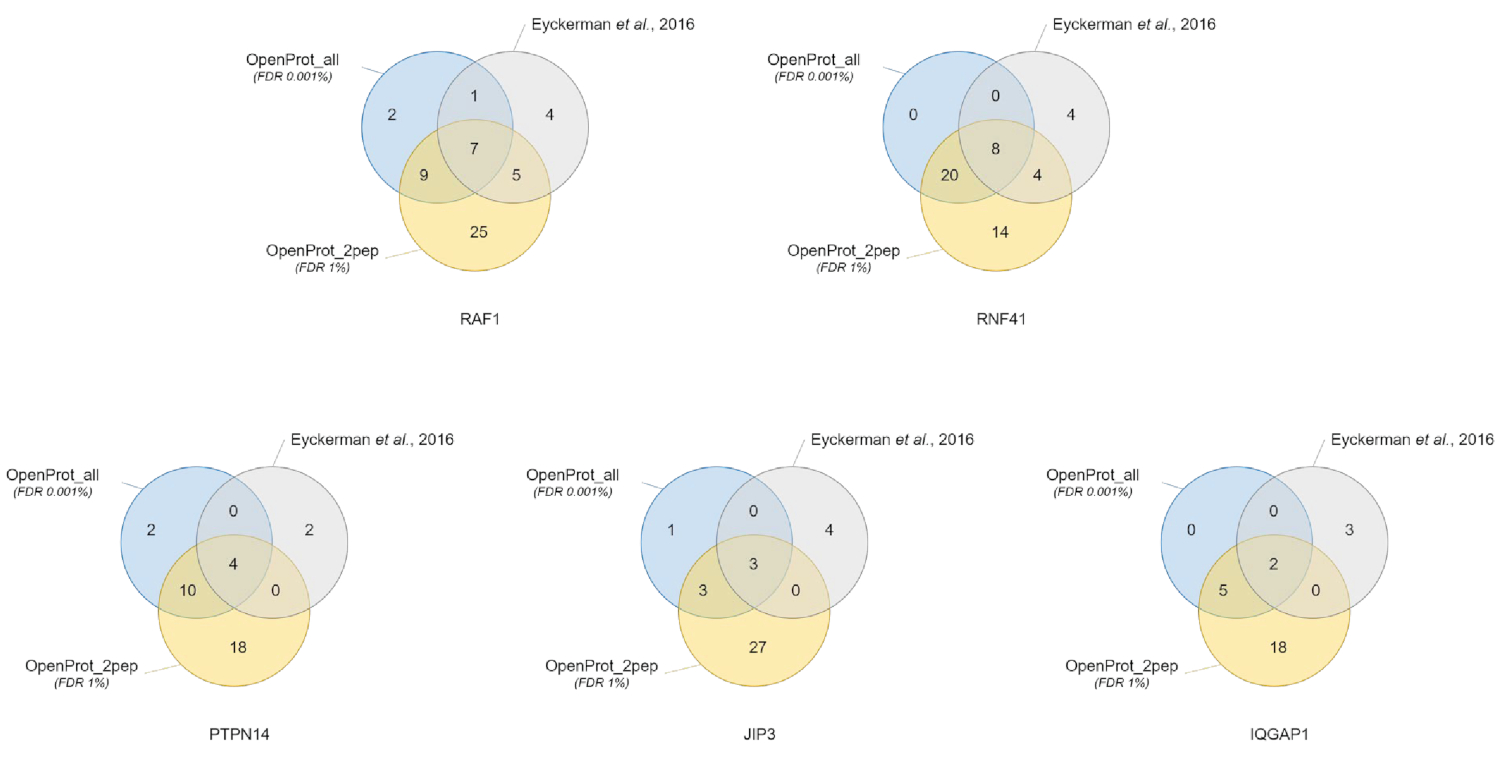
Quantificação e identificação de proteínas foram bons e podem ser reproduzidos através dos diferentes bancos de dados usados. Como mostrado na Figura 3, a maioria das proteínas identificadas no livro original também foram identificados usando o OpenProt_2pep ou OpenProt_all de banco de dados (uma lista detalhada está disponível em S5 de Material complementar). Este resultado mostra que o pipeline descrito aqui e o OpenProt de bancos de dados são capazes de produzir proteína identificação e quantificação comparável dos atuais procedimentos baseados na40UniProtKB bancos de dados. No entanto, o uso de bancos de dados de OpenProt tem a vantagem de permitir a deteção de romance e proteínas anteriormente indetectáveis, conforme demonstrado neste caso estudar.
11 proteínas bem suportadas (1 Isoform e 10 AltProts), no entanto, atualmente não anotados em bancos de dados, foram identificados através de todos os conjuntos de dados, com peptídeos confiantes, usando o banco de dados OpenProt_2pep (todas as adesões de proteína, juntamente com o número de apoio peptídeos, estão disponíveis em S5 de Material complementar). Este banco de dados permite o uso de um tradicional 1% FDR como o aumento do espaço de busca continua a ser moderado. Estas 11 proteínas não foram identificadas no estudo original, como eles estavam ausentes do banco de dados.
29 novas proteínas (16 isoformas e 13 AltProts) foram descobertas através de todos os conjuntos de dados, com peptídeos confiantes, usando o banco de dados OpenProt_all (todas as adesões de proteína, juntamente com o número de peptídeos de apoio, são S6 de Material complementar disponível em ). Como mostrado na Figura 3, o FDR rigorosa recomendado não afetou as identificações de proteína mais confiantes, embora ele diminuir o número total de proteínas identificadas. Comparativamente ao banco de OpenProt_2pep, um maior número de novas proteínas pode ser confiantemente identificado. Todas estas novas proteínas estão ausentes do banco de dados OpenProt_2pep. Isto ressalta o papel crucial do banco de dados escolhido para proteômica baseada em MS.
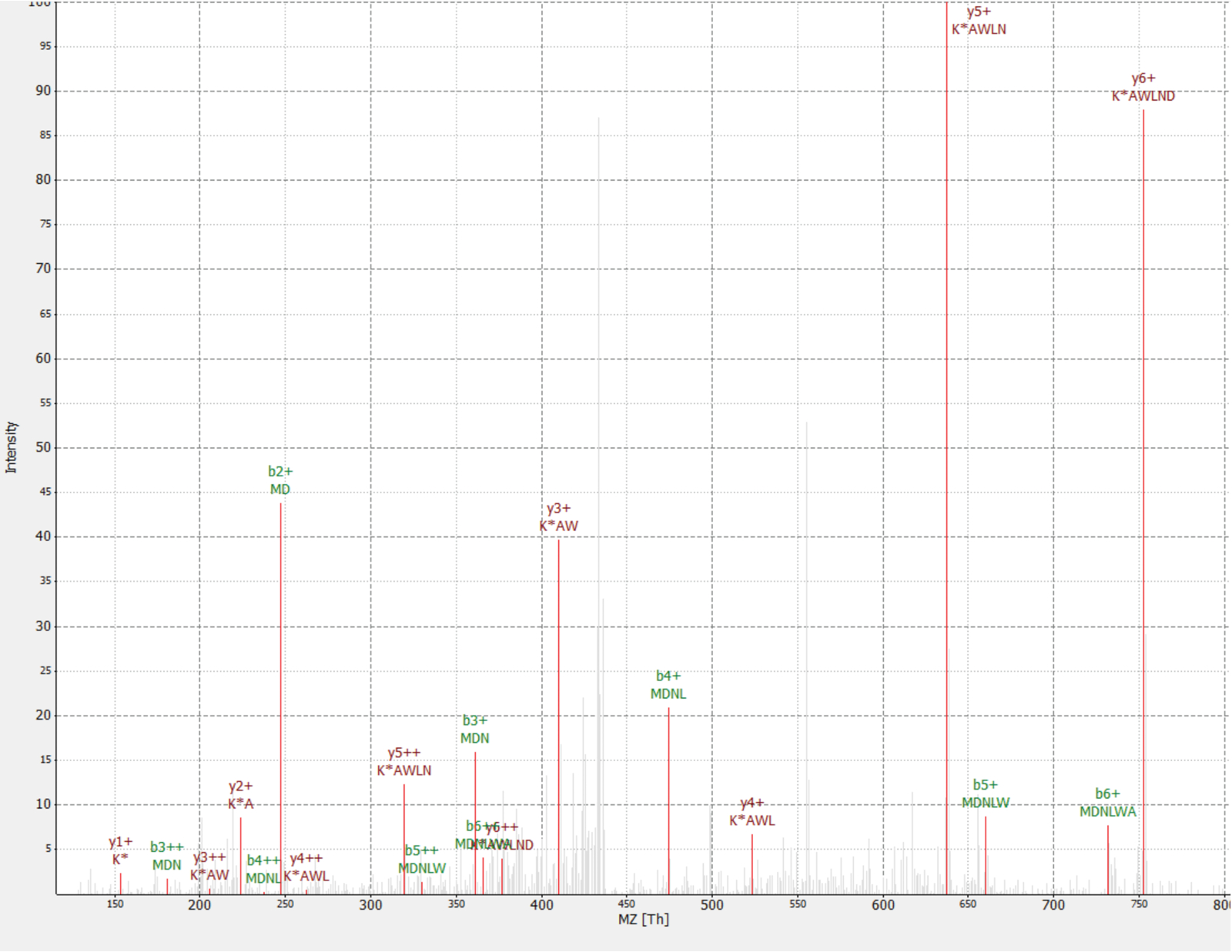
Uma nova proteína foi descoberta como um interactiano da proteína RAF1 (IP_637643). Usando o site OpenProt, pode-se ver esta proteína não foram detectada pelo MS nem Ribossoma perfilamento até agora (OpenProt v 1.3). A proteína é 46 aminoácidos longos e só pode dar dois peptídeos exclusivos mediante digestão tryptic. O peptídeo detectado em AP-MS a RAF1 dataset (fração 18) tinha um espectro de boa qualidade, como mostrado na Figura 4e exibido um rácio de pesados-à-luz de 1,09. A proteína é codificada no gene NANOGNBP1 , que é um pseudogene da NANOGNB. A transcrição (ENST00000448444), atualmente anotada como não-codificantes, foi detectada em vários tecidos de acordo com o portal GTEx40. A proteína contém um domínio funcional previsto associado com DNA de ligação (Gene Ontology GO: 0003677)41.

Figura 1 : Escolha para gráfico de análise proteômica do banco de dados. Análises de dados do MS, nomeadamente a escolha do banco de dados, dependem dos objectivos de investigação. Três objectivos comuns são descritos em azul (pipeline de proteomic clássico), verde (busca exaustiva proteomic) e laranja (descoberta proteomic). Cada objectivo depende de um banco de dados apropriado e pipeline. Uma ferramenta de identificação único pode ser utilizada para uma exaustiva e clássica proteomics pipelines. Para o pipeline de descoberta proteomic, recomendamos usar vários mecanismos de identificação. FDRs recomendados são indicados em vermelho, e tamanhos de banco de dados de proteínas são indicados nas caixas cinzentas. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2 : Representação gráfica do fluxo de trabalho do Galaxy usado. Passo a passo representação do fluxo de trabalho análise proteômica usado para re-análise de dados de Eyckerman et al.38. Arquivos de entrada, peptídeo pesquisa e quantificação de proteína são indicadas por caixas de laranja. Caixas azuis correspondem as ferramentas utilizadas e caixas cinzentas correspondem os arquivos de saída gerados. Os motores de busca diferentes (MS-GF + e X! Tandem) são indicados por cores diferentes (respectivamente vermelhas e roxas), bem como as setas indicando sua necessárias entradas e saídas. Caixa verde destaca a ferramenta para gerar uma lista de identificações de proteína. Quando são geradas várias saídas, usada para obter as etapas a jusante é indicada como o mais próximo para a seta. Este fluxo de trabalho está disponível gratuitamente no S2 de Material complementar. O X! Arquivo de configuração de parâmetros em tandem padrão está disponível em S4 de Material complementar. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3 : Comparação de identificação Interagente por isca usando diferentes bancos de dados. Diagramas de Venn de identificações de proteína usando o OpenProt mais confiante do banco de dados (em laranja, comprovativos de mínimos 2 peptídeos originais, OpenProt_2pep) com um 1% FDR, ou o OpenProt todo banco de dados (em azul, OpenProt_all) com um 0.001% FDR, ou conforme relatado o original de papel (em cinza)38. Cada diagrama corresponde a interactianos identificados para a isca mencionado: RAF1, RNF41, PTPN14, JIP3 e IQGAP1. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4 : MS/MS espectro de identificado MDNLWAK(6 de 13) peptídeo da proteína romance IP_637643. Intensidade é relativa (% de 0 a 100). Picos seleccionados são indicadas em vermelho, as anotações de íons de y são no escuro vermelho e b íons as anotações em verde. Extraído do TOPPview software34. Erro de precursor = 2,70 ppm, pontuação de PEP = 0,12. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Termo | Definição | Referência |
| ORF alternativo (AltORF) | não-canônicos ORF atualmente não anotados em anotações do genoma, mas anotados em OpenProt. | 15 |
| Referência ORF (RefORF) | ORF canônico anotado no genoma anotações e OpenProt. | 15 |
| Proteína alternativa (AltProt) | novela proteína codificada por um AltORF, com nenhuma similaridade significativa com um RefProt. Prefixo de adesão: IP_. | 15 |
| Proteína de referência (RefProt) | proteína atualmente anotada em bancos de dados de sequência de proteínas como UniProtKB, Ensembl ou RefSeq NCBI e também em OpenProt. | 15 |
| Isoform romance | novela proteína codificada por um AltORF, com uma semelhança significativa com um RefProt. Prefixo de adesão: II_. | 15 |
| OpenProt_2pep banco de dados | contém a sequência de todos os RefProts e novas proteínas previstas pela OpenProt, já detectado com um mínimo de 2 peptídeos exclusivos. | 15 |
| OpenProt_1pep banco de dados | contém a sequência de todos os RefProts e novas proteínas previstas pela OpenProt, já detectado com um mínimo de 1 único peptídeo. | 15 |
| OpenProt_all banco de dados | contém a sequência de todos os RefProts e novas proteínas previstas por OpenProt. | 15 |
Tabela 1: Definição de termos usados em OpenProt e em todo o protocolo
S1 de Material complementar: galáxia fluxo de trabalho para manipulação de banco de dados. Isto irá anexar as sequências CRAPome e engodo (reversos) no banco de dados de entrada. Saída é um arquivo Fasta. Clique aqui para baixar.
S2 de Material complementar: galáxia fluxo de trabalho para identificação de proteínas. Isto irá identificar as proteínas de um arquivo de dados de espectrometria de massa usando dois motores de busca (MS-GF + e X! Tandem). Cada parâmetro pode ser ajustado como desejado antes de executar o fluxo de trabalho. Clique aqui para baixar.
S3 de Material complementar: galáxia fluxo de trabalho para quantificação de proteína utilizando o isótopo estável rotulagem (SIL). Isto irá identificar e quantificar as proteínas de um arquivo de dados de espectrometria de massa usando dois motores de busca (MS-GF + e X! Tandem). Cada parâmetro pode ser ajustado como desejado antes de executar o fluxo de trabalho. Clique aqui para baixar.
S4 de Material complementar: X! Arquivo de configuração de parâmetros de padrão em tandem. XML este arquivo é necessário para executar o X! Ferramenta de TandemAdapter na plataforma da galáxia. Clique aqui para baixar.
S5 de Material complementar: quantificar proteínas de conjuntos de dados iMixPro. Arquivos de dados de Eyckerman et al. 201638 foram processados usando bancos de dados OpenProt e proteínas quantificadas são listadas para cada condição. As iscas são PTPN14, JIP3, IQGAP1, RAF1 e RNF41. Nomes de gene indicados em verde correspondem às proteínas também identificadas o papel original38. Nomes de gene indicados em laranja correspondem aos interactianos conhecidos de acordo com BioGrid que não foram relatados no livro original. Nomes de gene indicados em azul claro correspondem às novas proteínas identificadas como interactianos (o correspondente número de adesão de proteína é indicado entre parênteses). Nomes de gene indicaram em cinza claro e itálico corresponde aos prováveis contaminantes (proteínas de queratina). Clique aqui para baixar.
S6 de Material complementar: identificou novas proteínas de conjuntos de dados iMixPro. Arquivos de dados de Eyckerman et al. 201638 foram processados usando bancos de dados OpenProt e novas proteínas identificadas são listadas para cada condição. As iscas são PTPN14, JIP3, IQGAP1, RAF1 e RNF41. Números de adesão de proteína são listados, começando com II_ para romance isoformas de uma proteína conhecida e com IP_ para novas proteínas de uma alternativa ORF (AltProt). O número de peptídeos de apoio são indicadas entre parênteses. Clique aqui para baixar.
Discussão
Ao analisar dados de espectrómetros de massa, a qualidade da identificação de proteínas parcialmente depende da precisão do banco de dados usado6,20. As abordagens atuais usam tradicionalmente UniProtKB bancos de dados, no entanto, estas oferecem suporte o modelo de anotação do genoma de um único ORF por transcrição e um comprimento mínimo de 100 códons (com excepção dos exemplos previamente demonstrados)40. Vários estudos referem-se as deficiências de tais bancos de dados com a descoberta de ORFs funcionais de supostamente não-codificantes regiões8,11,12,13. Agora, OpenProt permite a identificação de proteína mais exaustiva como ele desenha sequências proteicas de múltiplas anotações transcriptome. OpenProt recupera RefSeq NCBI (GRCh38.p7) e transcriptomes de Ensembl (GRCh38.83) e anotações UniProtKB (UniProtKB-SwissProt, 2017-09-27)42,de40,43. Como anotações atuais apresentam pouca sobreposição, OpenProt, assim, exibe uma vista mais exaustiva da paisagem de proteomic potenciais do que quando limitada a uma anotação15.
Além disso, como OpenProt impõe um modelo policistrônico, permite múltiplas anotações de proteína por transcrição. Por motivos de estatísticos e computacionais, OpenProt mantém-se um limite de comprimento mínimo de 30 códons15. No entanto, prediz milhares de sequências de proteínas romance, assim, alargar o âmbito de possibilidades para identificação de proteínas. Com esta abordagem, OpenProt suporta proteomic descobertas de forma mais sistemática.
A qualidade da identificação de proteínas também pode ser afetada pelos parâmetros que são usados. MS-baseado proteomics análises normalmente mantenha um 1% de proteína FDR. No entanto, o banco de dados inteiro de OpenProt contém cerca de 6 vezes mais entradas (Figura 1). Para explicar este aumento substancial para o espaço, nós recomendamos usar um FDR mais rigorosa de 0,001%. Este parâmetro foi otimizado usando estudos de referência e avaliação manual dos espectros selecionado aleatoriamente15. Falso positivo são ainda uma possibilidade, embora, e nós encorajamos minuciosa inspeção e validação de provas para uma proteína de romance. Um padrão recomendado pode ser a identificação de uma proteína de duas execuções diferentes do MS, como os dados de fundo e falsos positivos variam entre conjuntos de dados15.
O gasoduto fornecidas aqui e utilizados para o estudo de caso pode ser modificado tão contente caber o delineamento experimental e parâmetros. Nós recomendaríamos usando vários motores de busca, como aumenta a sensibilidade e a sensibilidade do peptídeo identificação32. Além disso, incentivamos usando o banco de dados que melhor corresponde ao objectivo experimental (Figura 1). Como usar o OpenProt todo banco de dados vem com um FDR rigorosa, verdadeiras identificações podem ser perdidas. Assim, o banco de dados inteiro deve ser destinado a descoberta de novas proteínas, enquanto proteomics clássica de perfil deve estar usando os bancos de dados menores OpenProt (como OpenProt_2pep usado no estudo de caso acima).
OpenProt atualmente prevê sequências começando com um códon ATG, Considerando que diversos estudos destacou a iniciação da tradução em outros códons44,45. Quando uma proteína romance é identificada por um ou vários peptídeos exclusivos, é possível que o codão de iniciação verdadeira não é o presumível ATG. Os usuários podem procurar evidências de tradução no site OpenProt. Atualmente, OpenProt apenas relatórios de eventos de tradução se dizem respeito a toda proteína predita sequência (100% de sobreposição)15. Assim, ausência de evidência de tradução não significaria a proteína não é traduzida, mas que o códon de início pode não ser o suposta ATG.
Apesar de suas limitações atuais, o OpenProt oferece uma vista mais exaustiva do potencial de codificação dos genomas eucarióticos. OpenProt bancos de dados promover proteomic descobertas e a compreensão das funções de proteomic e interações. Desenvolvimentos futuros do banco de dados OpenProt irão incluir anotação de outras espécies, evidência de tradução do não-ATG começar códon e o desenvolvimento de um pipeline para incluir novas proteínas no genoma e estudos de sequenciamento exome.
Divulgações
Os autores declaram não há conflito de interesses.
Agradecimentos
Agradecemos a Vivian Delcourt por sua ajuda, discussões e conselhos sobre este trabalho. X.R. é um membro do Fonds de Recherche du Québec Santé FRQS-suporte do Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. Esta pesquisa foi apoiada por uma cadeira de pesquisa do Canadá em proteômica funcional e descoberta do romance proteínas X.R. e CIHR Grant MOP-137056. Agradecemos a equipe no cálculo Québec e Compute Canadá pelo seu apoio com o uso do mp2 supercomputador da Université de Sherbrooke. Operação do supercomputador mp2 é financiada pelo Canadá a Fundação de inovação (TPI), le ministère de l'Economie, de la science et de l'innovation du Québec (MESI) e les Fonds de Recherche du Québec - natureza et technologies (FRQ-NT). O servidor da galáxia que foi usado para alguns cálculos de proteômica é em parte financiado pelo colaborativo pesquisa centro 992 médica epigenética (grant DFG SFB 992/1 2012) e Ministério Federal alemão de educação e pesquisa (BMBF concede 031 A538A/A538C RBC, 031L0101B De /031L0101C. NBI-epi, de 0106 031L. ESCADA (de. NBI)).
Materiais
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
Referências
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados