Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Genotipado de ADN microsatélite y análisis de ploidía de citometría de flujo de tejidos molares de parafina incorporados en parafina-involuntaria-incrustadas
En este artículo
Resumen
Los lunares hidatidiformes son embarazos humanos anormales con etiologías heterogéneas que se pueden clasificar según sus características morfológicas y la contribución parental a los genomas molares. Aquí, los protocolos de genotipado de ADN microsatélite multiplex y citometría de flujo de tejidos molares de parafina incrustados fijados por formalina se describen en detalle, junto con la interpretación e integración de los resultados.
Resumen
El lunar hidatidiformo (HM) es un embarazo humano anormal caracterizado por una proliferación trofoblástica excesiva y un desarrollo embrionario anormal. Existen dos tipos de HM basados en la evaluación morfológica microscópica, HM completa (CHM) y HM parcial (PHM). Estos pueden subdividirse aún más en función de la contribución parental a los genomas molares. Tal caracterización de HM, por morfología y análisis de genotipos, es crucial para el manejo del paciente y para la comprensión fundamental de esta patología intrigante. Está bien documentado que el análisis morfológico de HM está sujeto a una amplia variabilidad interobservador y no es suficiente por sí solo para clasificar con precisión hm en CHM y PHM y distinguirlos de abortos hidropicos no molares. El análisis de genotipado se realiza principalmente en EL ADN y los tejidos de los productos de concepción de parafina fijados en formalina (FFPE), que tienen una calidad inferior a la óptima y, por lo tanto, pueden conducir a conclusiones erróneas. En este artículo, se proporcionan protocolos detallados para el genotipado multiplex y análisis de citometría de flujo de los tejidos molares FFPE, junto con la interpretación de los resultados de estos métodos, su solución de problemas y la integración con la evaluación morfológica , inmunohistoquímica p57KIP2 e hibridación in situ por fluorescencia (FISH) para llegar a un diagnóstico correcto y robusto. Aquí, los autores comparten los métodos y lecciones aprendidas en los últimos 10 años del análisis de aproximadamente 400 productos de concepción.
Introducción
Un lunar hidatidiformo (HM) es un embarazo humano anormal caracterizado por un desarrollo embrionario anormal, hiperproliferación del trofototo y degeneración hidrográfica de vellosidades coriónicas (CV). Históricamente, HM solía dividirse en dos tipos, HM completo (CHM) y HM parcial (PHM) basado únicamente en la evaluación morfológica1. Sin embargo, se ha demostrado que la evaluación morfológica por sí sola no es suficiente para clasificar HM en los dos subtipos (CHM y PHM) y distinguirlos de los abortos espontáneos no molares2,3,4.
Debido a que CHM y PHM tienen diferentes propensiones a las neoplasias malignas, por lo tanto, es importante determinar con precisión el tipo genotípico de HM para proporcionar un seguimiento y un manejo adecuados a los pacientes. En consecuencia, en las últimas décadas, se han desarrollado y evolucionado varias metodologías con el fin de identificar la contribución parental a los tejidos molares y alcanzar una clasificación correcta de HM. Estos incluyen análisis de cariotipo, polimorfismo de bandas cromosómicas, tilometría serológica de antígeno leucocito humano (HLA), polimorfismo de longitud de fragmento de restricción, número variable de repeticiones en tándem, genotipado de microsatélites, citometría de flujo y p57 Inmunohistoquímica KIP2. Esto ha permitido una subdivisión precisa de las concepciones hm basadas en la contribución de los padres a sus genomas, de la siguiente manera: CHM, que son diploides androgenéticos androgenéticos o diploides dispermicos androgenéticos, y PHM, que son triploide, disperménico en 99% y monobrímico en el 1% de los casos5,6,7,8. Además, hay otro tipo de HM genotípico que surgió en las últimas dos décadas, que es diploide biparental. Este último es en su mayoría recurrente y puede afectar a un solo miembro de la familia (casos simplex) o al menos a dos miembros de la familia (casos familiares). Estos lunares biparentales diploides son causados principalmente por mutaciones recesivas en NLRP7 o KHDC3L en los pacientes9,10,11,12. Diploid biparental HM en pacientes con mutaciones recesivas en NLRP7 puede ser diagnosticado como CHM o PHM por análisis morfológico y esto parece estar asociado con la gravedad de las mutaciones en los pacientes13,14. Además de la clasificación de HM según sus genotipos, la introducción y el uso de varios métodos de genotipado permitieron distinguir las diversas entidades molares de los abortos espontáneos no molares, tales como concepciones biparentales diploides aneuploides y otros tipos de concepciones5,15. Tales concepciones pueden tener alguna proliferación de trofoblastos y morfología villosa anormal que imitan, hasta cierto punto, algunas características morfológicas de HM.
El propósito de este artículo es proporcionar protocolos detallados para el genotipado multiplex y la citometría de flujo de los tejidos de parafina incrustada en formalina-fijación (FFPE), y análisis exhaustivos de los resultados de estos métodos y su integración con otros métodos para diagnóstico correcto y concluyente de los tejidos molares.
Protocolo
Este estudio de investigación fue aprobado por la Junta de Revisión Institucional de McGill. Todos los pacientes dieron su consentimiento por escrito para participar en el estudio y para que sus productos FFPE de concepción (POC) se recuperaran de varios departamentos de patología.
NOTA: Si bien existen varios métodos para el genotipado y la determinación de la ploidía por citometría de flujo, los protocolos proporcionados aquí describen un método de análisis utilizando una plataforma para cada uno.
1. Genotipado
-
Selección del mejor bloque FFPE
- Para cada producto FFPE de concepción (POC), prepare secciones teñidas de hematoxilina y eosina (H&E) de 4 m de espesor, tal como se describe en las secciones 1.2 y 1.3, una para cada bloque disponible, para la evaluación morfológica por microscopía.
- Usando las diapositivas H&E y un microscopio de luz, seleccione el bloque FFPE que tiene la mayor cantidad de vellosidades coriónicas (CV), y si es posible, el bloque que tiene CV separado de, y no entremezclado con, tejidos maternos.
-
Seccionamiento
- Coloque el bloque elegido sobre hielo durante 15 minutos para facilitar el seccionamiento.
- Ajustar el microtomo para cortar secciones de 4 m de espesor para la evaluación morfológica microscópica y de 10 m de espesor para la extracción de ADN.
- Coloque el bloque frío en el microtomo y corte una sección de cada bloque para la tinción de H&E y 10 a 30 secciones del bloque elegido, dependiendo de la cantidad de CV en el bloque, para la extracción de ADN.
NOTA: Para los bloques que están llenos de CV, 10 secciones son suficientes para la extracción de ADN. Si sólo alrededor del 10% del bloque contiene CV mientras que el resto son tejidos maternos, entonces se necesitan de 20 a 30 secciones para asegurar cantidades suficientes de ADN. - Con fórceps, transfiera cada sección a un baño de agua de 45 oC. Recoja la sección del baño de agua con una diapositiva cargada positivamente(Tabla de Materiales)que previamente está etiquetada con el número de identificación de la muestra usando un lápiz.
- Coloque los portaobjetos que contienen las secciones en un horno a 65 oC para permitir que las secciones se adhieran a los portaobjetos. Mantener los portaobjetos para H&E en el horno durante 25 min. Mantener los portaobjetos para la extracción de ADN en el horno durante 20 min.
NOTA: El menor tiempo de incubación hace que los tejidos sean ligeramente menos adherentes a las diapositivas y, en consecuencia, facilita la extracción de los tejidos maternos.
-
Tinción de H&E
- Deje que los portaobjetos se enfríen a temperatura ambiente (10 min).
- Preparación de reactivos
- Preparar la solución de trabajo de Eosin Y (0,25%) según el Cuadro 1. Mezclar bien y almacenar a temperatura ambiente.
- Preparar la solución de hematoxilina de trabajo diluyendo la solución de hematoxilina 5x en agua (es decir, mezclar 80 ml de agua con 20 ml de hematoxilina).
NOTA: Envuelva la solución de material de hematoxilina en papel de aluminio para su almacenamiento.
- Prepare los frascos de tinción con los reactivos correctos bajo una campana de humo según la Tabla 2.
- Realice la tinción H&E sumergiendo las diapositivas en los frascos de tinción apropiados durante el período de tiempo correcto de acuerdo con la Tabla 2.
- Montar las secciones de 4 m para el análisis morfológico con medio de montaje y cubrecondo con tapas de vidrio(Tabla de Materiales).
NOTA: No se deben cubrir las secciones de 10 m para el genotipado. - Deje las secciones de 10 m debajo de la campana de humos durante un mínimo de 3 h para que los olores tóxicos de xileno se disipe.
PRECAUCION: Todos los pasos de tinción deben realizarse bajo una campana de humo. Los productos de xileno deben mantenerse bajo el capó en todo momento porque los olores de xileno son tóxicos. Además, el xileno y la hematoxilina deben desecharse en recipientes especiales. Una vez que estos contenedores están llenos, deben ser desechados según lo recomendado por la organización de seguridad del laboratorio.
| Reactivo | Cantidad |
| Solución de stock de Eosin Y (1%) | 250 ml |
| 80% Etanol | 750 mL |
| Acido acético glacial (concentrado) | 5 mL |
Tabla 1: Solución de trabajo Eosin Y (0,25%) Preparación.
| Reactivo utilizado (100 ml por contenedor) | Duración |
| 1) Xileno | 5 min |
| 2) Xileno | 5 min |
| 3) 100% Etanol | 2 min |
| 4) 95% Etanol | 2 min |
| 5) 70% Etanol | 2 min |
| 6) 50% Etanol | 2 min |
| 7) Agua destilada | 5 min |
| 8) Hematoxilina | 4 min |
| 9) Agua destilada | 5 min |
| 10) Eosin | 1 min |
| 11) 95% Etanol | 5 min |
| 12) 100% Etanol | 5 min |
| 13) Xileno | 5 min |
| 14) Xileno | 5 min |
Tabla 2: Reactivos y duraciones para el protocolo de tinción H&E.
-
Aislamiento del CV
- Bajo un estereomicroscopio ligero, utilice fórceps y pequeños trozos de toallitas de papel humedecidas por agua(Tabla de materiales)para raspar los tejidos maternos no deseados de secciones de 10 m de espesor teñidas por H&E.
NOTA: El objetivo final es mantener nada más que CV o membranas fetales (cuando estén presentes) en las diapositivas y así eliminar todos los demás tejidos. Este paso puede necesitar mucho tiempo y paciencia, dependiendo del bloque, ya que requiere una atención meticulosa a los detalles. - Pida a una segunda persona que revise dos veces los portaobjetos después de la limpieza para asegurarse de que estén libres de tejidos maternos.
- Tome fotos de las diapositivas limpiadas o documente lo siguiente para ayudar con la interpretación de datos: 1) si el tejido era difícil de limpiar, hemorrágico o muy limpio, 2) el número de secciones utilizadas, y 3) la cantidad aproximada de tejidos limpios.
NOTA: La Figura 1 proporciona un ejemplo de una diapositiva que es fácil de limpiar. Para un bloque que contiene aproximadamente esta cantidad de CV, 10 secciones son suficientes para la extracción de ADN. La diapositiva de la Figura 2 tiene muy pocos CV que se entremezclan con tejidos maternos, lo que hace que sea muy difícil y lento limpiar. Para un bloque que contiene aproximadamente esta cantidad de CV, se necesitan 30 secciones para la extracción de ADN. - Recoja el CV con pequeñas toallitas de papel humedecidas. Usando los fórceps, arranca una pequeña pieza de las toallitas de papel humedecida y úsala para recoger el CV.
- Coloque las toallitas de papel con su CV adjunto en un tubo etiquetado de 1,5 ml.
- Minimice la cantidad de toallitas de papel utilizadas en este paso, ya que demasiado puede obstruir la columna de extracción de ADN y, en consecuencia, reducir la cantidad final de ADN recogido. En promedio, tenga como objetivo utilizar menos de siete pequeños trozos de toallitas de papel por muestra. Si eso no es posible debido a la presencia de grandes cantidades de CV, divida la muestra entre dos tubos para facilitar la extracción.
- Bajo un estereomicroscopio ligero, utilice fórceps y pequeños trozos de toallitas de papel humedecidas por agua(Tabla de materiales)para raspar los tejidos maternos no deseados de secciones de 10 m de espesor teñidas por H&E.

Figura 1: Diapositiva representativa para el genotipado. Arriba: Una diapositiva que necesita ser "limpiada" para liberarse de los tejidos maternos. Parte inferior: La misma diapositiva que se muestra después de que se ha limpiado y ahora no contiene nada más que CV para la extracción de ADN. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Diapositiva representativa para el genotipado. Arriba: Una diapositiva que necesita ser "limpiada" para liberarse de los tejidos maternos. Parte inferior: La misma diapositiva que se muestra después de que se ha limpiado y ahora no contiene nada más que CV para la extracción de ADN. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Siga el protocolo de la extracción de ADN del kit FFPE(Tabla de Materiales)para realizar la extracción de ADN.
NOTA: Algunos kits recomiendan el uso de 15 a 20 l de búfer de elución para la elución final. Por experiencia, la elución con 15 l de tampón de elución funciona bien para la mayoría de las muestras. Las diluciones se pueden preparar a partir del ADN de la acción según sea necesario.
-
Cuantificación del ADN
- Usando un dispositivo de espectrofotómetro de laboratorio, cargue 1 l de ADN y mida la absorbancia a 260 nm para la cuantificación.
- Cargue 1 l de ADN en un gel de agarosa del 2% y ejecute la electroforesis de gel a una tensión de 80 a 100 V para una evaluación cualitativa.
- Sobre la base de los resultados de los pasos 1.6.1 y 1.6.2, elija el volumen de ADN que se utilizará en la amplificación de la reacción en cadena de la polimerasa (PCR) de repetición en tándem corto multiplex (STR). Intenta utilizar un mínimo de 1000 ng de ADN en la amplificación de PCR que sigue.
NOTA: La Figura 3 muestra ejemplos representativos de geles junto con las concentraciones del ADN (basados en los resultados del espectrofotómetro), y el volumen de la solución de ADN que se recomienda para el multiplexS STR PCR que sigue.

Figura 3: Gel representativo para la cuantificación del ADN. Se incluyen las concentraciones de cada ADN, medida utilizando un espectrofotómetro, y las cantidades utilizadas para el PCR multiplex. Haga clic aquí para ver una versión más grande de esta figura.
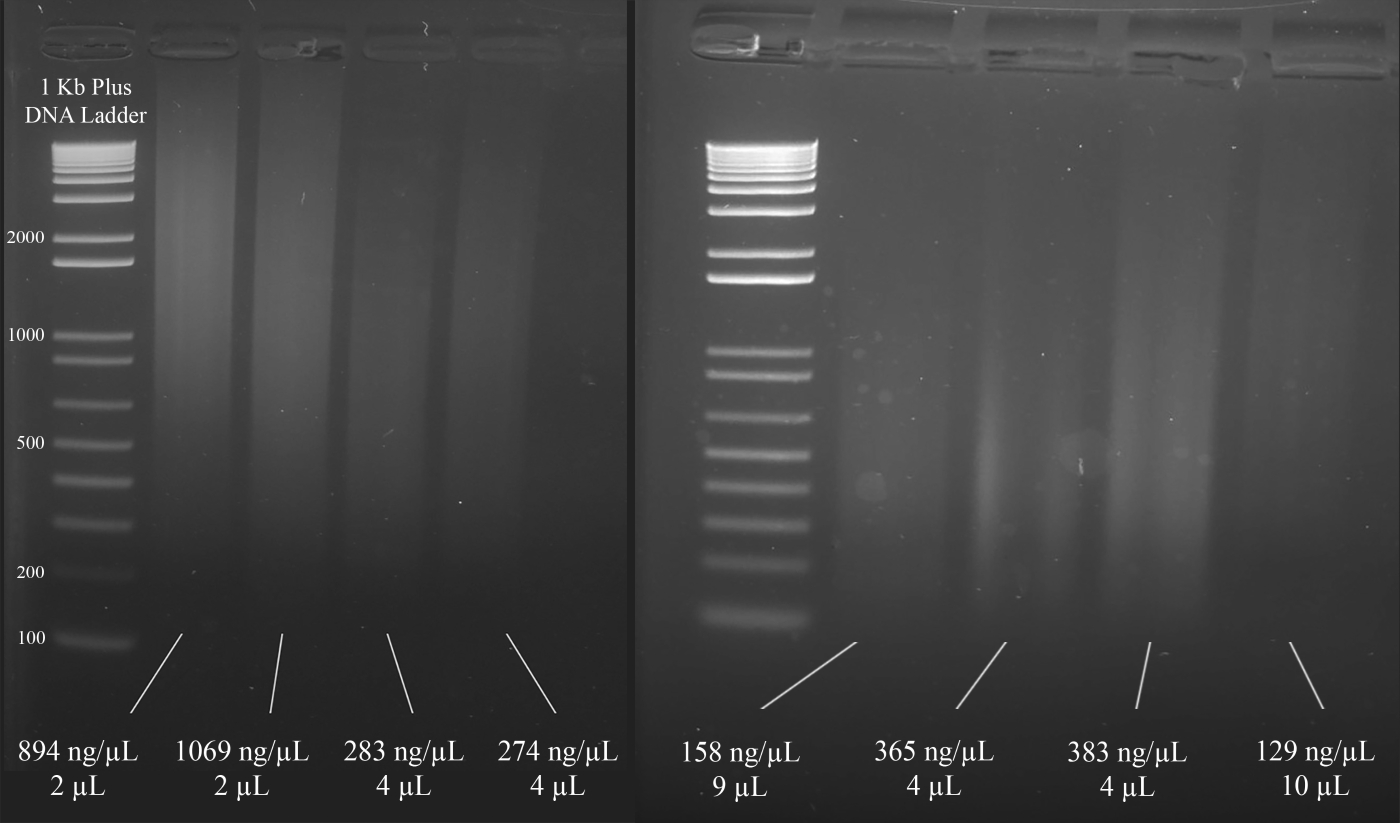{kind=link}
-
Amplificación de PCR
- Realizar genotipado de microsatélites fluorescentes utilizando un sistema STR multiplex(Tabla de materiales).
- Utilice las condiciones de PCR que se muestran en la Figura 4 para la amplificación de PCR utilizando el sistema STR multiplex(Tabla de materiales).
NOTA: Los siguientes imprimadores se utilizan en este sistema STR multiplex: D18S51, D21S11, TH01, D3S1358, Penta E, FGA, TPOX, D8S1179, vWA, Amelogenin, CSF1PO, D16S539, D7S820, D13S317, D5S818 y D.

Figura 4: Condiciones del ciclo PCR para el sistema STR multiplex. Haga clic aquí para ver una versión más grande de esta figura.
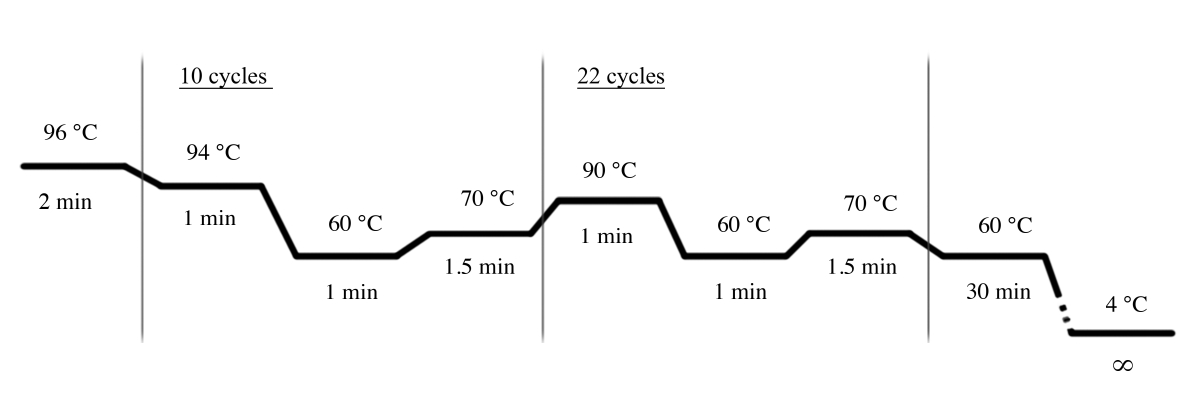{kind=link}
-
Resolver los productos PCR por electroforesis capilar.
- Suspenda 1 l de cada muestra amplificada en 0,5 l del carril estándar interno del sistema multiplex y 9,5 ml de formación altamente desionizada(Tabla de materiales).
- Ejecute muestras a través de un instrumento de electroforesis capilar(Tabla de materiales) utilizando una matriz de separación adecuada(Tabla de materiales)para el instrumento y el conjunto de tinte sin sistema multiplex.
- Análisis de datos
- Analice los datos con un software de análisis de fragmentos de ADN y compare los alelos POC con los alelos parentales para determinar su origen.
- Configure un estándar de tamaño.
NOTA: Esto permite que el software reconozca la escalera que se utiliza en el sistema STR multiplex, y asignar pares de bases a los amplicons basados en la escalera. Los siguientes pasos son para un software específico(Tabla de materiales),pero también pueden ser de ayuda para configurar otros tipos de software.- Abra el software. Haga clic en Iniciar nuevo proyecto y luego en Nuevo tamaño estándar.
- Asigne un nombre al estándar de tamaño (por ejemplo, ABI_600).
- En el cuadro llamado Introducir nueva definición estándar:escriba lo siguiente: 60, 80, 100, 120, 140, 160, 180, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 550, 600. A continuación, haga clic en Añadir tamaño(s).
NOTA: Los números introducidos aparecerán debajo del cuadro de la derecha, que se denomina Definición estándar de tamaño actual (consulte la figura 5). - Haga clic en Guardar.
- Para importar y analizar un archivo, haga clic en Agregar archivosy elija el archivo fsa que desea analizar. Haga clic en Agregar archivos seleccionados y luego en Aceptar. A continuación, siga estos pasos:
- Busque la columna Size Standard y elija ABI_600 (o cualquier nombre que se haya dado al estándar de tamaño).
- En Método de análisis, haga clic en Tamaño predeterminado - NPP y, a continuación, haga clic en el botón verde Analizar.
- El archivo ya está listo para su visualización. Ajuste las opciones de visualización para ver los datos como desee.
- Solución de problemas - método de análisis
NOTA: El software a veces puede fallar para identificar los picos y alinearlos correctamente. Esto sucede cuando los picos son demasiado bajos o demasiado altos. Los dos métodos de análisis siguientes pueden corregirse para esto y deben probarse antes de que se vuelva a probar una muestra.- Método de análisis 1 para picos altos:
- Haga clic en Nuevo método de análisis y asímócelo High Peaks (u otro nombre según sus preferencias personales).
- Haga clic en Rango y luego en Rango parcial para el análisis y el tamaño. A continuación, escriba 100 para el punto inicial y el tamañoinicial .
- Para el punto de parada, escriba 10.000. Para el tamaño de detención, escriba 1000.
- A continuación, haga clic en Alturas máximas mínimas y cambie los números de modo que el umbral máximo para los colores sea el siguiente: Azul: 50; Verde: 50; Amarillo: 20; Rojo: 100; Naranja: 5000.
- Guarde el nuevo método de análisis.
- Método de análisis 2 para picos bajos:
- Haga clic en Nuevo método de análisis y asímócelo Low Peaks (u otro nombre según sus preferencias personales).
- Haga clic en Rango y luego en Rango parcial para el análisis y el tamaño. A continuación, escriba 100 para el punto inicial y el tamañoinicial .
- Para el punto de parada, escriba 10.000. Para el tamaño de detención, escriba 1000.
- A continuación, haga clic en Indicadores de calidad y cambie el rango de paso de tal forma que se lee de 0.5 a 1. Cambie el rango de baja calidad de tal forma que lea de 0.0 a 0.0. Cambie La linealidad de suasto a lo siguiente: de (bp) 100.0 a (bp) 800.0.
- Guarde el nuevo método de análisis.
NOTA: Ahora es posible volver a analizar un archivo eligiendo Low Peaks o High Peaks en Analysis Method y luego haciendo clic en el botón verde Analyze.
- Método de análisis 1 para picos altos:

Figura 5: Captura de pantalla que muestra el Editor estándar de tamaño. Haga clic aquí para ver una versión más grande de esta figura.
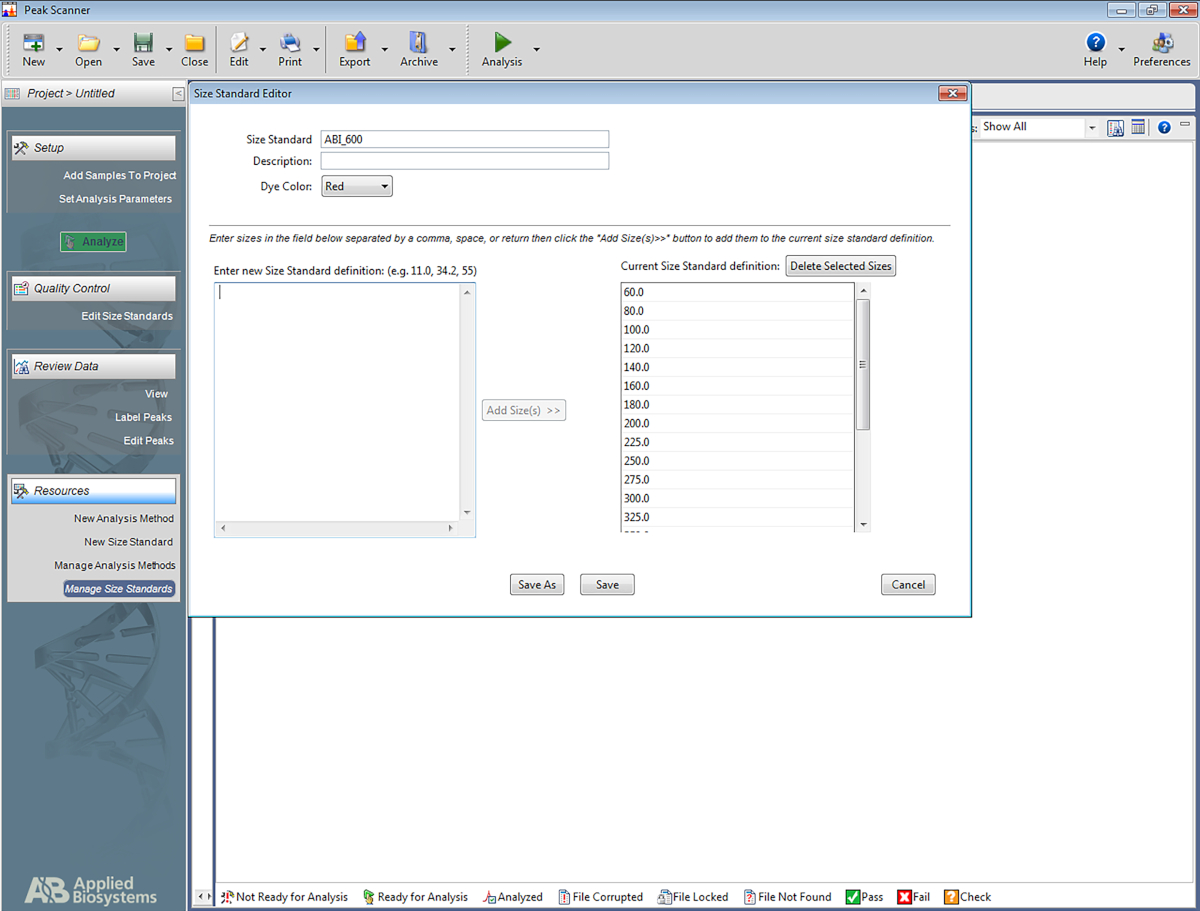{kind=link}
2. Citometría de flujo
-
Elegir el bloque FFPE ideal
- Usando diapositivas H&E y un microscopio de luz, seleccione un bloque FFPE que tenga alrededor del 50-70% de sus tejidos compuestos de CV.
NOTA: La Figura 6 es un ejemplo representativo de un bloque apropiado para el análisis de citometría de flujo, ya que se compone de aproximadamente 50% CV (mitad derecha de la sección) y 50% de tejidos maternos (mitad izquierda). La presencia de tejidos maternos es importante porque sirven como control interno para el pico diploide. - Para bloques que no tienen la cantidad ideal de CV, enriquecer para CV a medida que se realiza el seccionamiento. Para ello, identifique qué lado de las secciones recién cortadas contiene más CV de acuerdo con su diapositiva H&E correspondiente. Sobre la base de eso, utilice una cuchilla para cortar la otra mitad que necesita ser desechada para enriquecer para el CV.
NOTA: La Figura 7 muestra un bloque que no tiene suficiente CV para el análisis de citometría de flujo. Para bloques como éste, las secciones deben ser cortadas de tal manera que la mitad que contiene menos CV se desecha con el fin de aumentar las cantidades de CV con respecto a los tejidos maternos, como se muestra en la figura. Asegúrese de cortar más secciones para compensar lo que se descarta.
- Usando diapositivas H&E y un microscopio de luz, seleccione un bloque FFPE que tenga alrededor del 50-70% de sus tejidos compuestos de CV.

Figura 6: Sección H&E que representa un bloque POC que es ideal para la citometría de flujo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Sección H&E que representa un bloque más difícil para la citometría de flujo. Esta sección representativa de H&E muestra que sólo la mitad inferior de esta sección debe utilizarse para el análisis de citometría de flujo, con el objetivo de enriquecer el CV. El área esbozada, etiquetada como "CV", se compone principalmente de CV. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Seccionamiento
- Deje los bloques sobre hielo durante 15 minutos para facilitar el seccionamiento.
- Usando el mejor bloque FFPE posible, corte cuatro secciones que sean de 50 m de espesor (o dos secciones de 100 m de espesor) usando un microtome.
NOTA: Para la citometría de flujo es preferible tener secciones más gruesas. - En el caso de que no se disponga de un bloque FFPE ideal, tenga como objetivo mantener la relación de CV con el tejido materno. Por ejemplo, si sólo el 30% del bloque está compuesto por CV mientras el resto tiene tejidos maternos, retire al menos la mitad de la sección que contiene los tejidos maternos y use más secciones para compensar (ver Figura 7).
- Coloque las secciones en tubos etiquetados de 15 ml.
NOTA: Asegúrese de pegar sobre las etiquetas porque los reactivos orgánicos utilizados en el siguiente paso pueden disolverse y eliminar la tinta.
- Protocolo de citometría de flujo de los tejidos FFPE
- Desparafinación y rehidratación
- Realice los siguientes lavados (Tabla 3) bajo una campana de humos.
- Llene el tubo de 15 ml con 6 ml del reactivo apropiado, siguiendo el orden presentado en la Tabla 3, deje las secciones en los reactivos durante la duración respectiva y, a continuación, retire el reactivo con aspiración al vacío y una pipeta Pasteur de vidrio.
- Entre cada paso, sumerja la pipeta Pasteur primero en 70% de etanol, luego en agua destilada, y luego proceda al siguiente paso.
- Tenga mucho cuidado de no extraer trozos de tejido junto con el reactivo. Incline el tubo de 15 ml a un ángulo de 60 grados para facilitar la succión del reactivo líquido sin dibujar tejidos.
PRECAUCION: Los líquidos desechados contienen xileno y deben eliminarse en recipientes de residuos de xileno.
- Preparación de la solución
- Preparar la solución de citrato disolviendo 2 g de ácido cítrico en 1 L de agua destilada doble. Lleve el pH a 6. Conservar a 4oC.
- Preparar la solución de pepsina disolviendo 0,01 g de pepsina en 2 ml de 9 partes por mil NaCl, pH 1,64. Esto es para una muestra.
PRECAUCION: La pepsina es tóxica y puede dispersarse fácilmente y en el aire. Use una máscara cuando manipule la pepsina en su forma de polvo y limpie toda el área de trabajo después de usarla. - Propidium Iodida (PI)-ribonucleano Preparación de la solución para una muestra.
- Mezclar 50 s l de PI con 450 s de PBS (para diluir 10x).
- Añadir 50 ml de ribonucleasa A (1 mg/ml) a la mezcla. Mantener envuelto en papel de aluminio en todo momento.
- Digestión y tinción
- Añadir la solución de citrato de 4 oC a los tubos de 15 ml y luego colocar en un baño de agua de 80 oC durante 2 horas.
- Deje que la solución se enfríe a temperatura ambiente (15 min). Retire la solución de citrato.
- Agregue 6 mL de 1x PBS, vórtice, y espere 1 x 2 min para permitir que los tejidos se asienten en la parte inferior. Retire el 1x PBS con succión al vacío y una pipeta Pasteur de vidrio.
- Añadir 1 ml de solución de pepsina (precalentada a 37oC) y colocar en un baño seco de 37oC durante 30 min. Vortex cada 10 min. Preparar la solución PI-ribonuclea Una solución en los últimos 10 min de esta incubación.
- Agregue 6 mL de 1x PBS, vórtice, y espere 1 x 2 min para permitir que los tejidos se asienten en la parte inferior. Retire el 1x PBS con succión al vacío y una pipeta Pasteur de vidrio.
- Añadir 550 l de la solución PI-ribonuclease A y colocar las muestras en un baño seco de 37 oC durante 30 minutos.
NOTA: En este punto, las muestras se pueden envolver en papel de aluminio y dejar durante la noche a 4 oC hasta la mañana siguiente. - Filtre la solución a través de una malla de filtración de 48 m. Recoger el filtrado en tubos de fondo redondo de poliestireno, que se puede utilizar con el catómetro de flujo. Utilice fórceps para colocar una pieza de 5 cm por 5 cm de malla de filtración en la parte superior del tubo, de forma que el líquido se pueda canalizar a través de la malla y en el tubo.
NOTA: Las muestras ya están listas para ejecutarse con el catómetro de flujo. Manténgalos envueltos en papel de aluminio hasta que estén listos para ser ejecutados.
- Ejecute muestras con un catómetro de flujo con la ayuda del técnico de plataforma de citometría de flujo de la organización.
NOTA: El canal PE se utiliza para detectar el ADN manchado de PI y el caudal debe establecerse en Lento durante la adquisición. Asegúrese de que la tensión se elige de tal manera que el pico diploide está aproximadamente en 200 a lo largo del eje X PE-A para facilitar el análisis y la interpretación. Intenta registrar un mínimo de 20.000 eventos por muestra.
- Desparafinación y rehidratación
| Reactivo utilizado (6 ml cada uno) | Duración |
| 1) Xileno | 2 x 10 min |
| 2) 100% Etanol | 2 x 10 min |
| 3) 95% Etanol | 10 min |
| 4) 70% Etanol | 10 min |
| 5) 50% Etanol | 10 min |
| 6) Agua destilada | 2 x 10 min |
Tabla 3: Reactivos y duraciones para la desparafinación y rehidratación.
-
Análisis de datos de citometría de flujo
- Analizar datos con un software de análisis de citometría de flujo(Tabla de materiales).
NOTA: Los siguientes pasos son para un software específico(Tabla de materiales),pero también pueden ser de ayuda para configurar otros tipos de software.- Después de ejecutar las muestras en un citometro de flujo, descargue los archivos FCS 2.0 para su análisis.
- Abra el software de análisis de citometría de flujo, haga clic en Archivo . Nuevo documento.
- Haga clic en el
 icono Histograma ( ) y, a continuación, arrastre el puntero para crear un rectángulo.
icono Histograma ( ) y, a continuación, arrastre el puntero para crear un rectángulo. - Busque el archivo FCS y haga clic en Abrir. A lo largo del eje X, haga clic en FCS-A y, a continuación, seleccione PE-A.
- Haga clic en el
 icono Gráfica de puntos ( ) y, a continuación, arrastre el puntero para crear otro rectángulo debajo del trazado del histograma. A continuación, busque el mismo archivo FCS seleccionado para el histograma.
icono Gráfica de puntos ( ) y, a continuación, arrastre el puntero para crear otro rectángulo debajo del trazado del histograma. A continuación, busque el mismo archivo FCS seleccionado para el histograma. - Cambie el eje x de la gráfica de puntos a PE-A y el eje Y a PE-W.
NOTA: La Figura 8A muestra la apariencia de las gráficas en este punto. - Haga clic en el
 icono Región ( ) y dibuje un cuadro en la gráfica de puntos que comienza antes del pico diploide (alrededor de 100 en el eje X en la Figura 8B)y que termina alrededor de 700 en el eje X, como se muestra en la gráfica de puntos en la Figura 8 B.
icono Región ( ) y dibuje un cuadro en la gráfica de puntos que comienza antes del pico diploide (alrededor de 100 en el eje X en la Figura 8B)y que termina alrededor de 700 en el eje X, como se muestra en la gráfica de puntos en la Figura 8 B.
NOTA: El pico diploide en la Figura 8 es aproximadamente en 200 en el eje X. Esto se elige arbitrariamente ya que las muestras se registran a través del citómetro de flujo, simplemente para facilitar el análisis y la interpretación de los resultados. - Haga clic en Trazar Edite Regiones/Puertasy, a continuación, escriba R0 en la celda situada junto a la celda G0 en Estrategia. A continuación, haga clic en Cerrar.
- Haga clic en cualquier parte del histograma y, a continuación, en Gráfica de la zona de la zona de la zona de la zona de la zona de la Formato trazado/superposición. En Puerta, seleccione G0 á R0 y, a continuación, haga clic en Aceptar.
NOTA: Este es el paso de gating que permite visualizar mejor los picos de ploidy. El histograma ahora debería parecerse al histograma de la Figura 8B. Es posible jugar con la puerta creada (moviendo el cuadro dibujado en el paso 2.4.1.7) con el fin de centrarse en regiones específicas de la gráfica de puntos. - Para etiquetar los trazados, haga clic
 en el icono de área de texto ( ) y, a continuación, arrastre el puntero para crear un cuadro en la parte superior del documento y, a continuación, escriba la siguiente información: ID de paciente, ID de POC y el bloque utilizado (ya que puede haber varios bloques para un POC) , porcentaje CV presente en el bloque, voltaje utilizado para ejecutar la muestra y la fecha.
en el icono de área de texto ( ) y, a continuación, arrastre el puntero para crear un cuadro en la parte superior del documento y, a continuación, escriba la siguiente información: ID de paciente, ID de POC y el bloque utilizado (ya que puede haber varios bloques para un POC) , porcentaje CV presente en el bloque, voltaje utilizado para ejecutar la muestra y la fecha.
- Analizar datos con un software de análisis de citometría de flujo(Tabla de materiales).

Figura 8: Captura de pantalla que muestra un histograma y una gráfica de puntos de una muestra representativa que está desencerrada (A) y cerrada (B). Haga clic aquí para ver una versión más grande de esta figura.
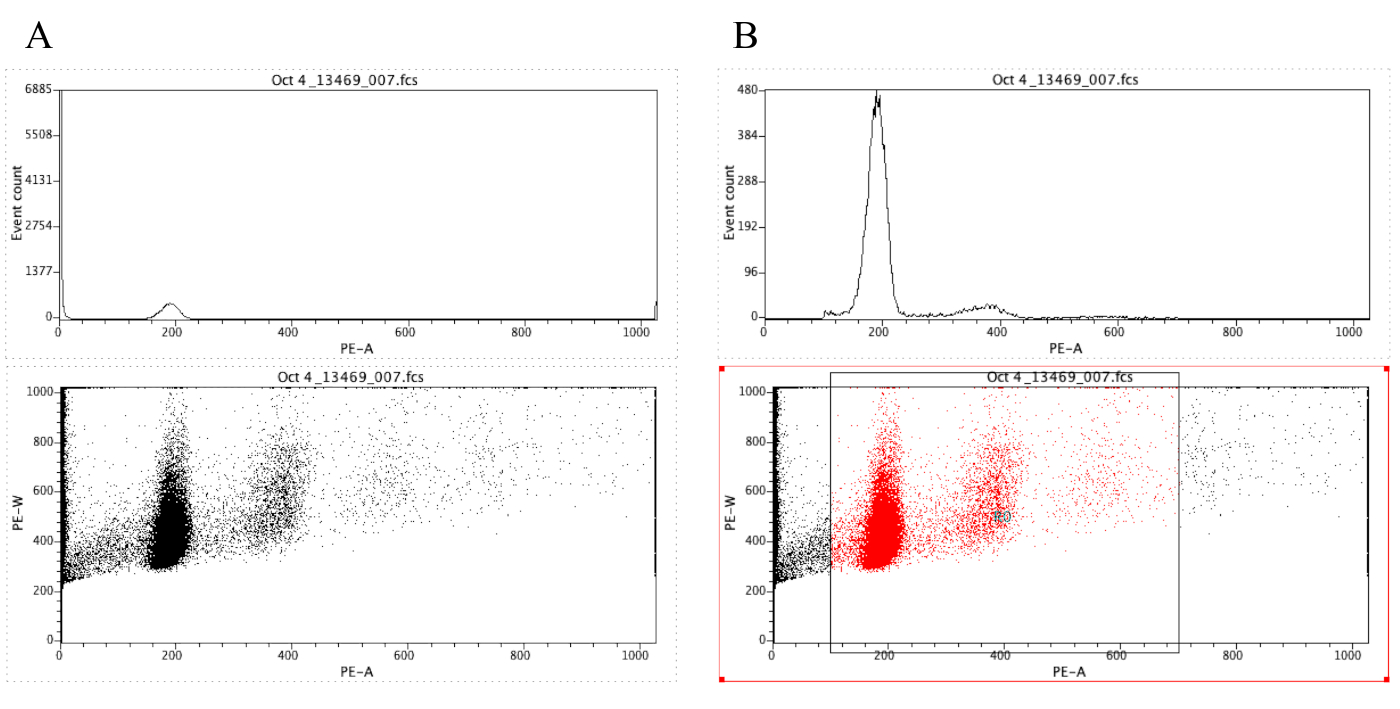{kind=link}
Resultados
La complejidad de los tejidos molares y el hecho de que puedan tener varios genotipos requieren un análisis riguroso y el uso de varios métodos como la evaluación morfológica, la inmunohistoquímica p57, el genotipado de microsatélites, la citometría de flujo y el FISH. Por ejemplo, un paciente (1790) fue referido con dos PHM que se encontró triploide por el análisis de microarray de los POC solamente. Por lo tanto, el paciente fue diagnosticado con PHM recurrente. El genotipado m...
Discusión
Los HM son embarazos humanos anormales con etiologías heterogéneas y tienen diferentes tipos histológicos y genotípicos, lo que hace que su clasificación y diagnóstico precisos sean desafiantes. La evaluación morfológica histopatológica a menudo se demostró inexacta y, por lo tanto, no es fiable por sí sola para clasificar a HM en CHM y PHM y distinguirlos de los abortos espontáneos no molares. Por lo tanto, un diagnóstico preciso de HM requiere el uso de otros métodos como el genotipado de ADN multix micro...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores agradecen a Sophie Patrier y Marianne Parésy por compartir el protocolo original de citometría de flujo, y Promega y Qiagen por proporcionar suministros y reactivos. Este trabajo fue apoyado por el Réseau Québécois en Reproduction y el Instituto Canadiense de Investigación Sanitaria (MOP-130364) a R.S.
Materiales
| Name | Company | Catalog Number | Comments |
| BD FACS Canto II | BD BioSciences | 338960 | |
| Capillary electrophoresis instrument: Genomes Applied Biosystems 3730xl DNA Analyzer | Applied biosystems | 313001R | Service offered by the Centre for Applied Genomics (http://www.tcag.ca) |
| Citric acid | Sigma | 251275 | |
| Cytoseal 60, histopathology mounting medium | Fisher | 23244257 | |
| Eosin Y stock solution (1%) | Fisher | SE23-500D | |
| FCSalyzer - flow cytometry analysis software | SourceForge | - | https://sourceforge.net/projects/fcsalyzer/ |
| FFPE Qiagen kit | Qiagen | 80234 | |
| Forceps | Fine Science Tools | 11295-51 | For sectioning and for the cleaning process |
| Glacial Acetic Acid (Concentrated) | Sigma | A6283-500mL | |
| Glass coverslips: Cover Glass | Fisher | 12-541a | |
| Hematoxylin | Fisher | CS401-1D | |
| Highly deionized formamide: Hi-Di Formamide | Thermofisher | 4311320 | |
| IHC platform: Benchmark Ultra | Roche | - | |
| Kimwipes | Ultident | 30-34120 | |
| Microtome | Leica | RM2135 | |
| Microtome blades | Fisher | 12-634-1C | |
| Nitex filtering mesh, 48 microns | Filmar | 74011 | http://www.filmar.qc.ca/index.php?filet=produits&id=51&lang=en ; any other filter is suitable, but this is an inexpensive and effective option from a non-research company |
| p57 antibody | Cell Marque | 457M | |
| Pasteur pipette | VWR | 53499-632 | |
| PCR machine | Perkin Elmer, Applied Biosystems | GeneAmp PCR System 9700 | |
| PeakScanner 1.0 | Applied Biosystems | 4381867 | Software for genotyping analysis. |
| Pepsin from porcine gastric mucosa | Sigma | P7012 | |
| Polystyrene round-bottom tubes | BD Falcon | 352058 | |
| Positively charged slides: Superfrost Plus 25x75mm | Fisher | 1255015 | |
| PowerPlex 16 HS System | Promega Corporation | DC2102 | |
| Propidium Iodide | Sigma | P4864 | |
| Ribonuclease A from bovine pancreas | Sigma | R4875 | |
| Separation matrix: POP-7 Polymer | Thermofisher | 4352759 | |
| UltraPure Agarose | Fisher | 16500-500 | |
| Xylene | Fisher | X3P1GAL |
Referencias
- Szulman, A. E., Surti, U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. American Journal of Obstetrics & Gynecology. 132 (1), 20-27 (1978).
- Fukunaga, M., et al. Interobserver and intraobserver variability in the diagnosis of hydatidiform mole. The American Journal of Surgical Pathology. 29 (7), 942-947 (2005).
- Gupta, M., et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis for both recently trained and experienced gynecologic pathologists. The American Journal of Surgical Pathology. 36 (12), 1747-1760 (2012).
- Howat, A. J., et al. Can histopathologists reliably diagnose molar pregnancy?. Journal of Clinical Pathology. 46 (7), 599-602 (1993).
- Banet, N., et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Modern Pathology. 27 (2), 238-254 (2014).
- Lipata, F., et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstetrics & Gynecology. 115 (4), 784-794 (2010).
- Buza, N., Hui, P. Partial hydatidiform mole: histologic parameters in correlation with DNA genotyping. International Journal of Gynecologic Pathology. 32 (3), 307-315 (2013).
- Fisher, R. A., et al. Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Human Genetics. 82 (3), 259-263 (1989).
- Murdoch, S., et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 38 (3), 300-302 (2006).
- Parry, D. A., et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. American Journal of Human Genetics. 89 (3), 451-458 (2011).
- Nguyen, N. M., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Current Obstetrics and Gynecology Reports. 3, 55-64 (2014).
- Sebire, N. J., Savage, P. M., Seckl, M. J., Fisher, R. A. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 34 (1), 50-56 (2013).
- Nguyen, N. M., et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. Journal of Medical Genetics. 51 (9), 623-634 (2014).
- Brown, L., et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either "partial" or "complete" hydatidiform moles. International Journal of Gynecologic Pathology. 32 (4), 399-405 (2013).
- Colgan, T. J., Chang, M. C., Nanji, S., Kolomietz, E. A Reappraisal of the Incidence of Placental Hydatidiform Mole Using Selective Molecular Genotyping. The International Journal of Gynecological Cancer. 26 (7), 1345-1350 (2016).
- Murphy, K. M., McConnell, T. G., Hafez, M. J., Vang, R., Ronnett, B. M. Molecular genotyping of hydatidiform moles: analytic validation of a multiplex short tandem repeat assay. The Journal of Molecular Diagnostics. 11 (6), 598-605 (2009).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados